A Concise Synthetic Strategy Towards the Novel Calcium-dependent Lipopeptide Antibiotic, Malacidin A and Analogues
 Nadiia Kovalenko1
Nadiia Kovalenko1  Georgina K. Howard1
Georgina K. Howard1  Jonathan A. Swain1
Jonathan A. Swain1  Yann Hermant1
Yann Hermant1  Alan J. Cameron1,2,3†
Alan J. Cameron1,2,3†  Gregory M. Cook3,4
Gregory M. Cook3,4  Scott A. Ferguson4
Scott A. Ferguson4  Louise A. Stubbing1
Louise A. Stubbing1  Paul W. R. Harris1,2,3*
Paul W. R. Harris1,2,3*  Margaret A. Brimble1,2,3*
Margaret A. Brimble1,2,3*- 1School of Chemical Sciences, The University of Auckland, Auckland, New Zealand
- 2School of Biological Sciences, The University of Auckland, Auckland, New Zealand
- 3Maurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Auckland, New Zealand
- 4Department of Microbiology and Immunology, School of Biomedical Sciences, University of Otago, Dunedin, New Zealand
Malacidin A is a novel calcium-dependent lipopeptide antibiotic with excellent activity against Gram-positive pathogens. Herein, a concise and robust synthetic route toward malacidin A is reported, employing 9-fluorenylmethoxycarbonyl solid-phase peptide synthesis of a linear precursor, including late-stage incorporation of the lipid tail, followed by solution-phase cyclization. The versatility of this synthetic strategy was further demonstrated by synthesis of a diastereomeric variant of malacidin A and a small library of simplified analogues with variation of the lipid moiety.
Introduction
The present COVID-19 pandemic shows how vulnerable society is to an infectious disease without access to an immediate effective treatment. Somewhat overshadowed by the current situation but equally as urgent, antimicrobial resistance (AMR) represents another ongoing global health crisis. According to World Health Organization (WHO) reports, as many as 2.8 million people contract infections caused by AMR pathogens in the U.S. alone, leading to more than 35,000 deaths annually (Centers for Disease Control and Prevention (U.S.), 2019; World Health Organization, 2019). Similar statistics are observed for Europe (Cassini et al., 2019). Although new stewardship programs and policies to increase AMR awareness and limit the use of existing antimicrobials are being introduced around the world, the existing clinical pipeline does not meet the demand to effectively combat increasing rates of AMR infections (World Health Organization, 2015). Thus, novel antimicrobial agents that can be developed into potential drug candidates are critically needed. Over the last 20 years antimicrobial peptides (AMPs) emerged as a rich yet underexplored source of such compounds. Development of alternative platforms for AMP discovery and methods for synthetic optimizations of natural scaffolds are yielding many promising examples of clinically relevant AMPs (Jin, 2020; Liu et al., 2021).
In 2018, Brady et al. reported the isolation of malacidin A (1) as part of an extensive metagenomic mining study of bacterial DNA obtained from soil samples in search of novel bioactive natural products (Owen et al., 2013; Hover et al., 2018). This novel AMP possesses potent bioactivity against a range of Gram-positive strains including multi-drug resistant pathogens such as methicillin-resistant Staphylococcus aureus (MRSA) (minimum inhibitory concentrations (MIC) 0.2–0.8 μg ml−1) and vancomycin-resistant Enterococcus faecium (VRE) (MIC 0.8–2.0 μg ml−1) (Hover et al., 2018). Malacidin A (1) belongs to a family of calcium-dependent lipopeptide antibiotics (CDLAs) that exhibit their activity upon binding to calcium ions. The CDLA family is represented by several sub-groups of potent antibiotics: A21978C complex, which includes the antibiotic of last resort, daptomycin; A54145 complex; calcium dependent antibiotics (CDAs); friulimicins, of which friulimicin B reached Phase I clinical trials; amphomycins, of which, MX-2401 (a semi-synthetic analogue), progressed to late-stage preclinical development; glycinocins and, recently, cadasides (Wood and Martin, 2019; Wu et al., 2019). Malacidin A (1) is structurally unique compared to other common CDLA members in that the canonical Asp-AA-Asp-Gly (AA = Gly or d-amino acid) calcium-binding motif lacks the spacer residue, AA, and the first Asp residue is replaced by an unusual 3-hydroxy aspartic acid (3-HyAsp) (Hover et al., 2018). Preliminary mechanistic studies of malacidin A revealed binding to lipid II, a different target compared to other CDLAs. Malacidin A (1) was also found to be non-cytotoxic and did not induce resistance after repeated exposure to S. aureus (Hover et al., 2018). These features render 1 an exciting target for development as a novel antibiotic. The key step toward this goal is design of robust synthetic routes that would enable facile access to the lead compound and analogues thereof to establish structure activity relationships (SARs).
