Increased TGFβ1 and SMAD3 Contribute to Age-Related Aortic Valve Calcification
 Mrinmay Chakrabarti1
Mrinmay Chakrabarti1  Aniket Bhattacharya1,2
Aniket Bhattacharya1,2  Mengistu G. Gebere1 John Johnson1 Zeeshan A. Ayub1 Ioulia Chatzistamou3
Mengistu G. Gebere1 John Johnson1 Zeeshan A. Ayub1 Ioulia Chatzistamou3  Narendra R. Vyavahare4
Narendra R. Vyavahare4  Mohamad Azhar1,5*
Mohamad Azhar1,5*- 1Department of Cell Biology and Anatomy, School of Medicine, University of South Carolina, Columbia, SC, United States
- 2Department of Neuroscience and Cell Biology, Child Health Institute of New Jersey Rutgers-Robert Wood Johnson Medical School, New Brunswick, NJ, United States
- 3Department of Pathology, Microbiology, and Immunology, School of Medicine, University of South Carolina, Columbia, SC, United States
- 4Biomedical Engineering, Clemson University, Clemson, SC, United States
- 5William Jennings Bryan Dorn VA Medical Center, Columbia, SC, United States
Aims: Calcific aortic valve disease (CAVD) is a progressive heart disease that is particularly prevalent in elderly patients. The current treatment of CAVD is surgical valve replacement, but this is not a permanent solution, and it is very challenging for elderly patients. Thus, a pharmacological intervention for CAVD may be beneficial. In this study, we intended to rescue aortic valve (AV) calcification through inhibition of TGFβ1 and SMAD3 signaling pathways.
Methods and Results: The klotho gene, which was discovered as an aging-suppressor gene, has been observed to play a crucial role in AV calcification. The klotho knockout (Kl–/–) mice have shorter life span (8–12 weeks) and develop severe AV calcification. Here, we showed that increased TGFβ1 and TGFβ-dependent SMAD3 signaling were associated with AV calcification in Kl–/– mice. Next, we generated Tgfb1- and Smad3-haploinsufficient Kl–/– mice to determine the contribution of TGFβ1 and SMAD3 to the AV calcification in Kl–/– mice. The histological and morphometric evaluation suggested a significant reduction of AV calcification in Kl–/–; Tgfb1± mice compared to Kl–/– mice. Smad3 heterozygous deletion was observed to be more potent in reducing AV calcification in Kl–/– mice compared to the Kl–/–; Tgfb1± mice. We observed significant inhibition of Tgfb1, Pai1, Bmp2, Alk2, Spp1, and Runx2 mRNA expression in Kl–/–; Tgfb1± and Kl–/–; Smad3± mice compared to Kl–/– mice. Western blot analysis confirmed that the inhibition of TGFβ canonical and non-canonical signaling pathways were associated with the rescue of AV calcification of both Kl–/–; Tgfb1± and Kl–/–; Smad3± mice.
Conclusion: Overall, inhibition of the TGFβ1-dependent SMAD3 signaling pathway significantly blocks the development of AV calcification in Kl–/– mice. This information is useful in understanding the signaling mechanisms involved in CAVD.
Introduction
Calcific aortic valve disease (CAVD) is a progressive heart disease in which aortic valve sclerosis progresses to aortic valve stenosis with severe calcification and impaired leaflet function (1–5). The aortic valve (AV) calcification affects 25% of the population over 65 years of age and about 50% of those over 85 years (2). Surgical valve replacement is the most effective treatment for valvular heart disease (6). Old age and chronic kidney disease (CKD) are important risk factors for CAVD (1). The serum levels of klotho (KL) decrease in CKD patients with heart valve calcification and therefore klotho is considered an independent risk factor for CAVD in CKD (7). The klotho gene (Kl) is identified as an anti-aging gene in mice that prolongs the life span (8). Since its serendipitous discovery, klotho has drawn significant attention owing to its potential role in aging, chronic kidney disease (CKD), and several cardiovascular diseases (9, 10). Earlier investigations indicated that klotho expresses predominantly in the distal tubular epithelial cells of the kidney (11, 12). Another study has confirmed klotho expression in human artery and cardiac myocytes (10). Functions of klotho include regulation of energy metabolism, anti-inflammatory and anti-oxidative effects, modulation of ion transport, and regulation of mineral metabolism (13). Several reports also endorsed the protective role of klotho in several organs as well as reversal of disease mechanisms including cardiovascular disease (14). The Kl encodes a single-pass transmembrane protein of 135 kDa. The klotho protein is clipped on the cell surface by membrane-anchored proteases and the entire extracellular domain (∼130 kDa) is released into the systemic circulation (8, 15, 16). Thus, klotho protein exists in two forms: membrane-bound klotho and secreted klotho (10, 17). More probably, membrane klotho solely serves as a co-receptor for the binding to fibroblast growth factor 23 (FGF23) (18). FGF23 is a bone-derived hormone that acts on kidney to stimulate phosphate elimination into urine and inhibit vitamin D synthesis, thereby modulating negative phosphate balance (19, 20). One significant characteristic of FGF23 is that it has very poor affinity to FGF receptors (21, 22). Binding of membrane klotho generates a constitutive binary complex with FGF receptors (FGFRs) which creates a de novo high-affinity binding site for FGF23 (22). Thus, FGF23 requires membrane klotho to bind to its cognate FGF receptors and exert its biological activity (22). The klotho (Kl–/–) and/or Fgf23 (Fgf23–/–) knockout mice exhibit many key aspects associated with human CAVD including, premature aging, kidney disease, increased serum phosphate levels (i.e., hyperphosphatemia), and increased osteogenic gene expression (7, 23, 24). The Kl–/– mice die by 12 weeks of age due to multiple age-related complications. The Kl–/– mice develop calcific nodules in the AV hinge and aortic annulus, but they do not show significant AV leaflet thickening, extracellular matrix (ECM) disorganization, or inflammation (25). In addition, the Kl–/– mice also develop ectopic calcification of aorta and kidneys (26). It has been reported that valve interstitial cells (VIC) in the calcified AV hinge in Kl–/– mice become activated and express several pro-calcific markers, including COX2, RUNX2, osteopontin (OPN1/SPP1), and alkaline phosphatase (ALP). Recently, Kl–/– mice have been successfully used in preclinical testing of potential new drugs for AV calcification (27). Since CKD is associated with klotho and human CAVD (28) and the AV calcification in Kl–/– mice resembles human CAVD (29), the Kl–/– mice are useful mouse models for investigating the mechanisms involved in pathogenesis of CAVD (29, 30).
Surgical specimens of the aortic valve (AV) obtained from older patients with CAVD have increased levels of transforming growth factor beta1 (TGFβ1) (31–37). Elevated TGFβ1 levels are frequently observed during vascular calcification and CKD (38–40) and contribute to the progression of calcification of cardiovascular tissues (41, 42). Both AV calcification and vascular calcification are regarded as the crucial risk factors in CKD and are associated with cardiovascular and all-cause morbidity and mortality (43) (44). The AV calcification is an active process with some parallels to physiological bone formation (3, 45). A critical role is attributed to vascular smooth muscle cells (VSMC) and VIC, which can differentiate and convert into myofibroblasts, osteoblast, and chondroblast-like phenotypes (46, 47). Osteoblasts or chondrogenic VSMCs/VICs actively promote valvular or vascular tissue mineralization (25, 48). Although the intracellular signaling pathways that control this trans-differentiation into myofibroblasts, osteoblasts, or chondrocytes are yet not understood completely (49). Human myxomatous valve disease (MMVD) is characterized by increased VIC proliferation, leaflet thickening, increased proteoglycan expression, and ECM remodeling (50). Cell proliferation is unaffected and αSMA expression is reduced in calcified AV of Kl–/– mice (25), suggesting that VIC activation or MMVD is not present in Kl–/– mice. TGFβ1 plays a crucial role in both myofibroblasts and osteogenic trans-differentiation of the VSMCs/VICs and induces cellular senescence through the upregulation of plasminogen activator inhibitor (Pai1) (38, 47). Moreover, TGFβ1-dependent osteoinductive signaling involves altered expression of the chondrogenic transcription factor SRY-Box 9 (SOX9), which is involved in AV calcification (51–54).
Increased levels of TGFβ ligands can contribute to cell degeneration, inflammation, metabolic malfunction, tissue fibrosis, and calcification. While the roles of TGFβ signaling depend on cellular contexts, age-related changes are also a potential context, and therefore the relationship between TGFβ signaling and klotho, which is involved in cellular senescence and aging-related diseases such as CAVD, warrants serious attention. There are many investigations that showed that Kl–/– mice can induce calcification in aortic valves (25, 27, 55, 56). The AV of Kl± mice do not show any calcification, although a high-fat diet resulted in collagen-I deposition and fibrosis of the aortic valve cusps on the aortic side (57). The signaling mechanisms of AV calcification in klotho deficient mice are still unclear and require further investigations. In this work, we investigated the impact of reduced TGFβ signaling on the AV calcification in Kl–/– mice. We observed that partial inhibition of TGFβ signaling through haploinsufficiency of Tgfb1 and Smad3 improves the pathological condition of the AV calcification in klotho-deficient mice.
Materials and Methods
Ethics Statement
All animal procedures were performed according to the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of South Carolina. Mice were euthanized by an overdose of isoflurane in a sealed container as approved by the IACUC.
Mouse Strains
Kl± (B6;129S5-KlTM 1Lex/Mmucd, Stock# 011732) mice were obtained from the Mutant Mouse Resource and Research Centers (MMRRC) supported by the NIH. These mice were first backcrossed on to C57BL/6 background for more than nine generations. First, the Kl±; Tgfb1± and Kl±; Smad3± mice were generated in our mouse facility by the genetic crossing of the Kl± (B6) to Tgfb1± (50% 129SvJ and 50% CF-1) (58) and Smad3± (129/SvJ) (59) mice. Self-crossing of Kl±; Tgfb1± and Kl±; Smad3± male and female mice resulted in the generation of wild-type control (Kl+/+, Kl+/+; Tgfb1+/+, and Kl+/+; Smad3+/+), Kl–/–, Kl–/–; Tgfb1±, and Kl–/–; Smad3± mice. Before starting each study, we collected the tail at the age of 3 weeks and genomic DNAs were extracted for each animal and confirmed the genotype using gene-specific primers for Kl, Tgfb1, and Smad3 (Table 1). PCR genotyping was done as described earlier (60–62).

