A pseudo-outbreak of Cyberlindnera fabianii funguria: Implication from whole genome sequencing assay
 Xin Fan
Xin Fan Rong-Chen Dai
Rong-Chen Dai Timothy Kudinha
Timothy Kudinha Li Gu1*
Li Gu1*- 1Department of Infectious Diseases and Clinical Microbiology, Beijing Institute of Respiratory Medicine and Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
- 2School of Public Health, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China
- 3School of Dentistry and Medical Sciences, Charles Sturt University, Leeds Parade, Oranges, NSW, Australia
- 4NSW Health Pathology, Regional and Rural, Orange hospital, Orange, NSW, Australia
Background: Although the yeast Cyberlindnera fabianii (C. fabianii) has been rarely reported in human infections, nosocomial outbreaks caused by this organism have been documented. Here we report a pseudo-outbreak of C. fabianii in a urology department of a Chinese hospital over a two-week period.
Methods: Three patients were admitted to the urology department of a tertiary teaching hospital in Beijing, China, from Nov to Dec 2018, for different medical intervention demands. During the period Nov 28 to Dec 5, funguria occurred in these three patients, and two of them had positive urine cultures multiple times. Sequencing of rDNA internal transcribed spacer (ITS) region and MALDI-TOF MS were applied for strain identification. Further, sequencing of rDNA non-transcribed spacer (NTS) region and whole genome sequencing approaches were used for outbreak investigation purpose.
Results: All the cultured yeast strains were identified as C. fabianii by sequencing of ITS region, and were 100% identical to the C. fabianii type strain CBS 5640T. However, the MALDI-TOF MS system failed to correctly identify this yeast pathogen. Moreover, isolates from these three clustered cases shared 99.91%-100% identical NTS region sequences, which could not rule out the possibility of an outbreak. However, whole genome sequencing results revealed that only two of the C. fabianii cases were genetically-related with a pairwise SNP of 192 nt, whilst the third case had over 26,000 SNPs on its genome, suggesting a different origin. Furthermore, the genomes of the first three case strains were phylogenetically even more diverged when compared to a C. fabianii strain identified from another patient, who was admitted to a general surgical department of the same hospital 7 months later. One of the first three patients eventually passed away due to poor general conditions, one was asymptomatic, and other clinically improved.
Conclusion: In conclusion, nosocomial outbreaks caused by emerging and uncommon fungal species are increasingly being reported, hence awareness must be raised. Genotyping with commonly used universal gene targets may have limited discriminatory power in tracing the sources of infection for these organisms, requiring use of whole genome sequencing to confirm outbreak events.
Introduction
Emerging fungal infections have become a global health concern in the past few decades due to their notable morbidity and mortality, especially among immunosuppressed patients admitted to intensive care units (ICUs), or undergoing invasive medical interventions (Pappas et al., 2018; Hoenigl et al., 2022; World Health Organization, 2022). Although Candida albicans remains the most predominant yeast pathogen, the incidence of uncommon yeast species causing human infections has increased enormously in recent years (Pappas et al., 2018; Chen et al., 2021). Uncommon yeast species often exhibit decreased susceptibility to commonly used antifungal agents, making them difficult to manage in clinical settings. Moreover, there are increasing incidences of nosocomial infections and outbreak events reported due to transmission of uncommon or emerging yeast species worldwide (Pappas et al., 2018; Chen et al., 2021). For instance, Candida auris, which was first described in 2009, has caused a number of outbreaks in different continents (Chow et al., 2018; Hoenigl et al., 2022; World Health Organization, 2022).
In the investigations and tracing of fungal nosocomial transmissions, molecular genotyping could provide essential genetic evidence. Hence, a wide variety of molecular typing assays have been evaluated and implemented in the study of outbreaks, including band-pattern-based DNA analysis like random amplified polymorphic DNA (RAPD) and pulsed field gel electrophoresis (PFGE), traditional DNA sequencing-based phylogenetic methods like single gene analysis, microsatellite analysis and multilocus sequence typing (MLST), protein spectrum analysis by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS), and whole genome sequencing (WGS) techniques (Reiss et al., 1998; Pulcrano et al., 2012; Mikosz et al., 2014; Xiao et al., 2014; Litvintseva et al., 2015; Oliveira and Azevedo, 2022). Of these methods, WGS has become increasingly used due to its outstanding discriminatory power (Litvintseva et al., 2015; Bougnoux et al., 2018; Desnos-Ollivier et al., 2020; Oliveira and Azevedo, 2022).
In this study, we report on three clustered funguria cases caused by a rare fungal pathogen, Cyberlindnera fabianii, which occurred over a two-week period within the same urology department, which was initially considered as a nosocomial outbreak event. Using WGS, this event was finally confirmed as a pseudo-outbreak caused by C. fabianii from two diverged genetic lineages.
Material and methods
Ethics
This study was approved by the Human Research Ethics Committee of the Beijing Chaoyang Hospital (No. KE332). Written informed consent was obtained from all participants involved.
Routine isolation of the microorganisms and MALDI-TOF MS identification.
C. fabianii strains were isolated from urine samples of three patients (number 1-3); on four different occasions for patient number 1, only once for patient number 2, and on three different occasions for patient number 3 (Figure 1 and Table 1). After these three cases, no further C. fabianii cases were detected in the same hospital for over seven months, till a new C. fabianii isolate, cultured from an ascites sample of a patient admitted to general surgery department (recorded as patient number 4), was detected, and this isolate was used as a comparator.
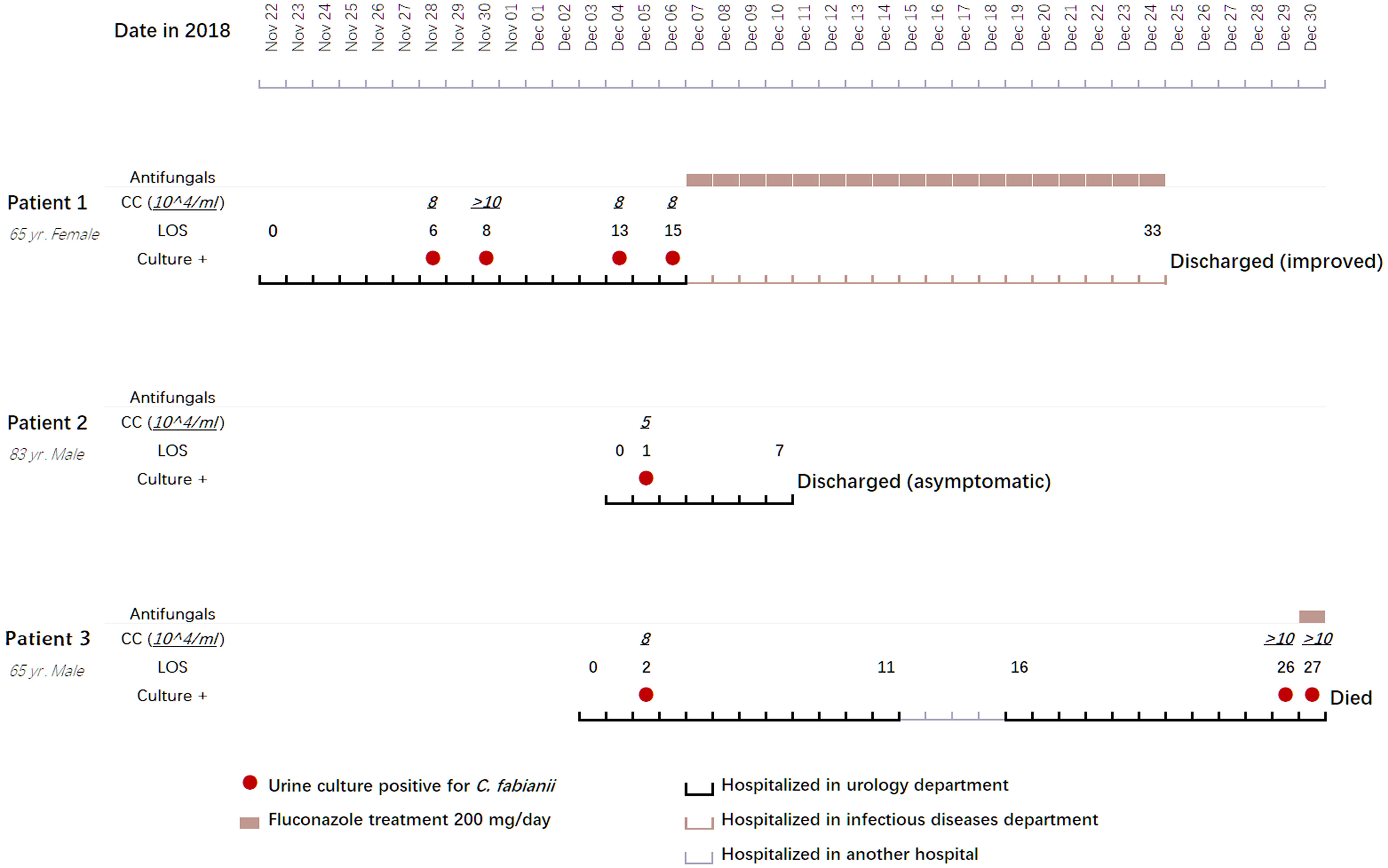
Figure 1 Clinical features, treatment regimens, and outcomes of three clustered cases with Cyberlindnera fabianii funguria. Abbreviations: CC, CFU (colony forming unit) count; LOS, length of stay; Culture +: culture positive for C. fabianii.
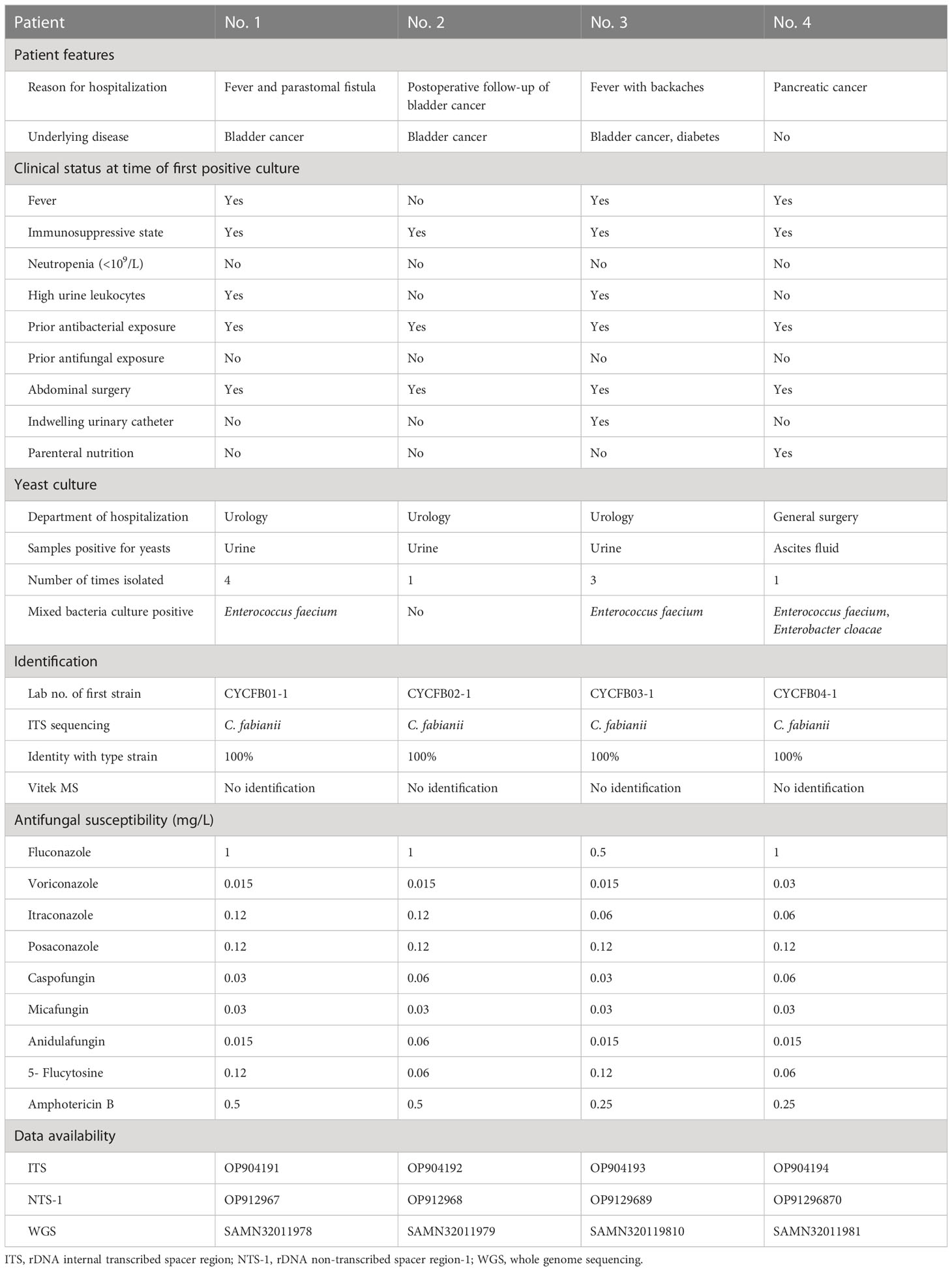
Table 1 Clinical features of four patients with Cyberlindnera fabianii funguria and microbiology characteristics of the strains.
Routine culture of specimens was carried out as per standard laboratory protocols. Generally, for urine samples, 1 μl of the sample was inoculated on Blood Agar media and incubated at 35 °C for 24 h. Thereafter, the number of colonies growing on the media plate was counted to ensure that they met the criterion for a urinary tract infection. A brief identification protocol revealed that the cultured isolates were yeast. Thus, Sabouraud glucose agar (SDA) was used to subculture these isolates for further identification testing. Attempts were made to identify the cultured yeasts by using a Vitek MS MALDI-TOF MS system (bioMérieux, Marcy l’Etoile, France, with IVD database version 2.1), following manufacturer’s instructions. For each run, Escherichia coli strain ATCC 8739 was used to calibrate and control the method. Unfortunately, this system was unable to identify the yeast strains.
Molecular identification and phylogenetic analysis by rDNA gene spacer regions
As all the suspected yeast isolates could not be identified using the MALDI-TOF MS systems, sequencing of rDNA internal transcribed spacer (ITS) regions was carried out. Briefly, DNA extraction of the isolates was performed using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The universal primer pair ITS1 and ITS4 was used for amplification and sequencing of the ITS region for each strain (Xiao et al., 2014), and a species identification was carried out by querying against the Westerdijk Fungal Biodiversity Institute’s database using a web-based pairwise alignment tool (https://wi.knaw.nl/page/Pairwise_alignment ).