Table 1. Primer sequence for PCR amplification.
Tissue Collection and Processing for Histology
Wild-type controls Kl–/–, Kl–/–; Tgfb1±, and Kl–/–; Smad3± mice were sacrificed, and aortic valve tissues were collected for histological, immunohistochemical, morphometric, and molecular analyses. The whole heart of each mouse was perfused with 1XPBS and later fixed in 4% paraformaldehyde in PBS (Fisher Scientific, Waltham, MA, United States) for 48 h. Then, tissues were dehydrated in 70, 95, and 100% ethanol (Fisher Scientific, Waltham, MA, United States) and cleared with xylene (Fisher Scientific, Waltham, MA, United States). Paraffin-embedded hearts were sectioned using an RM2245 Leica microtome (Leica Biosystems, Buffalo Grove, IL, United States) and 7 μm thick serial sections through the aortic roots including annulus, aortic leaflets and hinge, and a portion of the sinus of Valsalva were collected for various histological and morphometric evaluation. Multiple serial sections from each animal were used for quantifying structural or histological changes using the NIH Image J (Fiji) software.
Histology
All histological staining procedures were performed according to the protocol provided by the manufacturers. Before various histological staining procedures, the tissues were de-paraffinized and rehydrated. Hematoxylin and eosin staining (H&E staining) was carried out using Harris’ hematoxylin (Cat # HS-400) and Eosin (Sigma, Cat # HT110380). Tissue sections were kept in alizarin red stain (American MasterTech Scientific, Lodi, CA, United States) for 5 min for detection of calcium deposits. For alizarin red staining, tissue sections were fixed and dehydrated with –20°C cold acetone (Thermo Fisher Scientific, Waltham, MA, United States) and cleared with an acetone/xylene solution (50% acetone and 50% Safe Clear II Xylene substitute, both purchased from Thermo Fisher Scientific, Waltham, MA, United States). Alcian blue stain kit (catalog # KTABP2.5) was used to detect proteoglycans. The Verhoeff’s Elastin staining kit (Cat# KTVEL) was used for elastic fiber (black color) and collagens (red color) staining. Collagens were also detected by using the Manson’s Trichrome 2,000 stain kit (cat # KTMTR2PT). All kits for detecting proteoglycans, collagens, and elastin fibers were purchased from the American MasterTech Scientific (Lodi, CA, United States). At the end of each staining, the tissue slides were mounted with a permanent mounting medium (Vector Laboratories, Burlingame, CA, United States). All tissue sections were subsequently visualized and photographed in low and high magnifications under bright field optics on the Nikon Optiphot-2 (equipped with AxioCam MRc Camera) and EVOS TM FL Auto Imaging System (Thermo Fisher Scientific, Inc., Grand Island, NY, United States). Morphometric quantification of signal intensity was done on multiple serial sections from each animal per group by using the NIH- Image J (Fiji) software.
Immunohistochemistry
Paraformaldehyde-fixed paraffin-embedded serial sections representing annulus, aortic leaflets and hinge, and a portion of the sinus of Valsalva were used for immunohistochemistry. Sections were de-paraffined and hydrated with two changes in 1X PBS and one change in deionized water (5 min each). Heat-mediated antigen retrieval was performed by dipping the slides in a mildly boiling 1X citric acid buffer (catalog no. S1700; Agilent Dako, Santa Clara, CA, United States) for 10 min in a microwave, cooled to room temperature, and rinsed in PBS. Endogenous peroxidases were blocked with freshly prepared 0.5% H2O2/methanol for 30 min, followed by non-specific epitope blocking with 5% goat serum/0.1% Tween/0.02% sodium azide in PBS for 20 min. Avidin and Biotin blocking was done as per the manufacturer’s recommendation (Cat# SP-2001, Vector Labs, Burlingame, CA, United States), followed by overnight incubation at 4°C in anti-pSMAD2 (1:3000, Millipore, Burlington, MA, United States) (61). Slides were then washed and incubated with appropriate biotinylated secondary antibody (1:200) for 30 min, followed by Avidin-Biotin complex (Cat# PK-6100, Vectastain Elite ABC HRP kit) for 30 min, washed in PBS, and finally developed with DAB/H2O2. Nuclei were counterstained with hematoxylin and sections were dehydrated through graded ethanol series, cleared in xylene, and mounted. Both low- and high-magnification images were taken under bright field optics on the Nikon Optiphot-2 (equipped with AxioCam MRc Camera). Morphometric quantification of pSMAD2-stained average area (μm2) was done on multiple serial sections from each animal per group by using the NIH- Image J software.
RNA Isolation, cDNA Synthesis, and Quantitative PCR
The AV tissue was dissected manually from wild-type, Kl–/–, Kl–/–; Tgfb1±, and Kl–/–; Smad3± mice. The AV tissue contained aortic roots including annulus, aortic leaflets and hinge, and a portion of the sinus of Valsalva. Five individual biological samples from each group were analyzed for gene expression study. Total RNA was isolated from the AV tissue of wild-type and Kl–/–, Kl–/–; Tgfb1±, and Kl–/–; Smad3± mice using Trizol (Invitrogen/ThermoFisher, Grand Island, NY, United States) and miRNeasy micro kit (Qiagen, Germantown, MD, United States) according to the manufacturer’s protocols. cDNA was generated from 500 ng total RNA using an RT-PCR kit according to the instructions provided by the manufacturer (Bio-Rad Laboratories, Inc.). cDNA was then diluted 10 times and later subjected to quantitative PCR amplification (Bio-Rad CFX) using pre-validated gene-specific primers procured from the vendor (Bio-Rad Laboratories, Inc., Hercules, CA, United States) (Table 2). Approximately, 10 ng of cDNA was used for each 20 μl qPCR reaction. Following qPCR analyses, the cycle count threshold (Ct) was normalized to species-specific housekeeping genes (B2m; purchased from Bio-Rad, Inc.) and the 2^–ΔCt values were determined and graphically presented. Statistically significant differences in gene expression levels were determined using Student’s t-test, indicated in the figure legends, on at least three or more independent experiments with p < 0.05 considered significant.

Table 2. List of qPCR primers.
Western Blot Analysis
Three independent “pooled” samples of the AV tissue from each group of mice were used for western blotting. The AV tissues in each sample were “pooled” from two individual mice for each genotype (wild-type, Kl–/–, Kl–/–; Tgfb1±, and Kl–/–; Smad3±). Thus, each independent pooled sample consists of two biological replicates (n = 6 per genotype). The AV tissue contained aortic roots including annulus, aortic leaflets and hinge, and a portion of the sinus of Valsalva. The AV tissue was cut into small pieces and homogenized using Wheaton tapered tissue grinders (Thermo Fisher Scientific, Rockford, IL, United States) in M-PER mammalian protein extraction reagent (Thermo Fisher Scientific) with a complete mini protease inhibitor cocktail (Sigma-Aldrich, St. Louis, Louis, MO, United States) and Halt protease and phosphatase inhibitor single-use cocktail (Thermo Fisher Scientific, Rockford, IL, United States) as per the manufacturer’s protocol. Homogenized tissue lysates were subjected to brief sonication for 20 s on ice and then kept at room temperature for 20 min. Then, centrifugation was performed at 15,000 rpm for 20 min at 4°C and the supernatants were collected. Total protein concentration in the supernatant was determined using the Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL, United States). Samples were stored at –80°C until further use. Western blotting was performed with equal amounts of protein samples and the primary IgG antibodies against phospho-SMAD2 (Cell Signaling Technology, Danvers, MA, United States Cat #3108), SMAD2 (Cell Signaling, Cat #5339), phospho-SMAD3 (Cell Signaling, Cat #9520), SMAD3 (Cell Signaling, Cat #9523), phospho-SMAD1/5 (Cell Signaling, Cat #9516), SMAD1/5 (Cell Signaling, Cat #9743), phospho-p38 (Cell Signaling, Cat #4511), p38 (Cell Signaling, Cat #8690), phospho-ERK1/2 (Cell Signaling, Cat #4370), and ERK1/2 (Cell Signaling, Cat #4695) at a dilution of 1:1000. Primary IgG antibodies against all these proteins were purchased from Cell Signaling Technology, Inc. (Danvers, MA, United States). The horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary IgG antibody (Cell Signaling, Cat # 7074) was used at 1:5000 dilution to detect a primary IgG antibody. Western blots were incubated with Clarity western ECL detection reagents (Bio-Rad Laboratories, United States) and exposed to X-OMAT AR films (Eastman Kodak, Rochester, NY, United States) for autoradiography. The autoradiograms were scanned on an EPSON Scanner using Photoshop software (Adobe Systems, Seattle, WA, United States). β-actin, clone AC-15, monoclonal primary antibody (Sigma-Aldrich, St. Louis, MO, United States) was used as a loading control to compare equal loading in the SDS-PAGE. We quantified our results via densitometric analysis using NIH Image J (Fiji) software. Quantitative densitometric analysis normalized the expression levels of phosphorylated proteins with their non-phosphorylated/total forms or β-actin. Microsoft Excel was used for recording and managing the raw data. Statistics were performed using pair-wise comparisons between the groups, utilizing analysis of variance and unpaired two-tailed Student’s t-test (GraphPad Prism 9 Software, San Diego, CA). Data were reported as means ± SD of the mean. Probability values < 0.05 were considered significant.
Results
Increased Tgfb1 Expression and SMAD3 Activation Are Associated With the Calcification of the Aortic Valve in Klotho Knockout Mice
We collected tissues from the aortic valve from 8- to 10-week-old wild-type and Kl–/– mice and performed alizarin red staining of tissue sections to determine calcification in the aortic valve area. Our alizarin red staining confirmed the presence of calcific nodules in the AV hinge and aortic annulus of the Kl–/– mice (n = 12) (Figures 1A–D). There was no AV calcification seen in the control mice, which included Kl+/+ and Kl± mice. Immunohistochemistry analysis indicated that increased phosphorylated SMAD2 (pSMAD2) was associated with AV calcification in the hinge and aortic annulus (n = 6) (Figures 1E–I). We tested if the Tgfb1 mRNA level was upregulated in the AV tissue of Kl–/– mice by qPCR analysis. The data indicated significant upregulation of Tgfb1 mRNA expression (n = 5, p = 0.031) in Kl–/– mice compared to the wild-type control (Figure 1J). Next, we determined if the increased Tgfb1 mRNA expression level is consistent with the induction of the downstream TGFβ signaling molecules at the protein level. We analyzed the downstream TGFβ signaling pathway molecules in tissue samples collected from the AV tissue of wild-type and Kl–/– mice. Since phosphorylation of SMAD2 and SMAD3 is typically used as a surrogate for TGFβ signaling, western blot analysis was used to quantify both phosphorylated and total (no-phosphorylated) SMAD and SMAD3. The data showed statistically significant induction of activated forms of the SMAD2 (i.e., pSMAD2) and SMAD3 (i.e., pSMAD3) (TGFβ-specific SMADs), in Kl–/– mice when compared to wild-type mice (n = 6) (Figures 1K,L).