Further, to investigate the potential relatedness of these cases, the first yeast isolate of each patient case was chosen for further testing, and the rDNA non-transcribed spacer region 1 (NTS-1) was amplified with a forward primer NTS1-F (5’-GGGATAAATCATTTGTATACGAC-3’) and a reverse primer NTS1-R (5’-TTGCGGCCATATCCACAAGAAA-3’) as described previously (Al-Sweih et al., 2019), and sequenced from both directions. A phylogenetic tree of NTS-1 sequences was generated by Mega X (version 10.2, https://www.megasoftware.net/ ) using neighbor-joining method with bootstrap value of 1000. NTS-1 sequences from C. fabianii type strain CBS 5640T, and C. fabianii reference genome strain JOY008, were also downloaded from GenBank and included in the analysis. In addition, NTS-1 sequence extracted from the genome of Cyberlindnera jadinii strain NBRC 0988 was selected as an outgroup.
Whole genome sequencing and analysis of C. fabianii strains
Whole-genome sequencing was performed on each of the first yeast strain from each of patients 1 to 4. Generally, a 350-bp DNA library was prepared using NEB Next Ultra DNA library prep kits (NEW ENGLAND BioLabs Inc., MA, USA), following the manufacturer’s instructions. Library integrity was evaluated with an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Sequencing was performed on an Illumina NovaSeq using PE150 in a commercial company (Novogene Co., Ltd., Beijing, China).
For genome analysis, the complete reference genome of C. fabianii strain JOY008 (GenBank accession no. GCA_022641835.1) was used for read mapping. Single-nucleotide polymorphism (SNP) analysis was carried out by Burrows-Wheeler Aligner (version 0.7.7), SAMtools (version 1.2), and Genome Analysis Toolkit (GATK) (v.3.3-0) per GATK Best Practices (Li and Durbin, 2009; Li et al., 2009; Mckenna et al, 2010). The filtered reads were compared to the reference genome by SAMtools to generate BAM files. Then, variants were marked by GATK MarkDuplicates for each sample, and single-sample GVCF files were created by GATK HaplotypeCaller with the option –emitRefConfidence GVCF. The GVCF files were aggregated by GATK CombineGVCFs tool. After that, the GVCF files were jointly genotyped with the GATK GenotypeGVCFs to produce a single VCF file containing variants data on every strain. Finally, the VCF file was selected using GATK SelectVariants with the option -select-type SNP and filtered using the following parameters: VariantFiltration, QD < 2.0, ReadPosRankSum < −8.0, FS > 60.0, MQRankSum < −12.5, MQ < 40.0 and HaplotypeScore > 13.0.
Specifically, in this study, the term “pseudo-outbreak” was used to describe inappropriate artifactual clustering of real infections as an outbreak event, due to limitation of investigation tools.
Antifungal susceptibility testing
Minimum inhibitory concentrations (MICs) of all the isolates were determined by Sensititre YeastOne YO10 kits (Thermo Scientific, OH, USA) following the manufacturer’s instructions, and with Candida krusei ATCC 6258 and Candida parapsilosis ATCC 22019, used as quality control strains.
Data availability
DNA sequences of rDNA ITS and NTS-1 regions for each of the first yeast strain isolated from each individual has been deposited into NCBI GenBank database (accession nos. OP904191-OP904194 for ITS region and OP912967-OP912970 for NTS-1 region). Their WGS reads data is also now available in National Microbiology Data Center (NMDC) database (Bioproject accession no. PRJNA907923).
Results
Patients
Information pertaining to each of the 4 patients included in this case study is summarized in Figure 1 and Table 1.
Patient 1 was a 65-year-old female admitted to the urology department of Beijing Chao-Yang hospital on Nov 22, 2018, due to presence of fever for two weeks and a parastomal fistula after ileal replacement due to bladder cancer in 2016. Upon admission, the patient had fever for over a week. On day 6 after admission, a yeast strain was isolated from her urine sample and the colony count (CC) was 8×104 CFU/ml. The same urine culture also grew Enterococcus faecium (5×104 CFU/ml) as a mixed culture with the yeast. The routine MALDI-TOF MS identification system failed to identify the yeast isolate. Her urine samples collected on days 8, 13 and 15 after admission also yielded yeasts (CC of 8 to >10×104 CFU/ml). Follow-up ITS sequencing assigned all the strains as C. fabianii. The patient was given fluconazole at 200 mg/day for 18 days after which her condition improved notably, and she was finally discharged from the hospital on day 33 of admission.
Patient 2 was an 83-year-old male admitted to the urology department of the hospital on Dec 04, 2018, for follow-up of bladder cancer electrosurgery performed eight and four months before his admission. On day 1 after admission, a urine sample was collected for routine screening, which was reported positive for yeasts with a CC of 5×104 CFU/ml. The yeast strain was identified as C. fabianii by ITS sequencing. This patient didn’t present with any symptoms of infection, and hence antifungal therapy was not given. Later, he received a transurethral resection of bladder tumor on day 3, and was discharged on day 7 after admission.
Patient 3 was a 65-year-old male admitted to the urology department of the hospital on Dec 03, 2018, due to presence of high fever with backaches. Nine months before this admission, the patient hand undergone nephroureterectomy of the left kidney. He received nephrostomy on the right kidney immediately on day 1 after admission. His urine sample collected on day 2 was culture positive for a yeast (CC: 8×104 CFU/ml), which was identified as C. fabianii by ITS sequencing. However, no antifungal agents were prescribed for him and only a broad-spectrum antibiotic was given. On days 26 and 27, two urine samples were collected consecutively and both were positive for C. fabianii with a CC of > 10×104 CFU/ml. Of note, all his urine samples also grew E. faecium (>10×104 CFU/ml) as part of a mixed culture with the yeast. Though fluconazole therapy (200 mg/day) was initiated on day 27 after admission, the patient passed away on the same day.
Patient 4 was a 54-year-old female admitted to the general surgery department of the hospital on Jul 18, 2019, which was over seven months after the patient 1, 2 and 3 case clusters. She was hospitalized due to pancreatic cancer, and received radical pancreatoduodenectomy on day 12 after admission; later with pancreatic intestinal anastomotic fistula. The patient’s ascites sample collected on day 20 was reported positive for C. fabianii and E. faecium. However, she didn’t have any other culture positive results for fungi after that, nor received any antifungal treatment, and was discharged from the hospital on day 52.
C. fabianii identification
All the yeast strains could not be identified using the Vitek MS MALDI-TOF MS system IVD 2.0 database, nor were the isolates misidentified as something else (identification confidence values <60.0). This is not surprising as C. fabianii is not currently included in the system’s spectrum database.
By sequencing of the ITS region, all the yeast strains from the four patients were unambiguously assigned to C. fabianii, with their respective ITS sequences 100% (602/602 bp) identical to that of C. fabianii type strain CBS 5640T.
Phylogenetic analysis by rDNA NTS-1 region
Since C. fabianii is a rare yeast species identified in human patients, and the fact that the clustered cases (patients 1 to 3) described here were identified within a two-week period from the same department, an investigation was carried out to assess the possibility of this being a nosocomial outbreak. Owing to the high sequence similarity of the ITS region among the strains, sequence analysis based on rDNA NTS-1 region was further carried out, which was assumed to have higher discriminatory power and has previously been used to confirm a C. fabianii outbreak in Kuwait (Al-Sweih et al., 2019).
Using C. jadinii (strain NBRC 0988) as an outgroup, the phylogenetic tree based on the NTS-1 region clustered all the C. fabianii isolates together (Figure 2), and inter-species variation between C. jadinii and C. fabianii in NTS-1 region was >43.5%. Amongst C. fabianii strains, some intra-species variation was observed (Figure 2). However, sequence variations amongst the strains from patients 1 to 3 was inconspicuous, as these strains exhibited the same sequence type, while the strain from patient 2 had only one SNP (identity 1179/1180, 99.92%). In contrast, strains from the three clustered cases were quite diverged from the strain from patient 4 which was isolated seven months later, which had an overall 7-bp insertions and 4 additional SNPs in its NTS-1 region (identity 99.07%).
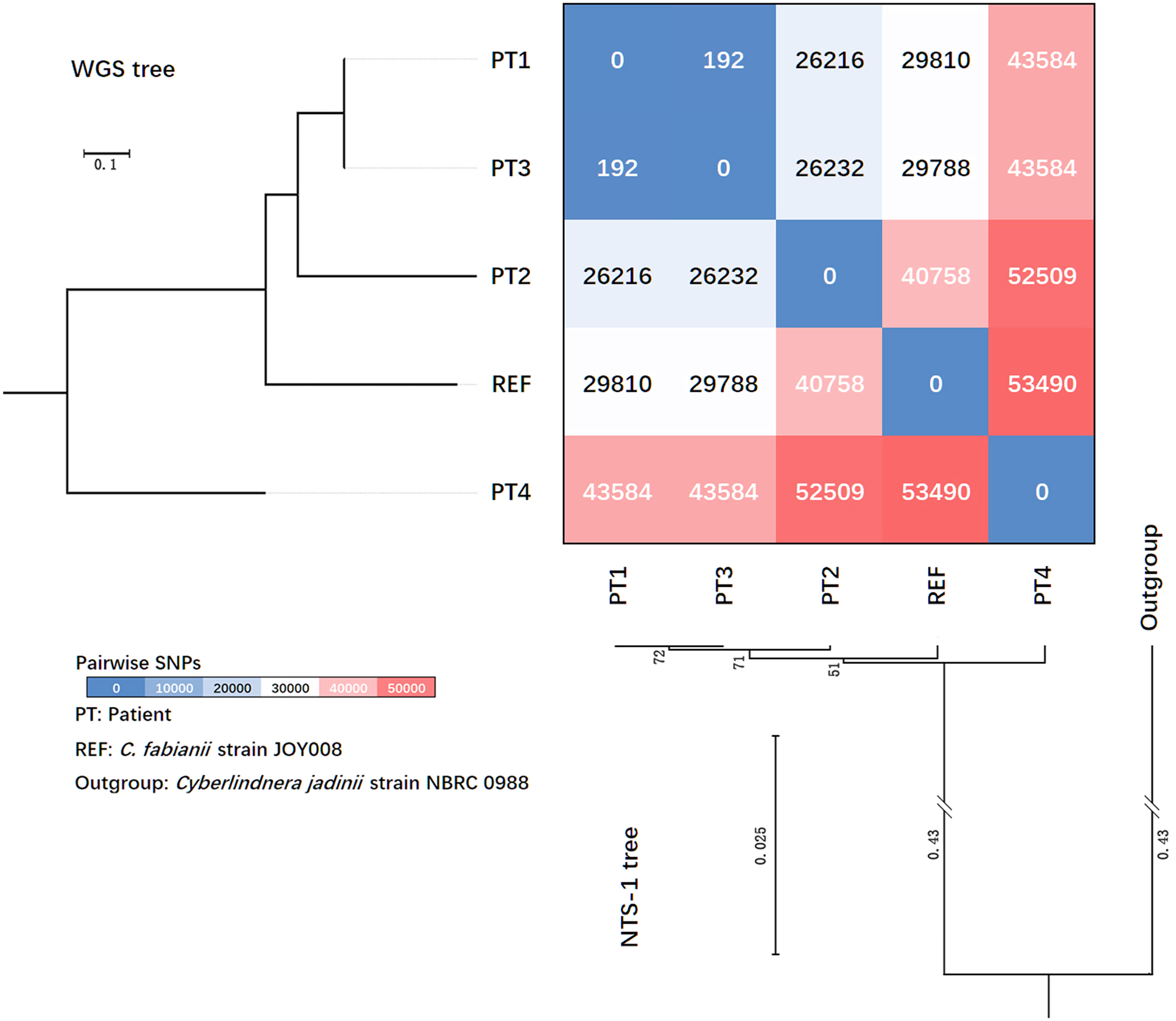
Figure 2 Phylogenetic trees generated based on rDNA non-transcribed spacer (NTS) region-1 sequences and whole genome sequencing (WGS) SNPs, and heatmaps revealing pairwise differences of SNPs amongst four patients’ strains collected in this study.
Whole genome sequencing results
Genome sequencing of yeast strains from patients 1 to 4 produced 2.6 to 3.7 Gb of clean data, and average depths of sequencing were all above 200×. The average genome size obtained was 12.94 Mb. Their genomes had an average GC content of 44.4% to 45.1%, with N50 of 13,739 bp to 202,514 bp. Comparative genomic analysis was performed for all strains. The pairwise differences between genome reference strain JOY008, which originated from a soil environment in USA, and our four patients’ clinical strains, were 29,810-53,490 SNPs (Figure 2).
We carried out a review of previous outbreak reports caused by different yeast species, and the number of pairwise SNPs described varied from less than ten to several hundred (Table 2). The yeast strains from patient 1 and 3 had only 192 SNPs identified, suggesting that they were probably closely related (Figure 2). However, there were over 26,000 SNPs identified between the strain from patient 2 and strains from patients 1 and 3 (Figure 2), which was considered as a high-level genomic variation. These findings suggested that the C. fabianii strain from patient 2 was from a different origin. In addition, the yeast strain from patient 4 was even more diverged, with >43,000 of SNPs compared to all strains from patients 1 to 3, and the reference genome (Figure 2). Lastly, the phylogenetic tree constructed based on whole genome SNPs also support the same conclusion (Figure 2).