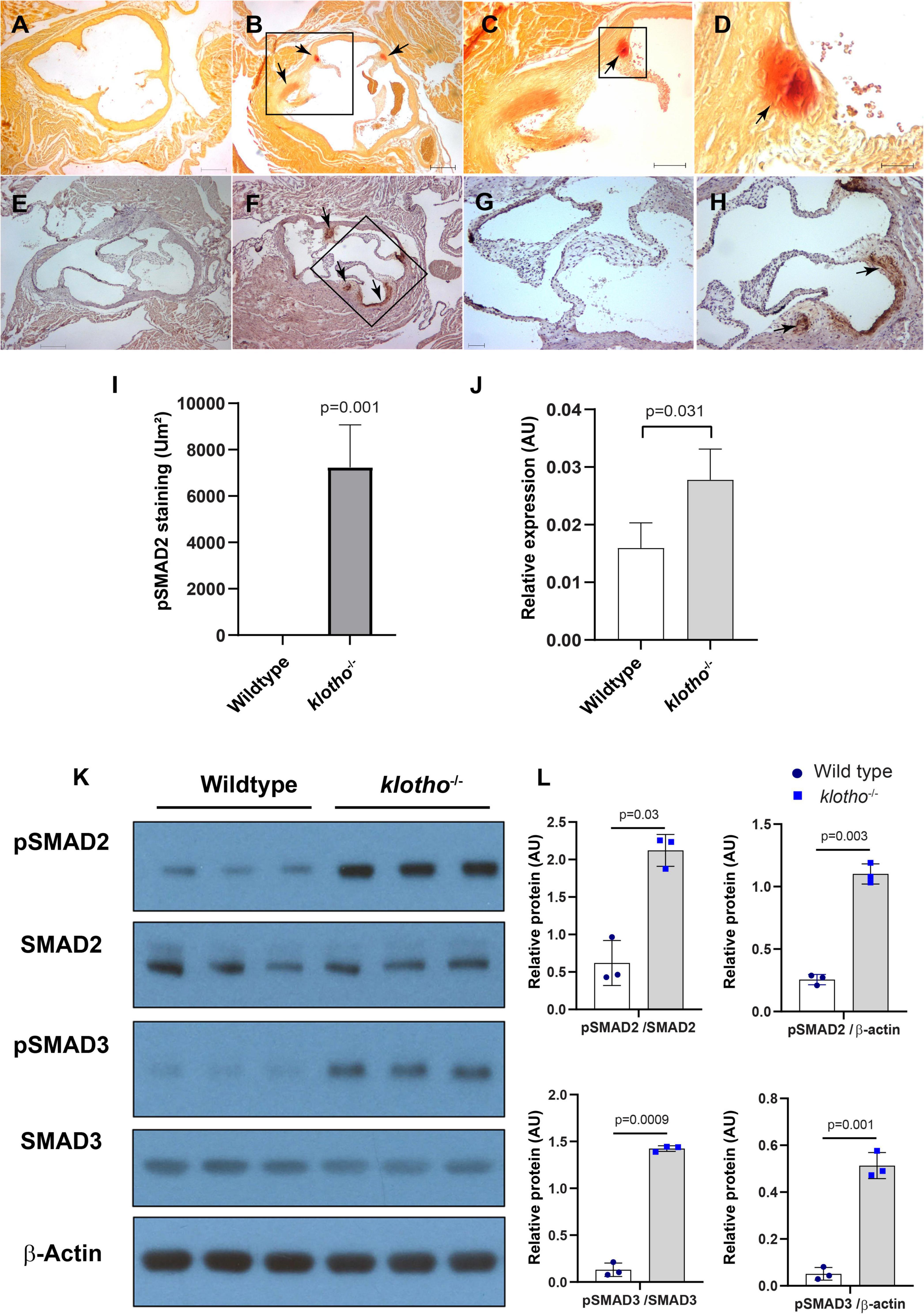
Figure 1. The klotho (Kl) genetic deletion leads to calcification in the aortic valve hinge and annulus with upregulation of Tgfb1 and TGFβ-dependent SMAD2 signaling in 10–12-week-old mice. (A–D) Alizarin red staining of wild-type (A) and Kl–/– (B–D) mice. Scale bars = (A–B) 200 μm; (C) 100 μm; (D) 50 μm. (E–I) Immunohistochemistry showing localized pSMAD2 levels in AV hinge and aortic annulus of wild-type (E,G) and Kl–/– (F,H). The pSMAD2-stained area was quantified using NIH Image J software (I). (J) qPCR study to quantify Tgfb1 expression in the pooled and micro-dissected tissue samples of AV and annulus from wild-type and Kl–/– mice. (K) Western blotting analyses showing levels of phosphorylated SMADs (p SMAD2, pSMAD3), total SMADs (SMAD2, SMAD3), and β-actin in micro-dissected and pooled tissue samples containing AV and annulus from wild-type and Kl–/– mice. (L) Densitometric analysis quantifying band intensities of western blots. The densitometry graphs of pSMAD2 and pSMAD3 were normalized with total or non-phosphorylated form of the proteins or β-actin. Values indicate mean ± SD, and significant “p-values” between wild-type and Kl–/– groups were given on the top of histograms.
Genetic Inactivation of TGFβ Signaling Pathway Components Improved Structural Features and Extracellular Matrix Organization of Aortic Valve in Klotho-Deficient Mice
Histological staining was carried out to examine the morphological, cellular, and structural changes in the AV tissue of 8–10-week-old Kl–/– mice following the partial genetic deletion of TGFβ1 and SMAD3 genes (Figures 2A–H). Histological examination of H&E-stained serial tissue sections confirmed calcified nodules in the AV hinge and annulus in the Kl–/– mice (n = 12) (Figures 2A,B). Morphometric quantitative analysis indicated no significant changes in the overall thickening of AV leaflets in Kl–/– mice compared to wild-type mice (n = 6) (Figure 2I). Histological and morphometric evaluation of Kl–/–;Tgfb1± mice showed partial improvement in the overall tissue structure of the AV hinge and annulus compared to Kl–/– mice (n = 6) (Figures 2A–C,I). Similar analysis of Kl–/–;Smad3± mice revealed a complete rescue of the AV hinge and aortic annulus (Figures 2A–D,I). Alcian blue staining was used to observe chondrogenic differentiation and proteoglycans or glycosaminoglycans (GAGs) distribution in the AV hinge and aortic annulus in all four groups of mice (n = 6–12). The data indicated abnormal proteoglycans in the AV hinge and aortic annulus in both Kl–/– (n = 12) and Kl–/–;Tgfb1± (n = 6) mice compared to wild-type or Kl–/–;Smad3± mice (n = 12), but morphometric quantification did not reveal any significant changes in the overall proteoglycans content (n = 6) (Figures 2E–H,J). Finally, we determined collagen and elastic fibers’ organization through the VVG staining of serial sections (n = 6–12) (Figures 3A–J). The quantitative analysis did not reveal any significant changes in overall collagen content (Figure 3I). Histological assessment of high-powered images and morphometric evaluation showed dysregulated elastic fiber organization in the AV hinge of Kl–/– mice (n = 12), which was significantly rescued by partial genetic deletion of Smad3 (n = 12) but not Tgfb1 (n = 6) (Figures 3A–H,J).