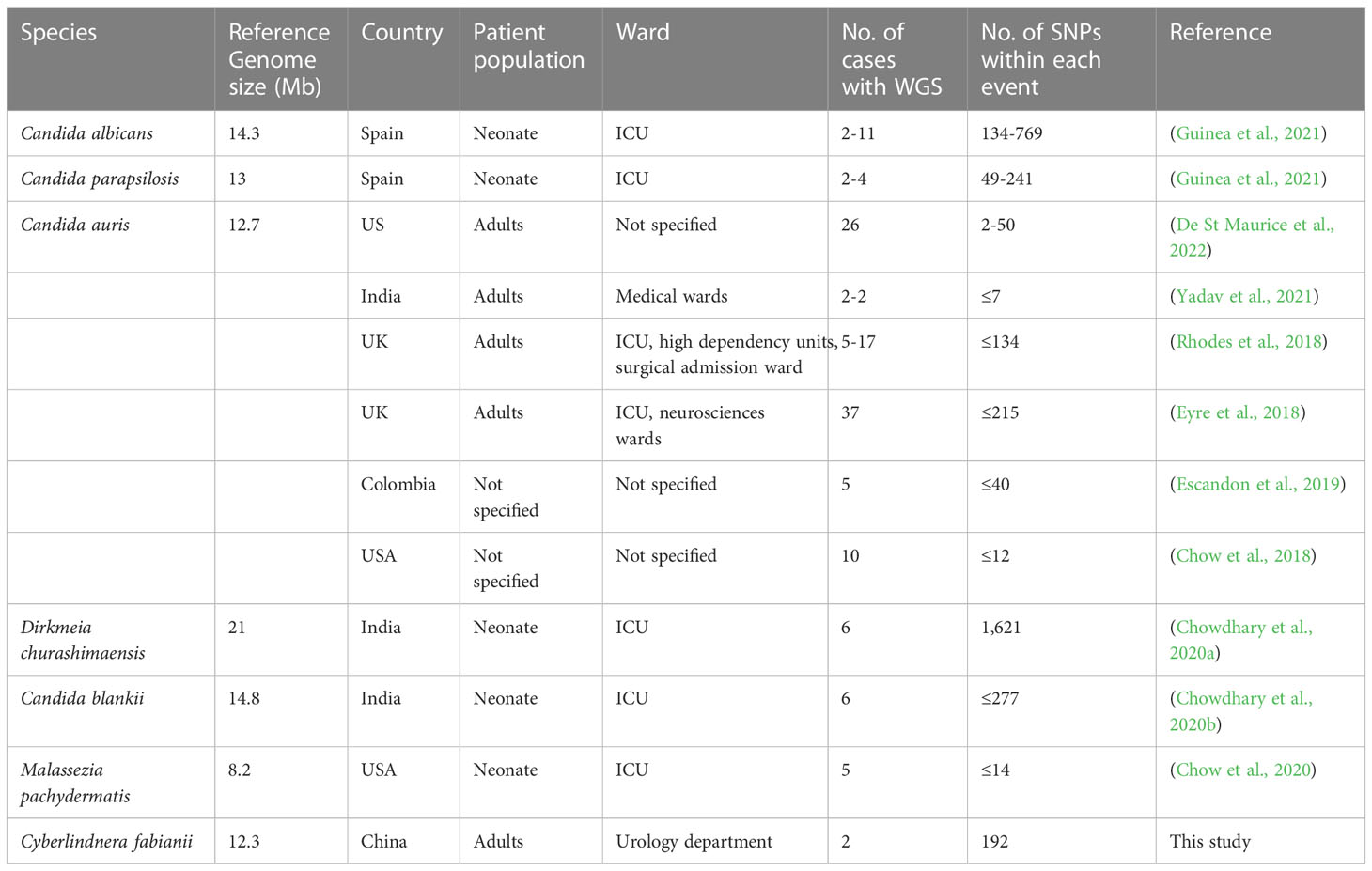
Table 2 Review of outbreak events caused by yeast species that were characterized by whole genome sequencing in previous studies.
Antifungal susceptibilities
All the C. fabianii strains isolated in this study showed good susceptibility to all the nine antifungal agents tested (Table 1), with geometric minimum inhibitory concentration (GM MIC) of 0.84 mg/L to fluconazole, 0.02 mg/L to voriconazole, 0.08 mg/L to itraconazole, 0.12 mg/L to posaconazole, 0.04 mg/L to caspofungin, 0.03 mg/L to micafungin, 0.02 mg/L to anidulafungin, 0.08 mg/L to 5-flucytosine, and finally, 0.35 mg/L to amphotericin B. If using clinical breakpoints or epidemiological cut-off values of C. albicans as references, all these strains could be classified as susceptible or of wild-type phenotype to all antifungal agents tested.
Discussion
C. fabianii, basionym Hansenula fabianii, homotypic synonyms Candida fabianii, Lindnera fabianii and Pichia fabianii, is an ascomycetous yeast that has a close relationship with human activities (Kato et al., 1997; Arastehfar et al., 2019; Van Rijswijck et al., 2019). This yeast species is commonly seen in fermented food products like alcohols (Arastehfar et al., 2019; Van Rijswijck et al, 2019), and has also been used for treatment of waste water with a long history (Kato et al., 1997). The species has now been assigned within the Wickerhamomycetaceae clade (Kidd et al., 2023). Within this clade, there are several other species that have been reported to cause human infections, such as Wickerhamomyces anomalus and Cyberlindnera jadinii (Treguier et al., 2018; Zhang et al., 2021).
Generally, detection of C. fabianii in clinical settings is rare. According to previous surveillance reports on human fungal diseases, the prevalence of C. fabianii is generally <0.1% (Pfaller et al., 2019; Xiao et al., 2020). However, this yeast species is also an opportunistic pathogen that can cause a broad-range of infections, including lethal fungemia (Al-Sweih et al., 2019; Arastehfar et al., 2019). A previous research suggests that C. fabianii only has low virulence attributes (Arastehfar et al., 2019), although Nouraei et al. observed that this fungal species was one of the uncommon yeasts with high-level production of hemolysin, phospholipase and proteinase (Nouraei et al., 2020). In addition, C. fabianii has been observed to have a strong capacity for biofilm formation, which may contribute to its persistence and resistance to antifungal therapies (Hamal et al., 2008; Nouraei et al., 2020).
Of note, several studies have revealed difficulties in the accurate identification of C. fabianii using conventional methods, which may influence precision clinical recognition and management of infections caused by this organism (Svobodova et al., 2016; Al-Sweih et al., 2019). MALDI-TOF MS has been reported as a powerful tool for identification of yeasts, but the system’s identification capacity largely relies on the peptide mass fingerprint database that is incorporated into the system. Some of the MALDI-TOF MS systems, such as Biotyper (Bruker Daltonics, Germany, with IVD library version 8) and MicroIDSys (ASTA, Korea, with database version 1.23.2), have demonstrated capacity to accurately identify C. fabianii strains (Park et al., 2019; Teke et al., 2021). In contrast to this, C. fabianii is still absent from the Vitek MS IVD database (up to version 3.2), hence this system failed to identify any of C. fabianii isolates in this study. Similar findings were also reported by Teke et al. (Teke et al., 2021).
Although nosocomial transmission of fungal pathogens is less frequently encountered in clinical practice compared to bacterial pathogens, fungal outbreaks are more common than publicly appreciated, and are mostly associated with medical products or contamination of the hospital environment (Kanamori et al., 2015; Litvintseva et al., 2015; Magill et al., 2018). For instance, Candida parapsilosis, one of the most prevalent human pathogenic yeast species, is well-known for causing catheter-related bloodstream infections. There have been a large number of nosocomial outbreaks caused by Candida parapsilosis worldwide, including several recently reported cases caused by fluconazole-resistant clones that raised more public health concerns (Arastehfar et al., 2020; Zhang et al., 2020; Thomaz et al., 2022). Moreover, reports of outbreaks caused by unusual fungal pathogens, such as the recently emerged C. auris, are increasing (Litvintseva et al., 2015; Chow et al., 2018). Of note, during the COVID-19 pandemic period, fungal outbreaks caused higher medical burdens to healthcare facilities and patients (Hoenigl et al., 2022; Thomaz et al., 2022), and hence are beginning to receive more attention.
Of note, a recent outbreak of C. fabianii in Kuwait was described by Al-Sweih et al., which involved a total of 10 fungemia cases in neonates (Al-Sweih et al., 2019). Furthermore, previous reviews on C. fabianii cases have demonstrated that the elderly population is the second most vulnerable population after neonates overall, with funguria being the first to second commonest clinical symptom (Al-Sweih et al., 2019; Arastehfar et al., 2019; Park et al., 2019). This agrees with our three-clustered C. fabianii funguria cases which all occurred in elderly patients, and with funguria as the common clinical symptom, though not every patient had symptomatic urinary tract infection.
Published literature have emphasized that presence of indwelling urinary catheter is the most important risk factor and transmission route for nosocomial urinary tract infections, especially when catheter care quality is poor. However, a variety of additional risk factors have also been described, including female gender, increased age, diabetes, bladder instrumentation, urinary outflow obstruction, amongst others. (Pearson-Stuttard et al., 2016; Mody et al., 2017; Odabasi and Mert, 2020). Of the four patients described in this study, only one carried an indwelling urinary catheter, and all of them had undergone abdominal surgeries prior to the onset of funguria. Besides, three of the four patients had E. faecium detected concurrently with C. fabianii in the same urine sample. Enterococcus species, including E. faecium, are well-known ubiquitous inhabitants of the human gut microbiota and could lead to urinary tract infections (Magruder et al., 2019). Moreover, C. fabianii has also been identified in the human intestinal microbiota (Zhai et al., 2020), and previously Mathy et al. hypothesized that translocation of C. fabianii from the gut was responsible for a ventriculoperitoneal shunt case (Mathy et al., 2020). Therefore, it is possible that C. fabianii funguria cases identified in our study may have resulted from gut microbiota translocations, and abdominal surgeries might serve as triggers or risk factors.
As widely-acknowledged, application of ITS sequencing could allow accurate identification of yeast species but with insufficient discriminatory power for intra-species typing (Stielow et al., 2015; Al-Sweih et al., 2019). Al-Sweih et al. applied sequencing of NTS-1 regions, a gene locus that is considered to have a higher discriminatory power, in C. fabianii outbreak investigation, and found that all outbreak strains in Kuwait shared 100% identical NTS-1 sequences (Al-Sweih et al., 2019). In comparison, we found a single SNP within NTS-1 region on patient 2’s strain versus strains from patients 1 and 3 in this study. However, further solid evidence was still needed to rule out the possibility of a potential outbreak.
To address concerns on readiness and limitations in discriminatory power of molecular typing methods in outbreak investigations, WGS has been recommended as a valuable alternative (Litvintseva et al., 2015; Bougnoux et al., 2018; Desnos-Ollivier et al., 2020). In this study, SNP-based analysis based on results acquired from WGS data clearly suggested that the genome of patient 2’s strain was quite divergent amongst the three clustered cases, which indicated a pseudo-outbreak event. Of note, the phrase “pseudo-outbreaks” could refer to either clustering of false infections, or artifactual clustering of real infections (Wallace et al., 1998). Clustering of false infections was more widely-noted, which may be associated with e.g. medical device or clinical laboratory contaminations (Kirby et al., 2017; Abdolrasouli et al., 2021). However, as indicated in our study, artifactual misinterpretation of “outbreaks” due to limitation of investigation methodologies (such as inadequate discriminatory power of molecular typing assays) should also be avoided.
Although WGS has made significant contributions in epidemiological studies, some limitations still remain. One major issue, as noted in outbreak investigations of all microbes including bacteria and fungi, is lack of consensus for data interpretation. Specifically, setting-up pairwise SNP-based cut-off values for assigning transmission events is still cumbersome, which has limited the wide utility of WGS in epidemiological studies (Coll et al., 2020; Guinea et al., 2021). In review of previous reports for outbreaks caused by yeast species that were characterized by WGS, it can be seen that the number of pairwise SNPs described in different studies of diverged species varied significantly, from less than ten to over hundreds. In the present study, genomic evidence clearly supported that patient 2’s C. fabianii strain was from a different origin, compared to others (with >26,000 SNPs compared to strains from patients 1 and 3). However, the 192 pairwise-SNP between strains from patient 1 and 3 may suggest that these two patients could have acquired the yeasts from a common source in the same ward but through different routes, rather than a direct transmission between the 2 patients, in which case the number of SNPs would be expected to be much less. But the hypothesis needs additional evaluation in a larger population and with more cases.
Due to the possibility of nosocomial transmission of this yeast in the described ward, surveillance infection control cultures were obtained to screen for C. fabianii in the department’s environment and amongst related health-care staff, but no C. fabianii was detected. Additional infection control strategies implemented further included enhancing environmental cleaning and hand hygiene practices, as well as providing education of fungal nosocomial infections to all healthcare staff.
One limitation of the study is that, antifungal susceptibility testing was not carried out using the standard broth microdilution methods, though YeastOne has proved equally efficient with good correlation in testing of yeasts (Cuenca-Estrella et al., 2010). Furthermore, with the limited number of cases studied, our base-line understanding for intra-species variation of C. fabianii genomes was still limited. Interpretation of any outbreak events shouldn’t simply rely on WGS result alone. It warrants a comprehensive analysis of different aspects of the cases, including patients’ clinical characteristics and epidemiological data, as well as the pathogens’ phenotypic and molecular characteristics.
In conclusion, as there are increasing reports of nosocomial outbreaks caused by emerging and uncommon fungal species, increased awareness of these rare organisms is warranted in public health. Conventional genotyping methods may have limited discriminatory power in investigating outbreaks due to these rare organisms; WGS has proven to be a good typing method for supporting investigation of such rare outbreak events.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Genbank: OP904191-OP904194 for ITS region, OP912967-OP912970 for NTS-1 region. WGS reads data can be found in NCBI database under Bioproject accession no. PRJNA907923.
Ethics statement
The studies involving human participants were reviewed and approved by Human Research Ethics Committee of the Beijing Chaoyang Hospital. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
XF, R-CD and LG conceived the work. XF, and R-CD performed the experiments and data analysis. XF, TK, and LG drafting the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Beijing Hospitals Authority Youth Program (grant no. QML20190301) and Reform and Development Program of Beijing Institute of Respiratory Medicine (XF and LG).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdolrasouli, A., Gibani, M. M., De Groot, T., Borman, A. M., Hoffman, P., Azadian, B. S., et al. (2021). A pseudo-outbreak of rhinocladiella similis in a bronchoscopy unit of a tertiary care teaching hospital in London, united kingdom. Mycoses 64, 394–404. doi: 10.1111/myc.13227
Al-Sweih, N., Ahmad, S., Khan, S., Joseph, L., Asadzadeh, M., Khan, Z. (2019). Cyberlindnera fabianii fungaemia outbreak in preterm neonates in Kuwait and literature review. Mycoses 62, 51–61. doi: 10.1111/myc.12846
Arastehfar, A., Daneshnia, F., Hilmioglu-Polat, S., Fang, W., Yasar, M., Polat, F., et al. (2020). First report of candidemia clonal outbreak caused by emerging fluconazole-resistant candida parapsilosis isolates harboring Y132F and/or Y132F+K143R in Turkey. Antimicrob. Agents Chemother. 64, e01001–20. doi: 10.1128/AAC.01001-20
Arastehfar, A., Fang, W., Al-Hatmi, A. M. S., Afsarian, M. H., Daneshnia, F., Bakhtiari, M., et al. (2019). Unequivocal identification of an underestimated opportunistic yeast species, cyberlindnera fabianii, and its close relatives using a dual-function PCR and literature review of published cases. Med. Mycol 57, 833–840. doi: 10.1093/mmy/myy148
Bougnoux, M. E., Brun, S., Zahar, J. R. (2018). Healthcare-associated fungal outbreaks: New and uncommon species, new molecular tools for investigation and prevention. Antimicrob. Resist. Infect. Control 7, 45. doi: 10.1186/s13756-018-0338-9