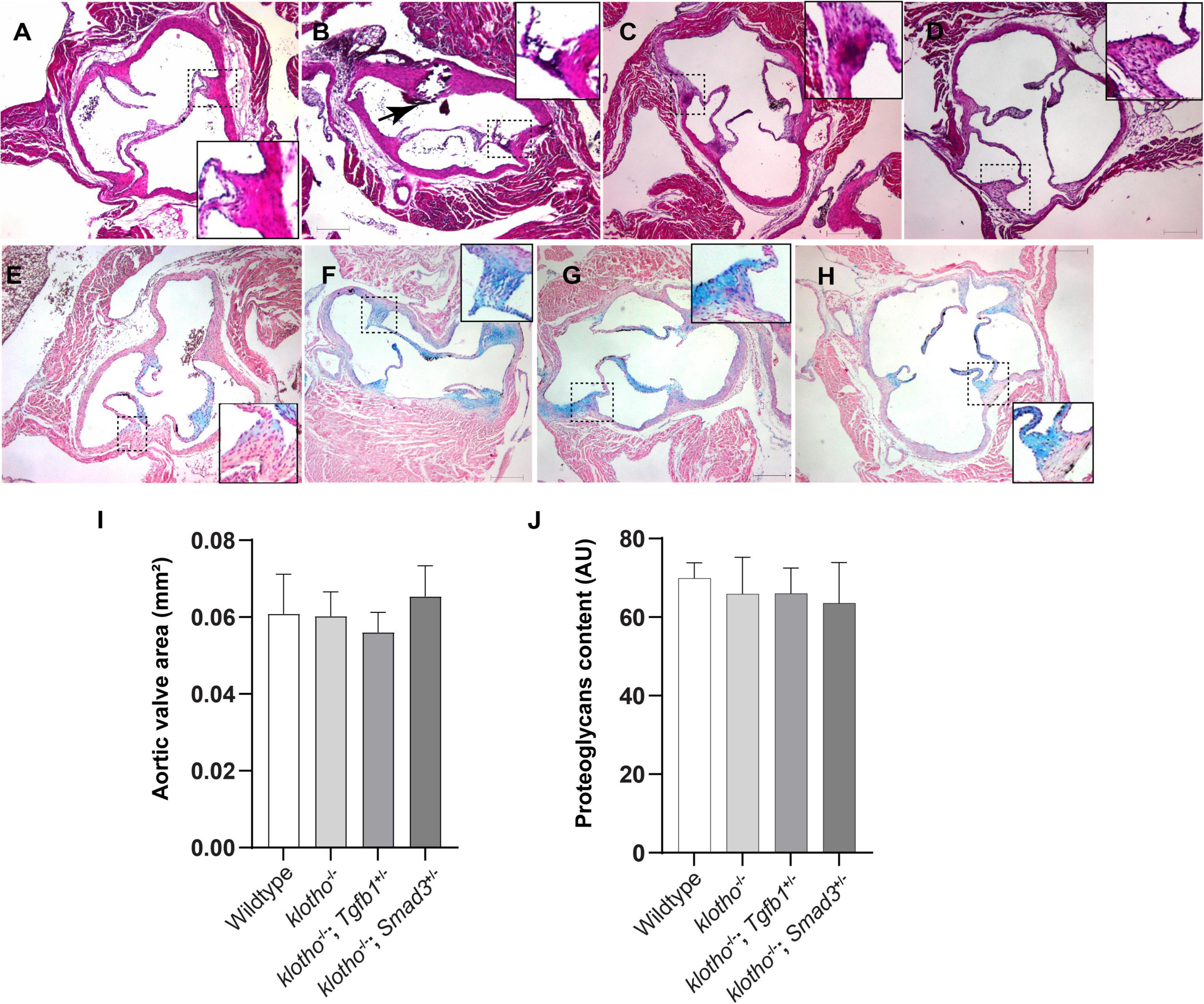
Figure 2. Effect of lowering of TGFβ1 and SMAD3 on AV structure and proteoglycan (GAG) distribution in 10- to 12-week-old Kl–/– mice. (A–D) Hematoxylin and eosin (H&E) staining and (E–H) Alcian blue staining of wild-type, Kl–/–, Kl–/–;Tgfb1±, and Kl–/–;Smad3± mice showing aortic valve histology and proteoglycans distribution. A magnified view showing further details of the boxed region is presented for each image. (I–J) Quantitative analysis of AV area in H&E-stained (I) and proteoglycans content in alcian blue-stained (J) images. Quantification of AV area and GAG content was done on multiple serial sections from each animal per group by using NIH- Image J software. Values indicate mean ± SD (n = 5). Scale bars = (A–H) 200 μm.
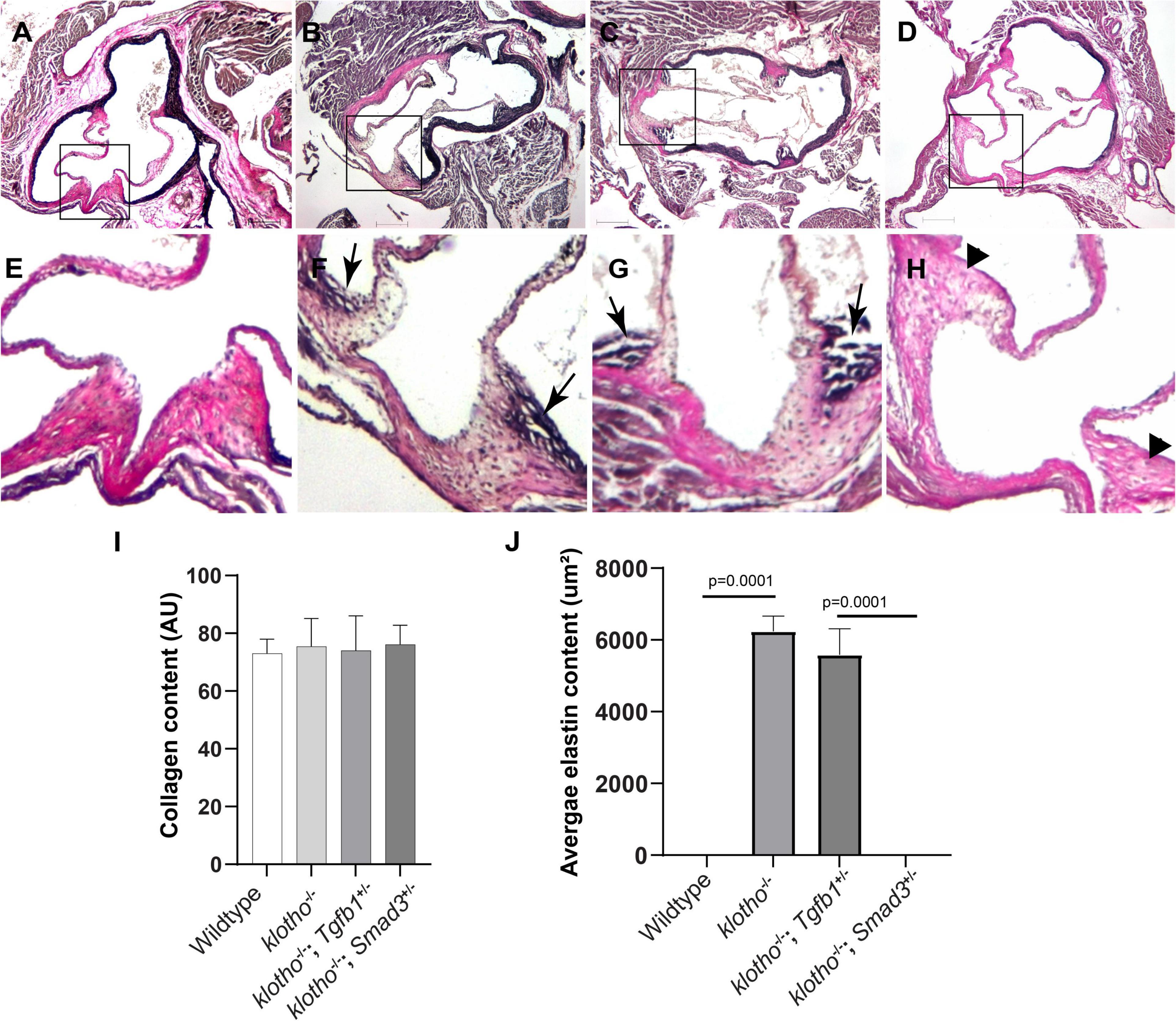
Figure 3. Effect of Tgfb1 and Smad3 heterozygous deletion on collagen and elastin fibers in the aortic valve of 10–12-week-old Kl–/– mice. (A–H) Verhoeff-Van Gieson (VVG) staining of tissue sections through the aortic valve and annulus of wild-type (A,E), Kl–/– (B,F), Kl–/–;Tgfb1± (C,G), and Kl–/–;Smad3± (D,H) mice. Magnified images of the boxed area (A–D) are given in (E–H). Arrows indicate the presence of abnormal elastin fibers in Kl–/– (F) and Kl–/–;Tgfb1± (G) mice. VVG stains elastin in black and collagen fibers in red color. (I) Histogram showing total collagen content in the aortic valve, including leaflets, annulus, and AV hinge. Quantification of collagen fibers content was done by measuring the intensity of collagen staining on multiple serial sections from each animal per group by using NIH-Image J software. Values indicate mean ± SD (n = 5). (J) Histogram showing the average area containing the elastin fibers in the AV hinge. Quantification of elastin fibers content was done by measuring the average area in the AV hinge with elastin staining on multiple serial sections from each animal per group by using NIH-Image J software. Values indicate mean ± SD (n = 5), and significant “p-values” were given on the top of histograms. Scale bars: (A–H) 200 μm.
Haploinsufficiency of Tgfb1 and Smad3 Significantly Reduced Vascular Calcification in Klotho-Deficient Mice
We generated Kl–/–;Tgfb1± and Kl–/–;Smad3± mice to confirm the effect of TGFβ1 and canonical SMAD3-dependent TGFβ signaling pathways on AV calcification in klotho-deficient mice. Histological and quantitative analyses of alizarin red-stained serial sections through aortic roots including annulus, aortic leaflets and hinge, and a portion of the sinus of Valsalva of the 8- to 10-week-old wild-type, Kl–/–, Kl–/–;Tgfb1± and Kl–/–;Smad3± were performed (n = 6–12) (Figures 4A–I). There was no AV calcification in wild-type mice (Figures 4A,E). The Kl–/– mice showed calcific nodules in the AV hinge (n = 12, p = 0.002) (Figures 4A,B,E,F,I). Interestingly, the overall extent of the calcification in the AV hinge was less in Kl–/–;Tgfb1± mice compared to the Kl–/– mice (n = 6, p = 0.01) (Figures 4A–C,E–G,I). Importantly, the AV calcification was almost completely rescued in Kl–/–;Smad3± mice (n = 12, p = 0.002) (Figures 4A–I).
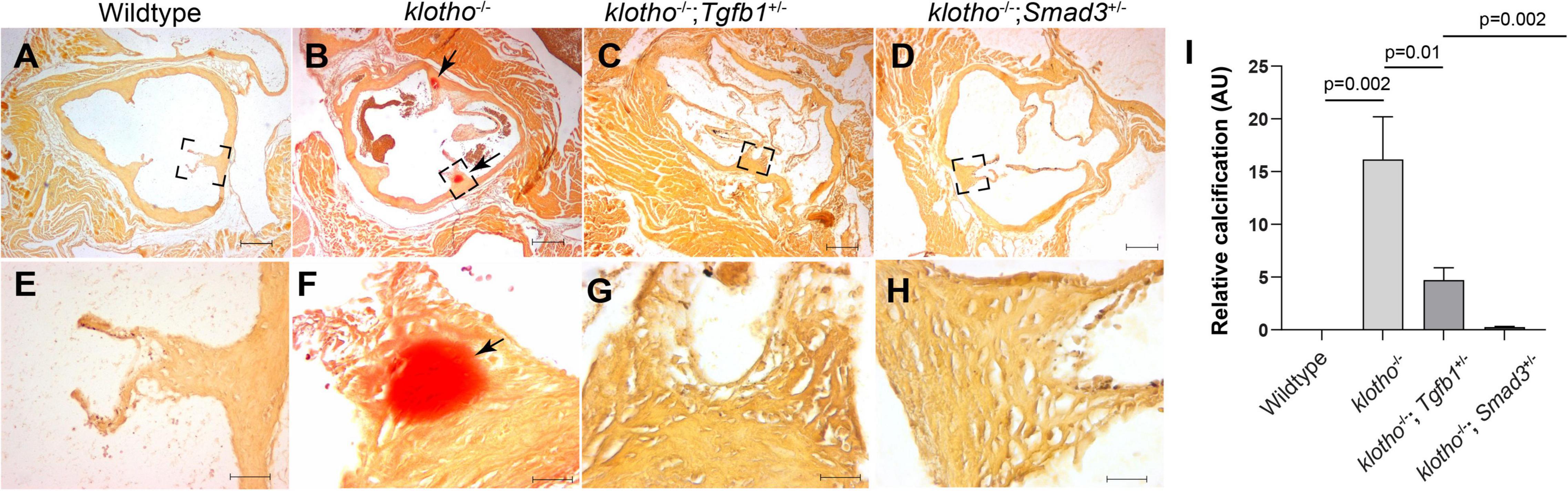
Figure 4. Partial genetic inactivation of Tgfb1 and Smad3 inhibits AV calcification in 10–12-week-old Kl–/– mice. (A–H) Alizarin red staining of aortic valve in wild-type (A,E), Kl–/– (B,F), Kl–/–;Tgfb1± (C,G), and Kl–/–;Smad3± (D,H) mice. Images (E–H) are the magnified views of the corresponding boxed region (A–D). Scale bars = (A–D), 200 μm; (E–F), 50 μm. (I) Quantitative analysis of calcification. Quantification of calcification was done on multiple serial sections from each animal per group by using NIH-Image J software. Values indicate mean ± SD, and significant “p-values” were given on the top of histograms.