Chen, S. C., Perfect, J., Colombo, A. L., Cornely, O. A., Groll, A. H., Seidel, D., et al. (2021). Global guideline for the diagnosis and management of rare yeast infections: an initiative of the ECMM in cooperation with ISHAM and ASM. Lancet Infect. Dis. 21, e375–e386. doi: 10.1016/S1473-3099(21)00203-6
Chow, N. A., Chinn, R., Pong, A., Schultz, K., Kim, J., Gade, L., et al. (2020). Use of whole-genome sequencing to detect an outbreak of malassezia pachydermatis infection and colonization in a neonatal intensive care unit-california-2016. Infect. Control Hosp Epidemiol. 41, 851–853. doi: 10.1017/ice.2020.73
Chow, N. A., Gade, L., Tsay, S. V., Forsberg, K., Greenko, J. A., Southwick, K. L., et al. (2018). Multiple introductions and subsequent transmission of multidrug-resistant candida auris in the USA: a molecular epidemiological survey. Lancet Infect. Dis. 18, 1377–1384. doi: 10.1016/S1473-3099(18)30597-8
Chowdhary, A., Sharada, K., Singh, P. K., Bhagwani, D. K., Kumar, N., De Groot, T., et al. (2020a). Outbreak of dirkmeia churashimaensis fungemia in a neonatal intensive care unit, India. Emerg. Infect. Dis. 26, 764–768. doi: 10.3201/eid2604.190847
Chowdhary, A., Stielow, J. B., Upadhyaya, G., Singh, P. K., Singh, A., Meis, J. F. (2020b). Candida blankii: an emerging yeast in an outbreak of fungaemia in neonates in Delhi, India. Clin. Microbiol. Infect. 26 648, e645–648e648. doi: 10.1016/j.cmi.2020.01.001
Coll, F., Raven, K. E., Knight, G. M., Blane, B., Harrison, E. M., Leek, D., et al. (2020). Definition of a genetic relatedness cutoff to exclude recent transmission of meticillin-resistant staphylococcus aureus: a genomic epidemiology analysis. Lancet Microbe 1, e328–e335. doi: 10.1016/S2666-5247(20)30149-X
Cuenca-Estrella, M., Gomez-Lopez, A., Alastruey-Izquierdo, A., Bernal-Martinez, L., Cuesta, I., Buitrago, M. J., et al. (2010). Comparison of the vitek 2 antifungal susceptibility system with the clinical and laboratory standards institute (CLSI) and European committee on antimicrobial susceptibility testing (EUCAST) broth microdilution reference methods and with the sensititre YeastOne and etest techniques for in vitro detection of antifungal resistance in yeast isolates. J. Clin. Microbiol. 48, 1782–1786. doi: 10.1128/JCM.02316-09
Desnos-Ollivier, M., Maufrais, C., Pihet, M., Aznar, C., Dromer, F., French Mycoses Study, G. (2020). Epidemiological investigation for grouped cases of trichosporon asahii using whole genome and IGS1 sequencing. Mycoses 63, 942–951. doi: 10.1111/myc.13126
De St Maurice, A., Parti, U., Anikst, V. E., Harper, T., Mirasol, R., Dayo, A. J., et al. (2022). Clinical, microbiological, and genomic characteristics of clade-III candida auris colonization and infection in southern california-2022. Infect. Control Hosp Epidemiol. 1-9. doi: 10.1017/ice.2022.204
Escandon, P., Chow, N. A., Caceres, D. H., Gade, L., Berkow, E. L., Armstrong, P., et al. (2019). Molecular epidemiology of candida auris in Colombia reveals a highly related, countrywide colonization with regional patterns in amphotericin B resistance. Clin. Infect. Dis. 68, 15–21. doi: 10.1093/cid/ciy411
Eyre, D. W., Sheppard, A. E., Madder, H., Moir, I., Moroney, R., Quan, T. P., et al. (2018). And its control in an intensive care setting. N Engl. J. Med. 379, 1322–1331. doi: 10.1056/NEJMoa1714373
Guinea, J., Mezquita, S., Gomez, A., Padilla, B., Zamora, E., Sanchez-Luna, M., et al. (2021). Whole genome sequencing confirms candida albicans and candida parapsilosis microsatellite sporadic and persistent clones causing outbreaks of candidemia in neonates. Med. Mycol 60. doi: 10.1093/mmy/myab068
Hamal, P., Ostransky, J., Dendis, M., Horvath, R., Ruzicka, F., Buchta, V., et al. (2008). A case of endocarditis caused by the yeast pichia fabianii with biofilm production and developed in vitro resistance to azoles in the course of antifungal treatment. Med. Mycol 46, 601–605. doi: 10.1080/13693780802078180
Hoenigl, M., Seidel, D., Sprute, R., Cunha, C., Oliverio, M., Goldman, G. H., et al. (2022). COVID-19-associated fungal infections. Nat. Microbiol. 7, 1127–1140. doi: 10.1038/s41564-022-01172-2
Kanamori, H., Rutala, W. A., Sickbert-Bennett, E. E., Weber, D. J. (2015). Review of fungal outbreaks and infection prevention in healthcare settings during construction and renovation. Clin. Infect. Dis. 61, 433–444. doi: 10.1093/cid/civ297
Kato, M., Iefuji, H., Miyake, K., Iimura, Y. (1997). Transformation system for a wastewater treatment yeast, hansenula fabianii J640: isolation of the orotidine-5’-phosphate decarboxylase gene (URA3) and uracil auxotrophic mutants. Appl. Microbiol. Biotechnol. 48, 621–625. doi: 10.1007/s002530051105
Kidd, S. E., Abdolrasouli, A., Hagen, F. (2023). Fungal nomenclature: Managing change is the name of the game. Open Forum Infect. Dis. 10, ofac559. doi: 10.1093/ofid/ofac559
Kirby, J. E., Branch-Elliman, W., Lasalvia, M. T., Longhi, L., Mackechnie, M., Urman, G., et al. (2017). Investigation of a candida guilliermondii pseudo-outbreak reveals a novel source of laboratory contamination. J. Clin. Microbiol. 55, 1080–1089. doi: 10.1128/JCM.02336-16
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Litvintseva, A. P., Brandt, M. E., Mody, R. K., Lockhart, S. R. (2015). Investigating fungal outbreaks in the 21st century. PloS Pathog. 11, e1004804. doi: 10.1371/journal.ppat.1004804
Magill, S. S., O’leary, E., Janelle, S. J., Thompson, D. L., Dumyati, G., Nadle, J., et al. (2018). Changes in prevalence of health care-associated infections in U.S. hospitals. N Engl. J. Med. 379, 1732–1744. doi: 10.1056/NEJMoa1801550
Magruder, M., Sholi, A. N., Gong, C., Zhang, L. S., Edusei, E., Huang, J., et al. (2019). Gut uropathogen abundance is a risk factor for development of bacteriuria and urinary tract infection. Nat. Commun. 10, 5521. doi: 10.1038/s41467-019-13467-w
Mathy, V., Chousterman, B., Munier, A. L., Cambau, E., Jacquier, H., De Ponfilly, G. P. (2020). First reported case of postneurosurgical ventriculoperitonitis due to kocuria rhizophila following a ventriculoperitoneal shunt placement. Infect. Dis. Clin. Pract. 28, 169–170. doi: 10.1097/IPC.0000000000000829
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mikosz, C. A., Smith, R. M., Kim, M., Tyson, C., Lee, E. H., Adams, E., et al. (2014). Fungal endophthalmitis associated with compounded products. Emerg. Infect. Dis. 20, 248–256. doi: 10.3201/eid2002.131257
Mody, L., Greene, M. T., Meddings, J., Krein, S. L., Mcnamara, S. E., Trautner, B. W., et al. (2017). A national implementation project to prevent catheter-associated urinary tract infection in nursing home residents. JAMA Intern. Med. 177, 1154–1162. doi: 10.1001/jamainternmed.2017.1689
Nouraei, H., Pakshir, K., Zareshahrabadi, Z., Zomorodian, K. (2020). High detection of virulence factors by candida species isolated from bloodstream of patients with candidemia. Microb. Pathog. 149, 104574. doi: 10.1016/j.micpath.2020.104574
Odabasi, Z., Mert, A. (2020). Candida urinary tract infections in adults. World J. Urol 38, 2699–2707. doi: 10.1007/s00345-019-02991-5
Oliveira, M., Azevedo, L. (2022). Molecular markers: An overview of data published for fungi over the last ten years. J. Fungi (Basel) 8, 803. doi: 10.3390/jof8080803
Pappas, P. G., Lionakis, M. S., Arendrup, M. C., Ostrosky-Zeichner, L., Kullberg, B. J. (2018). Invasive candidiasis. Nat. Rev. Dis. Primers 4, 18026. doi: 10.1038/nrdp.2018.26
Park, J. H., Oh, J., Sang, H., Shrestha, B., Lee, H., Koo, J., et al. (2019). Identification and antifungal susceptibility profiles of cyberlindnera fabianii in Korea. Mycobiology 47, 449–456. doi: 10.1080/12298093.2019.1651592
Pearson-Stuttard, J., Blundell, S., Harris, T., Cook, D. G., Critchley, J. (2016). Diabetes and infection: assessing the association with glycaemic control in population-based studies. Lancet Diabetes Endocrinol. 4, 148–158. doi: 10.1016/S2213-8587(15)00379-4
Pfaller, M. A., Diekema, D. J., Turnidge, J. D., Castanheira, M., Jones, R. N. (2019). Twenty years of the SENTRY antifungal surveillance program: Results for Candida species from 1997-2016. Open Forum Infect. Dis. 6, S79–S94. doi: 10.1093/ofid/ofy358
Pulcrano, G., Roscetto, E., Iula, V. D., Panellis, D., Rossano, F., Catania, M. R. (2012). MALDI-TOF mass spectrometry and microsatellite markers to evaluate candida parapsilosis transmission in neonatal intensive care units. Eur. J. Clin. Microbiol. Infect. Dis. 31, 2919–2928. doi: 10.1007/s10096-012-1642-6
Reiss, E., Tanaka, K., Bruker, G., Chazalet, V., Coleman, D., Debeaupuis, J. P., et al. (1998). Molecular diagnosis and epidemiology of fungal infections. Med. Mycol 36 Suppl 1, 249–257.
Rhodes, J., Abdolrasouli, A., Farrer, R. A., Cuomo, C. A., Aanensen, D. M., Armstrong-James, D., et al. (2018). Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen candida auris. Emerg. Microbes Infect. 7, 43. doi: 10.1038/s41426-018-0098-x
Stielow, J. B., Levesque, C. A., Seifert, K. A., Meyer, W., Iriny, L., Smits, D., et al. (2015). One fungus, which genes? development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 35, 242–263. doi: 10.3767/003158515X689135
Svobodova, L., Bednarova, D., Ruzicka, F., Chrenkova, V., Dobias, R., Mallatova, N., et al. (2016). High frequency of candida fabianii among clinical isolates biochemically identified as candida pelliculosa and candida utilis. Mycoses 59, 241–246. doi: 10.1111/myc.12454
Teke, L., Baris, A., Bayraktar, B. (2021). Comparative evaluation of the bruker biotyper and vitek MS matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) systems for non-albicans candida and uncommon yeast isolates. J. Microbiol. Methods 185, 106232. doi: 10.1016/j.mimet.2021.106232
Thomaz, D. Y., Del Negro, G. M. B., Ribeiro, L. B., Da Silva, M., Carvalho, G., Camargo, C. H., et al. (2022). A Brazilian inter-hospital candidemia outbreak caused by fluconazole-resistant candida parapsilosis in the COVID-19 era. J. Fungi (Basel) 8, 100. doi: 10.3390/jof8020100
Treguier, P., David, M., Gargala, G., Camus, V., Stamatoullas, A., Menard, A. L., et al. (2018). Cyberlindnera jadinii (teleomorph candida utilis) candidaemia in a patient with aplastic anaemia: a case report. JMM Case Rep. 5, e005160. doi: 10.1099/jmmcr.0.005160
Van Rijswijck, I. M. H., Van Mastrigt, O., Pijffers, G., Wolkers-Rooijackers, J. C. M., Abee, T., Zwietering, M. H., et al. (2019). Dynamic modelling of brewers’ yeast and cyberlindnera fabianii co-culture behaviour for steering fermentation performance. Food Microbiol. 83, 113–121. doi: 10.1016/j.fm.2019.04.010
Wallace, R. J., Jr., Brown, B. A., Griffith, D. E. (1998). Nosocomial outbreaks/pseudo-outbreaks caused by nontuberculous mycobacteria. Annu. Rev. Microbiol. 52, 453–490. doi: 10.1146/annurev.micro.52.1.453
World Health Organization (2022). WHO fungal priority pathogens list to guide research, development and public health action. WHO. Available at: https://www.who.int/publications/i/item/9789240060241.
Xiao, M., Chen, S. C., Kong, F., Xu, X. L., Yan, L., Kong, H. S., et al. (2020). Distribution and antifungal susceptibility of candida species causing candidemia in China: An update from the CHIF-NET study. J. Infect. Dis. 221, S139–S147. doi: 10.1093/infdis/jiz573
Xiao, M., Wang, H., Lu, J., Chen, S. C., Kong, F., Ma, X. J., et al. (2014). Three clustered cases of candidemia caused by candida quercitrusa and mycological characteristics of this novel species. J. Clin. Microbiol. 52, 3044–3048. doi: 10.1128/JCM.00246-14
Yadav, A., Singh, A., Wang, Y., Haren, M. H. V., Singh, A., De Groot, T., et al. (2021). Colonisation and transmission dynamics of candida auris among chronic respiratory diseases patients hospitalised in a chest hospital, Delhi, India: A comparative analysis of whole genome sequencing and microsatellite typing. J. Fungi (Basel) 7, 81. doi: 10.3390/jof7020081
Zhai, B., Ola, M., Rolling, T., Tosini, N. L., Joshowitz, S., Littmann, E. R., et al. (2020). High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat. Med. 26, 59. doi: 10.1038/s41591-019-0709-7
Zhang, L., Xiao, M., Arastehfar, A., Ilkit, M., Zou, J., Deng, Y., et al. (2021). Investigation of the emerging nosocomial wickerhamomyces anomalus infections at a Chinese tertiary teaching hospital and a systemic review: Clinical manifestations, risk factors, treatment, outcomes, and anti-fungal susceptibility. Front. Microbiol. 12, 744502. doi: 10.3389/fmicb.2021.744502
Zhang, L., Yu, S. Y., Chen, S. C., Xiao, M., Kong, F., Wang, H., et al. (2020). Molecular characterization of candida parapsilosis by microsatellite typing and emergence of clonal antifungal drug resistant strains in a multicenter surveillance in China. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01320
Keywords: uncommon fungal pathogen, molecular typing, nosocomial transmission, Cyberlindnera fabianii, whole genome sequencing (WGS)
Citation: Fan X, Dai R-C, Kudinha T and Gu L (2023) A pseudo-outbreak of Cyberlindnera fabianii funguria: Implication from whole genome sequencing assay. Front. Cell. Infect. Microbiol. 13:1130645. doi: 10.3389/fcimb.2023.1130645
Received: 23 December 2022; Accepted: 21 February 2023;
Published: 07 March 2023.
Edited by:
Shangxin Yang, University of California, Los Angeles, United StatesReviewed by:
Marie Desnos-Ollivier, Institut Pasteur, FranceAndres Ceballos-Garzon, Université Clermont Auvergne, France
Copyright © 2023 Fan, Dai, Kudinha and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Gu, guli2013227@foxmail.com
†These authors have contributed equally to this work