Heterozygous Deletion of Tgfb1 and Smad3 Blocks the Expression of Genes Involved in Aortic Valve Calcification
We have investigated the impact of partial genetic deletion of Tgfb1 and Smad3 on the expression of genes involved in TGFβ (Tgfb1, Pai1) and BMP (Bmp2, Alk2) signaling and osteoblast differentiation (Spp1, Runx2) during CAVD development and progression in Kl–/– mice. The qPCR analysis was performed on aortic valve tissue containing aortic roots including annulus, sinus of Valsalva, and aortic leaflets and hinge from individual 8- to 10-week-old wild-type, Kl–/–, Kl–/–;Tgfb1±, and Kl–/–;Smad3± mice (n = 5) (Figure 5). We observed significant increase in the Tgfb1 (p = 0.03), Pai1 (p = 0.0001), Bmp2 (p = 0.002), Alk2 (p = 0.0004), Spp1 (p = 0.0003), and Runx2 (p = 0.01) mRNA expression in the AV tissue from the Kl–/– mice compared to wild-type mice (Figure 5). Partial genetic inhibition of Tgfb1 and Smad3 significantly inhibited the expression of all these genes. Heterozygous Smad3 deletion was observed to be more potent in reducing the expression of Pai1 and Spp1 genes in Kl–/– mice compared to partial Tgfb1 ligand inhibition (Figures 5C,D).
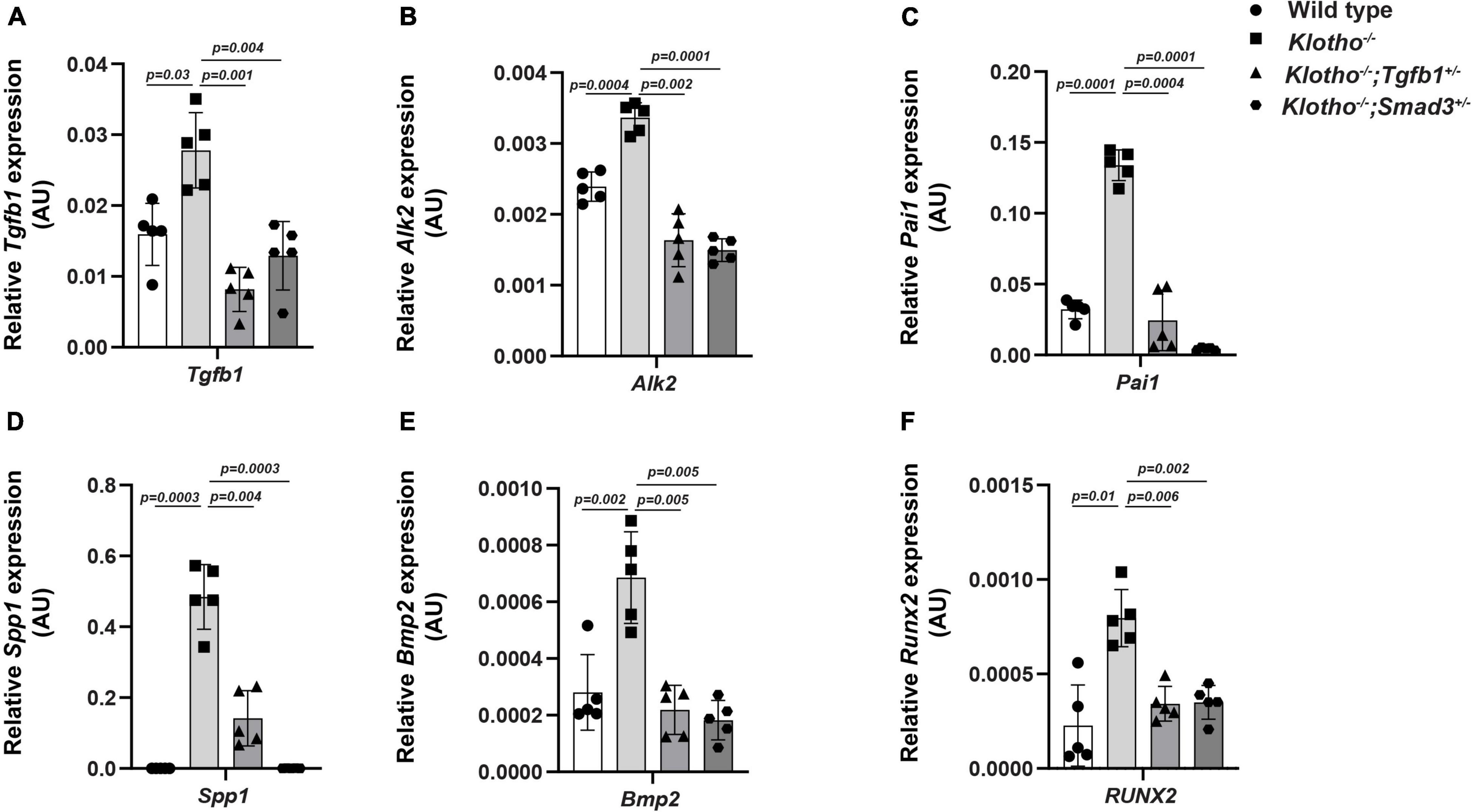
Figure 5. Effect of partial genetic deletion of Tgfb1 and Smad3 on key genes involved in TGFβ and BMP signaling, and AV calcification. The qPCR analysis of selected genes in micro-dissected pooled tissue samples representing AV leaflets and hinge and aortic annulus of wild-type, Kl–/–, Kl–/–;Tgfb1±, and Kl–/–;Smad3± mice. Expression levels of Tgfb1 (TGFβ ligand) (A), Alk2 (BMP Type I receptor) (B), Pai1 (TGFβ target gene) (C), Spp1 (calcification marker) (D), Bmp2 (BMP ligand) (E), and Runx2 (osteoblast differentiation marker) (F) were quantified. Triplicate samples from each of the five different biological replicates (n = 5) were analyzed. Quantitative data are shown as mean ± SD (n = 5). Two-sided Student’s t-test (unpaired) was used. Significant comparisons are indicated on the top of each histogram.
Partial Genetic Deletion of Tgfb1 and Smad3 Blocks Aortic Valve Calcification by Reducing Both Canonical and Non-canonical TGFβ Signaling Pathways
We observed significant induction of TGFβ receptor-dependent phosphorylation of serine/threonine residues of TGFβ SMADs (i.e., pSMAD2 and pSMAD3), TGFβ/BMP SMAD1/5 (i.e., pSMAD1/5), and non-SMAD pathways (i.e., p38 MAPK, ERK1/2 MAPK) in the AV tissue from the Kl–/– mice (n = 6) (Figure 6). Our immunoblot analyses of AV tissues from the wild-type control, Kl–/–, Kl–/–;Tgfb1±, and Kl–/–;Smad3± mice indicated that haploinsufficiency of Tgfb1 and Smad3 reduced the activation of these molecules in the Kl–/– mice (Figure 6A). There was no significant change in the total amount of the SMAD2, SMAD3, SMAD1/5, p38, ERK1/2 (ERK1 (top band), 44 KDa; ERK2 (bottom band), 42 KDa), and β-actin proteins in the wild-type control, Kl–/–, Kl–/–;Tgfb1±, and Kl–/–;Smad3± mice (Figure 6A). Quantitative densitometric analysis comparing the expression levels of phosphorylated proteins with their non-phosphorylated/total forms or β-actin revealed similar results (Figure 6B). Collectively, partial Smad3 deletion was more potent than heterozygous deletion of Tgfb1 in reducing the activation of SMAD2/3/5, p38, and ERK1/2 MAPK during aortic valve calcification in klotho-deficient mice (Figures 6A,B).
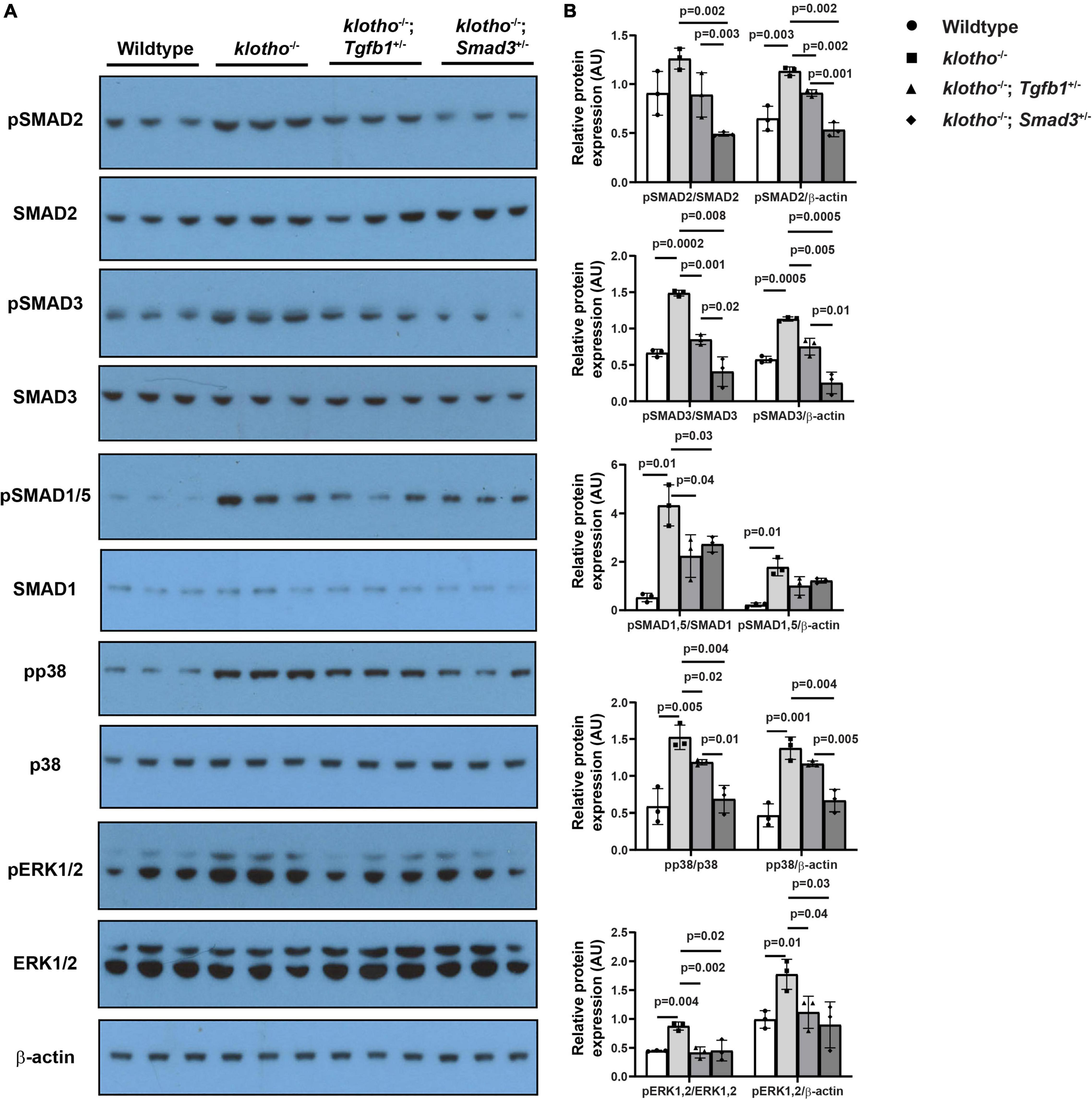
Figure 6. Haploinsufficiency of Tgfb1 and Smad3 inhibits canonical and non-canonical TGFβ signaling in the aortic valve of Kl–/– mice. (A) Western blot analysis on three different micro-dissected pooled tissues, from two to three hearts/sample, representing AV leaflets and hinge area and aortic annulus of 10–12-week-old wild-type, Kl–/–, Kl–/–;Tgfb1±, and Kl–/–;Smad3± mice showing protein levels of the phosphorylated forms of canonical and non-canonical pathway molecules (pSMAD2, pSMAD3, pSMAD1/5, pp38, and pERK1/2) and their non-phosphorylated/total forms (SMAD2, SMAD3, SMAD1/5, p38, and ERK1/2). A common and independent β-actin blot (bottom) was also used for normalizing the data. (B) Densitometric quantification of phosphorylated proteins after normalization to total non-phosphorylated proteins or β-actin. Numerical data (mean ± SD) from three pooled samples are presented as scatter plots with bar. The p-values are shown on top of the histograms.
Discussion
The klotho deficiency has been shown to cause age-related calcification of AV hinge and aortic annulus in mice (25). The Kl–/– mice also exhibit increased expression of many key osteogenic genes involved in human CAVD (25, 63). We demonstrated that upregulation of TGFβ1 and SMAD3 are involved in AV calcification in klotho-deficient mice. The data also indicate that calcific nodule formation in the aortic valve of klotho-deficient mice is associated with increased Tgfb1 expression and elevated levels of activated forms of SMAD2, SMAD3, SMAD1/5, p38, and ERK1/2 MAPK.
Importantly, partial genetic ablation of Tgfb1 or Smad3 significantly decreases the expression of Tgfb1 and both canonical (SMAD-dependent) and non-canonical (MAPK-mediated) TGFβ signaling pathways and blocks the pathological progression of the AV calcification in Kl–/– mice. Collectively, while Tgfb1 haploinsufficiency significantly improved the structural and ECM features, Smad3 haploinsufficiency almost fully reversed pathological structural changes and disrupted elastin fiber organization in the aortic valve of klotho-deficient mice. Dysregulated elastin fibers in the AV leaflet hinge in Kl–/– mice are consistent with a recent report by Gomez-Stallons et al. (64) showing increased elastin fragmentation and loss of elastin integrity in postmortem leaflet patients with CAVD.
Haploinsufficiency of Smad3 has been observed to be more potent in inhibiting aortic valve calcification compared to Tgfb1 ligand inhibition. This is consistent the notion that signaling from TGFβ1 and/or other TGFβ ligands could be effectively blocked by partial deletion of Smad3. Thus, the effect of lowering TGFβ1 on SMAD2/3 activation and AV calcification rescue may be somewhat less pronounced in Kl–/–;Tgfb1± mice as compared to Kl–/–;Smad3± mice. This may be due to functional compensation of the loss of TGFβ1 by other TGFβ ligands (i.e., TGFβ2, 3) and SMAD2. All three TGFβ ligands (TGFβ1, TGFβ2, and TGFβ3) interact with TGFβ Type I and II receptors and lead to activation of SMAD3 and SMAD2. Activated SMAD2/3 with SMAD4 translocate to the nucleus and regulate TGFβ target genes (e.g., Pai1) (36, 65). Increased levels of TGFβ1 (31) and pSMAD2 and PAI1 (66) are associated with CAVD. PAI1 is a known TGFβ target, and therefore, its expression was reduced along with the AV calcification rescue in response to partial inactivation of Tgfb1 (Kl–/–;Tgfb1± mice) or Smad3 (Kl–/–;Smad3± mice) in klotho-deficient mice. Wirrig et al. have reported increased expression of Spp1 and Runx2 in AV tissue of the Kl–/– mice (55). Gomez-Stallons et al. (56) (ATVB) have reported that Spp1, Bmp2, and Runx2 are increased in Kl–/– AV tissue (56). Our results indicating significant rescue of CAVD and expression of these genes in Kl–/– mice by partial genetic deletion of Tgfb1 or Smad3 are consistent with these published findings. It remains unclear if SMAD2 and SMAD3 play a redundant or unique role in AV calcification in CAVD.
There are several experimental limitations in this study. The effect of sex is not specifically addressed. The levels of active and latent TGFβ1 protein are not determined. Since most widely used anti-TGFβ1 antibodies (2G7, 1D11) cross-react to TGFβ3 and not TGFβ2 ligands (67), this study only demonstrated upregulation of Tgfb1 transcript levels in the aortic valve of Kl–/– mice. Regardless of these limitations, the molecular data indicate that both TGFβ1 and SMAD3 contribute to the expression of critical genes involved in the pathogenesis of CAVD. Collectively, our findings represent novel genetic approaches for blocking the progression of CAVD.
Another important piece of information that emerged from this study is the effect of lowering TGFβ1 or SMAD3 on BMP signaling pathway in CAVD. TGFβs and the BMPs are members of the TGFβ family (68). It is well established that increased BMP signaling contributes to AV calcification in Kl–/– mice. It has been reported that both Bmp2 and pSMAD1/5 levels are increased in AV tissue of Kl–/– mice and that genetic inactivation of BMP receptor Bmpr1a in the aortic valve interstitial cells can prevent AV calcification (56). It is remarkable to find that lowering of TGFβ1 as well as SMAD3 has significantly reduced the expression of BMP ligand (Bmp2) and BMP Type I receptor (Alk2) and activation of BMP-dependent SMAD (pSMAD1/5). Constitutively active mutants of ALK2 have been identified as causative of Fibrodysplasia Ossificans Progressiva (FOP), which is an extremely rare heritable disorder of connective tissues characterized by progressive heterotopic ossification in various skeletal sites (69). It is known that TGFβ1 via TGFβ Type I receptor can interact with ALK2 resulting in the simultaneous activation of SMAD2/3 and SMAD1/5 (70, 71). Also, Alk2 deletion can reduce both SMAD2/3 and SMAD1/5 in cushion mesenchymal cells during heart development (72). Importantly, lowering pSMAD3 (this study) or pSMAD1/5 (56) has a similar effect in blocking AV calcification in Kl–/– mice, suggesting crosstalk between TGFβ and BMP pathways in maintaining AV homeostasis (Figure 7). It is noteworthy that SMAD7, an inhibitory SMAD and TGFβ target gene, binds to and blocks all type I receptors in the TGFβ family, whereas SMAD6 shows preferential binding and inhibitory activity toward the BMP type I receptors ALK3 and ALK6 (73). Both SMAD 6 and 7 cooperate in suppressing the physiological BMP signaling during the differentiation of mesenchymal progenitor cells to osteoblasts (74). This is due to rapid and direct induction of Smad6 expression by the primary BMP stimulus, followed by activation of autocrine TGFβ signaling, which then induces a second wave of Smad7 expression that, together with the preexisting SMAD6, shuts down BMP receptor activity in a more sustained manner and limits the rate of differentiation to osteoblasts (74). Whether the negative regulation of TGFβ and BMP pathway via SMAD7 is failed in Kl–/– mice remains a potential area of future investigation.
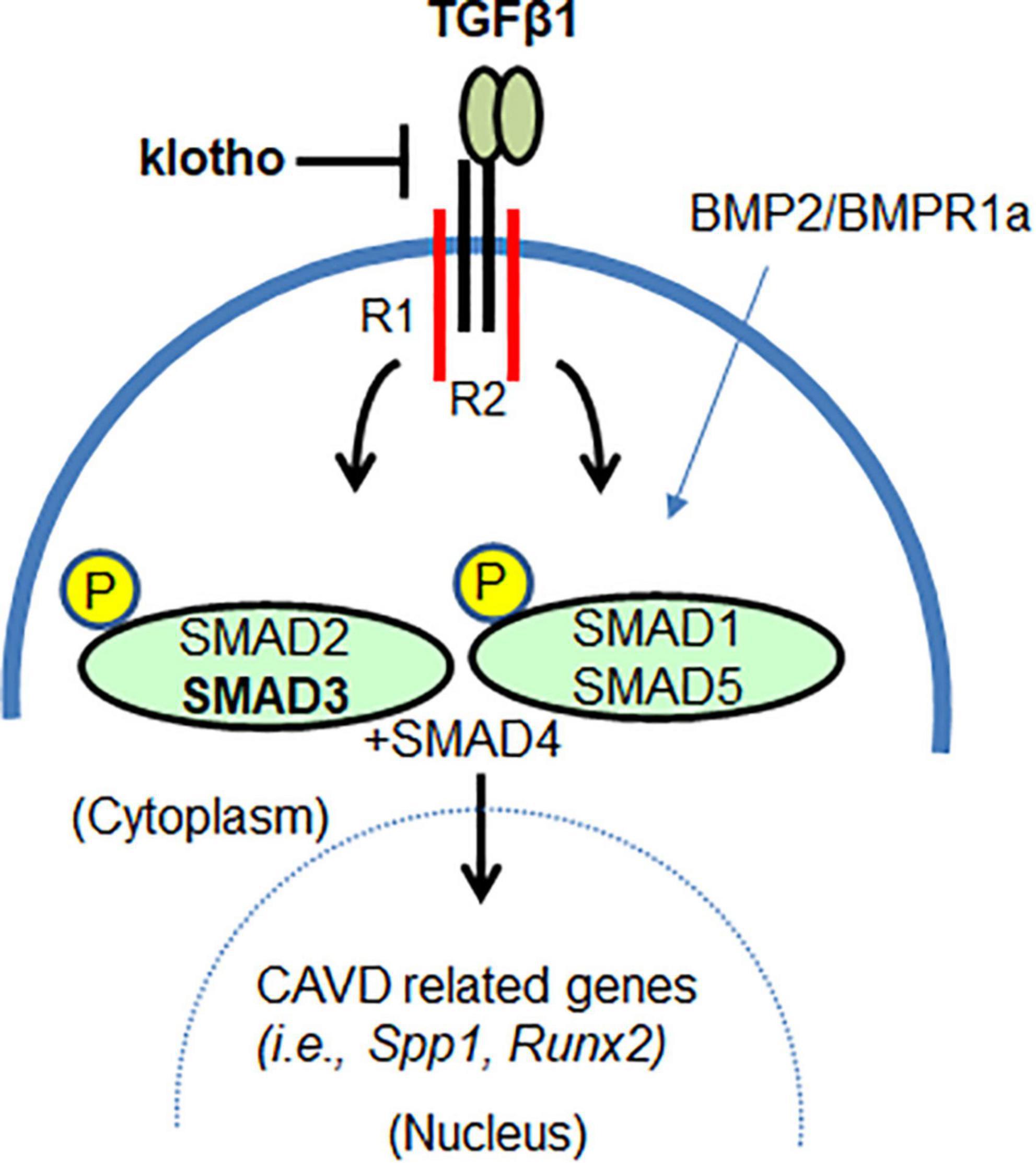
Figure 7. The klotho as a mediator of TGFβ1-dependent SMAD3 signaling in AV calcification. SMAD3 can mediate klotho-dependent TGFβ1 signaling. Lowering TGFβ1-dependent SMAD3 signaling also lowers SMAD1/5 signaling. Thus, klotho functions, in part, as a mediator by restraining or attenuating the augmented TGFβ/BMP signaling and TGFβ-regulated transcription of genes involved in CAVD.
This study indicates that the haploinsufficiency of Smad3 is more effective than Tgfb1 heterozygous deletion in decreasing the activation of p38 MAPK in AV tissue of klotho-deficient mice. Improper activation of p38 or ERK MAPKs is a precursor of constitutive SMAD2/3 signaling associated with aortopathy (75, 76). TGFβ-activation of the SMAD2/3 requires the kinase activity of the TGFβRI, while the TGFβRI kinase activity is not required for the TGFβ-induced oligomerization of the TGFβRII/TGFβRI complex, where the TRAF6 binding to the TGFβRI results in the activation of TAK1-p38 MAPK pathway. Thus, TGFβ activation of TGFβRII/TGFβRI-complex leads to (a) the TGFβRI-kinase-dependent activation of SMAD2/3 and (b) the TGFβRII/TGFβRI-hetero-oligomerization which will cause a rapid activation of TRAF6 resulting in p38 MAPK pathway activation. It is possible that changes in the non-canonical TGFβ pathway may be a primary determinant of AV calcification in Kl–/– mice. Thus, the exact contribution of the non-canonical (non-SMAD) TGFβ pathway in the AV calcification requires further investigation.
Overall, our findings suggest that klotho is an important mediator of TGFβ signaling in vivo in adult aortic valves (Figure 7). Both TGFβ1 and TGFβ-dependent canonical (pSMAD2/3) and non-canonical (p38 MAPK, ERK1/2 MAPK) pathways are activated in the absence of klotho. This suggests that klotho restrains TGFβ ligand-receptor-dependent TGFβ signaling and that increased TGFβ-induced activation of SMAD2/3 and/or SMAD1/5 somehow drives the AV calcification in klotho-deficient mice. This is consistent with the observation that partial genetic inactivation of TGFβ1 and SMAD3 (this study), and BMPR1A (56) can block the development and progression of AV calcification in klotho-deficient animals. Our observations are further supported by in vitro evidence that klotho can bind to the TGFβ Type II receptor complex, thereby contributing to the inhibition of TGFβ1 signaling (22, 77). In conclusion, our results demonstrated that inhibition of TGFβ signaling by selectively targeting TGFβ1 or SMAD3 could inhibit AV calcification. This information will be useful in designing safer treatments for CAVD.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the IACUC University of South Carolina.
Author Contributions
MA: conceptualization and supervision. MC, AB, MG, IC, ZA, and JJ: methodology. MC, AB, IC, and MA: formal analysis. MC and MA: writing—original draft preparation. IC and MA writing—review and editing. MA and NV: funding acquisition. All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was supported by the Transformative Research Seed Grant Initiative, Bob Price Instrumentation Resource Facility Endowment Fund, and ASPIRE-II (Advanced Support Program for Integration of Research Excellence-II- Office of the Vice President for Research) from the University of South Carolina, and the National Institutes of Health (grant nos. R01HL126705, R01HL145064, and 1P20GM103641) (Center for Dietary Supplements and Inflammation; Prakash Nagarkatti and Mitzi Nagarkatti).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Instrumentation Resource Facility at the University of South Carolina School of Medicine and the Dorn VA Medical Center for the instrumentation support.
References
1. Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, et al. Calcific aortic stenosis. Nat Rev Dis Primers. (2016) 2:16006. doi: 10.1038/nrdp.2016.6
2. Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, et al. Calcific aortic valve disease: a consensus summary from the alliance of investigators on calcific aortic valve disease. Arterioscler Thromb Vasc Biol. (2014) 34:2387–93. doi: 10.1161/ATVBAHA.114.302523
3. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease-2011 update. Circulation. (2011) 124:1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767
4. Dutta P, Lincoln J. Calcific aortic valve disease: a developmental biology perspective. Curr Cardiol Rep. (2018) 20:21. doi: 10.1007/s11886-018-0968-9
5. Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental Mechanisms of Aortic Valve Malformation and Disease. Annu Rev Physiol. (2016) 79:21–41. doi: 10.1146/annurev-physiol-022516-034001
6. Bui HT, Khair N, Yeats B, Gooden S, James SP, Dasi LP. Transcatheter heart valves: a biomaterials perspective. Adv Healthc Mater. (2021) 10:e2100115. doi: 10.1002/adhm.202100115
7. Chen Y, Chen YX, Huang C, Duan ZB, Xu CY. The clinical value of klotho and FGF23 in cardiac valve calcification among patients with chronic kidney disease. Int J Gen Med. (2021) 14:857–66. doi: 10.2147/IJGM.S299197
8. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone Klotho. Science. (2005) 309:1829–33. doi: 10.1126/science.1112766
9. Jadhav S, Tripathi S, Chandrekar A, Waikar SS, Hsiao LL. A novel antibody for the detection of alternatively spliced secreted KLOTHO isoform in human plasma. PLoS One. (2021) 16:e0245614. doi: 10.1371/journal.pone.0245614
10. Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. (2012) 125:2243–55. doi: 10.1161/CIRCULATIONAHA.111.053405
11. Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. (1998) 242:626–30. doi: 10.1006/bbrc.1997.8019
12. Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, et al. Establishment of the anti-Klotho monoclonal antibodies and detection of Klotho protein in kidneys. Biochem Biophys Res Commun. (2000) 267:597–602. doi: 10.1006/bbrc.1999.2009
13. Hu MC, Kuro-o M, Moe OW. Renal and extrarenal actions of Klotho. Semin Nephrol. (2013) 33:118–29. doi: 10.1016/j.semnephrol.2012.12.013
14. Kuro OM. The Klotho proteins in health and disease. Nat Rev Nephrol. (2019) 15:27–44. doi: 10.1038/s41581-018-0078-3
15. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. (2007) 104:19796–801. doi: 10.1073/pnas.0709805104
16. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, et al. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. (2009) 583:3221–4. doi: 10.1016/j.febslet.2009.09.009
17. Donate-Correa J, Mora-Fernandez C, Martinez-Sanz R, Muros-de-Fuentes M, Perez H, Meneses-Perez B, et al. Expression of FGF23/KLOTHO system in human vascular tissue. Int J Cardiol. (2013) 165:179–83. doi: 10.1016/j.ijcard.2011.08.850
18. Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, et al. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. (2004) 565:143–7. doi: 10.1016/j.febslet.2004.03.090
19. Consortium A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. (2000) 26:345–8. doi: 10.1038/81664
20. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. (2004) 19:429–35. doi: 10.1359/JBMR.0301264
21. Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, et al. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology. (2005) 146:4647–56. doi: 10.1210/en.2005-0670
22. Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. (2011) 286:8655–65. doi: 10.1074/jbc.M110.174037
23. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. (1997) 390:45–51.
24. Linefsky JP, O’Brien KD, Katz R, de Boer IH, Barasch E, Jenny NS, et al. Association of serum phosphate levels with aortic valve sclerosis and annular calcification: the cardiovascular health study. J Am Coll Cardiol. (2011) 58:291–7. doi: 10.1016/j.jacc.2010.11.073
25. Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol. (2012) 52:689–700. doi: 10.1016/j.yjmcc.2011.12.013
26. Hu MC, Shi M, Zhang J, Quinones H, Griffith C, Kuro-o M, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. (2011) 22:124–36.
27. Dutta P, Kodigepalli KM, LaHaye S, Thompson JW, Rains S, Nagel C, et al. KPT-330 prevents aortic valve calcification via a novel C/EBPbeta signaling pathway. Circ Res. (2021) 128:1300–16. doi: 10.1161/CIRCRESAHA.120.318503
28. Piers LH, Touw HR, Gansevoort R, Franssen CF, Oudkerk M, Zijlstra F, et al. Relation of aortic valve and coronary artery calcium in patients with chronic kidney disease to the stage and etiology of the renal disease. Am J Cardiol. (2009) 103:1473–7. doi: 10.1016/j.amjcard.2009.01.396
29. Thubrikar MJ, Aouad J, Nolan SP. Patterns of calcific deposits in operatively excised stenotic or purely regurgitant aortic valves and their relation to mechanical stress. Am J Cardiol. (1986) 58:304–8.
30. Menon V, Lincoln J. The genetic regulation of aortic valve development and calcific disease. Front Cardiovasc Med. (2018) 5:162. doi: 10.3389/fcvm.2018.00162
31. Jian B, Narula N, Li QY, Mohler ER III, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. (2003) 75:457–65. doi: 10.1016/s0003-4975(02)04312-6
32. Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER III, et al. Transforming growth factor-beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg. (2007) 83:946–53. doi: 10.1016/j.athoracsur.2006.10.026
33. Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. (1985) 316:701–5.
34. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. (2012) 11:790–811.
36. Heldin CH, Moustakas A. Signaling receptors for TGF-beta family members. Cold Spring Harb Perspect Biol. (2016) 8:a022053.
37. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. (1992) 359:693–9.
38. Luong TTD, Estepa M, Boehme B, Pieske B, Lang F, Eckardt KU, et al. Inhibition of vascular smooth muscle cell calcification by vasorin through interference with TGFbeta1 signaling. Cell Signal. (2019) 64:109414. doi: 10.1016/j.cellsig.2019.109414
39. Musial K, Zwolinska D. Novel indicators of fibrosis-related complications in children with chronic kidney disease. Clin Chim Acta. (2014) 430:15–9. doi: 10.1016/j.cca.2013.12.031
40. Vianna HR, Soares CM, Silveira KD, Elmiro GS, Mendes PM, de Sousa Tavares M, et al. Cytokines in chronic kidney disease: potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatr Nephrol. (2013) 28:463–9. doi: 10.1007/s00467-012-2363-x
41. Voelkl J, Lang F, Eckardt KU, Amann K, Kuro OM, Pasch A, et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. (2019) 76:2077–91.
42. Voelkl J, Cejka D, Alesutan I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr Opin Nephrol Hypertens. (2019) 28:289–96.
43. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. (2011) 109:697–711.
44. Hoevelmann J, Mahfoud F, Lauder L, Scheller B, Bohm M, Ewen S. Valvular heart disease in patients with chronic kidney disease. Herz. (2021) 46:228–33.
45. Alves RD, Eijken M, van de Peppel J, van Leeuwen JP. Calcifying vascular smooth muscle cells and osteoblasts: independent cell types exhibiting extracellular matrix and biomineralization-related mimicries. BMC Genomics. (2014) 15:965. doi: 10.1186/1471-2164-15-965
46. Voelkl J, Luong TT, Tuffaha R, Musculus K, Auer T, Lian X, et al. SGK1 induces vascular smooth muscle cell calcification through NF-kappaB signaling. J Clin Invest. (2018) 128:3024–40. doi: 10.1172/JCI96477
47. Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. (2004) 95:253–60.
48. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. (2001) 89:1147–54. doi: 10.1161/hh2401.101070
49. Alesutan I, Feger M, Tuffaha R, Castor T, Musculus K, Buehling SS, et al. Augmentation of phosphate-induced osteo-/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc Res. (2016) 110:408–18. doi: 10.1093/cvr/cvw062
50. Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. (2001) 104:2525–32.
51. Leibrock CB, Alesutan I, Voelkl J, Pakladok T, Michael D, Schleicher E, et al. NH4Cl treatment prevents tissue calcification in klotho deficiency. J Am Soc Nephrol. (2015) 26:2423–33.
52. Alesutan I, Musculus K, Castor T, Alzoubi K, Voelkl J, Lang F. Inhibition of phosphate-induced vascular smooth muscle cell osteo-/chondrogenic signaling and calcification by bafilomycin a1 and methylamine. Kidney Blood Press Res. (2015) 40:490–9. doi: 10.1159/000368524
53. Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD, et al. Valve endothelial cell-derived Tgfbeta1 signaling promotes nuclear localization of sox9 in interstitial cells associated with attenuated calcification. Arterioscler Thromb Vasc Biol. (2016) 36:328–38. doi: 10.1161/ATVBAHA.115.306091
54. Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. (2010) 106:712–9.
55. Wirrig EE, Gomez MV, Hinton RB, Yutzey KE. COX2 inhibition reduces aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol. (2015) 35:938–47. doi: 10.1161/ATVBAHA.114.305159
56. Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, Conway SJ, Yutzey KE. Bone morphogenetic protein signaling is required for aortic valve calcification. Arterioscler Thromb Vasc Biol. (2016) 36:1398–405.
57. Chen J, Lin Y, Sun Z. Deficiency in the anti-aging gene Klotho promotes aortic valve fibrosis through AMPKalpha-mediated activation of RUNX2. Aging Cell. (2016) 15:853–60. doi: 10.1111/acel.12494
58. Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, et al. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev Dyn. (2009) 238:431–42. doi: 10.1002/dvdy.21854
59. Daniel SG, Ball CL, Besselsen DG, Doetschman T, Hurwitz BL. Functional changes in the gut microbiome contribute to transforming growth factor beta-deficient colon cancer. mSystems. (2017) 2:e00065–17. doi: 10.1128/mSystems.00065-17
60. Chakrabarti M, Al-Sammarraie N, Gebere MG, Bhattacharya A, Chopra S, Johnson J, et al. Transforming growth factor beta3 is required for cardiovascular development. J Cardiovasc Dev Dis. (2020) 7:19.
61. Bhattacharya A, Al-Sammarraie N, Gebere MG, Johnson J, Eberth JF, Azhar M. Myocardial TGFbeta2 is required for atrioventricular cushion remodeling and myocardial development. J Cardiovasc Dev Dis. (2021) 8:26.
62. Azhar M, Yin M, Bommireddy R, Duffy JJ, Yang J, Pawlowski SA, et al. Generation of mice with a conditional allele for transforming growth factor beta 1 gene. Genesis. (2009) 47:423–31.
63. Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. (2011) 50:561–9. doi: 10.1016/j.yjmcc.2010.12.005
64. Gomez-Stallons MV, Tretter JT, Hassel K, Gonzalez-Ramos O, Amofa D, Ollberding NJ, et al. Calcification and extracellular matrix dysregulation in human postmortem and surgical aortic valves. Heart. (2019) 105:1616–21. doi: 10.1136/heartjnl-2019-314879
65. Azhar M, Ware SM. Genetic and developmental basis of cardiovascular malformations. Clin Perinatol. (2016) 43:39–53.
66. Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. (2006) 114:2065–9.
67. Angelov SN, Hu JH, Wei H, Airhart N, Shi M, Dichek DA. TGF-beta (Transforming Growth Factor-beta) signaling protects the thoracic and abdominal aorta from angiotensin II-induced pathology by distinct mechanisms. Arterioscler Thromb Vasc Biol. (2017) 37:2102–13. doi: 10.1161/ATVBAHA.117.309401
68. Morikawa M, Derynck R, Miyazono K. TGF-beta and the TGF-beta family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. (2016) 8:a021873.
69. Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells. (2014) 32:1289–300. doi: 10.1002/stem.1633
70. Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. (2008) 28:6889–902. doi: 10.1128/MCB.01192-08
71. Flanders KC, Heger CD, Conway C, Tang B, Sato M, Dengler SL, et al. Brightfield proximity ligation assay reveals both canonical and mixed transforming growth factor-beta/bone morphogenetic protein Smad signaling complexes in tissue sections. J Histochem Cytochem. (2014) 62:846–63. doi: 10.1369/0022155414550163
72. Wang J, Sridurongrit S, Dudas M, Thomas P, Nagy A, Schneider MD, et al. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol. (2005) 286:299–310.
73. Goto K, Kamiya Y, Imamura T, Miyazono K, Miyazawa K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J Biol Chem. (2007) 282:20603–11. doi: 10.1074/jbc.M702100200
74. Maeda S, Hayashi M, Komiya S, Imamura T, Miyazono K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. (2004) 23:552–63. doi: 10.1038/sj.emboj.7600067
75. Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. (2009) 284:5630–6. doi: 10.1074/jbc.M806962200
76. Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. (2011) 332:358–61.
Keywords: Tgfb = transforming growth factor beta, Smad3, Klotho, CAVD (calcific aortic valve disease), aortic valve calcification
Citation: Chakrabarti M, Bhattacharya A, Gebere MG, Johnson J, Ayub ZA, Chatzistamou I, Vyavahare NR and Azhar M (2022) Increased TGFβ1 and SMAD3 Contribute to Age-Related Aortic Valve Calcification. Front. Cardiovasc. Med. 9:770065. doi: 10.3389/fcvm.2022.770065
Received: 03 September 2021; Accepted: 20 June 2022;
Published: 19 July 2022.
Edited by:
Katherine Yutzey, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Robert J. Levy, Children’s Hospital of Philadelphia, United StatesBrendan Martin Corcoran, University of Edinburgh, United Kingdom
Copyright © 2022 Chakrabarti, Bhattacharya, Gebere, Johnson, Ayub, Chatzistamou, Vyavahare and Azhar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamad Azhar, Mohamad.Azhar@uscmed.sc.edu