Herein, we report a concise synthetic strategy toward malacidin A (1) as demonstrated by the synthesis of a diastereomeric variant and simplified analogues thereof. The key steps involve preparation of the key linear precursor by 9-fluorenylmethoxycarbonyl (Fmoc)-solid-phase peptide synthesis (SPPS), followed by tail-to-side chain solution-phase cyclisation.
Structurally, malacidin A (1) consists of a 9-mer cyclic core and a single exocyclic amino acid, 3-methylaspartic acid (3-MeAsp1). The lipopeptide is acylated at the N-terminus with an unusual polyunsaturated lipid tail, (2E,4Z)-8-methylnona-2,4-dienoic acid (Figure 1). The macrolactam bond is formed between the side chain of 3-methyldiaminopropinoic acid (3-MeDap2) and the C-terminal carboxyl group of (4R)-4-methylproline ((4R)-4-MePro10). The sequence of malacidin A (1) is unusually rich in non-canonical amino acids that include the aforementioned 3-MeAsp1, 3-MeDap2, 3-HyAsp5, (4R)-4-MePro10, as well as d-Val3 and d-3-MeAsp8. Upon discovery of malacidin A (1), the exact configuration of the β-carbon centers in 3-MeAsp1, 3-MeDap2, 3-HyAsp5 and d-3MeAsp8 could not be determined, thus giving rise to 16 possible diastereomers (Hover et al., 2018). Therefore, it was decided to concentrate synthetic efforts on a diastereomer of malacidin A (1a) that contained (2S,3S)-3-MeAsp1, (2S,3S)-3-MeDap2, (2S,3S)-3-HyAsp5 and (2R,3R)-d-3-MeAsp8. This choice was based on structural and biosynthetic gene cluster similarities of malacidin A (1) and friulimicin B (Müller et al., 2007; Hover et al., 2018).
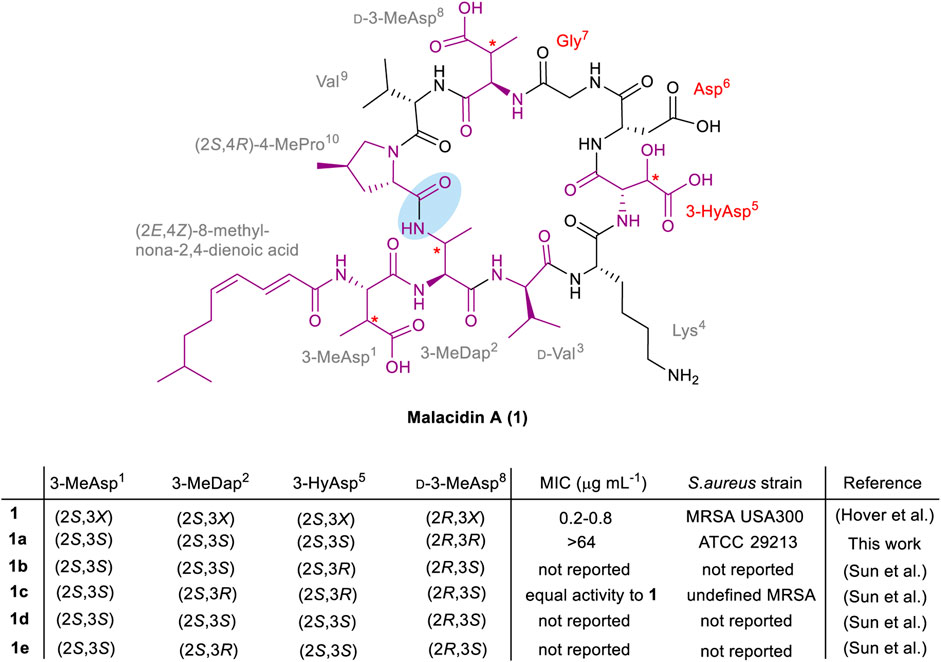
FIGURE 1. Structure of malacidin A (1); non-canonical amino acids and the lipid tail are highlighted in purple; the undefined stereochemistry of β-carbon centers is indicated with an asterisk; the calcium-binding region residues are specified in red; the macrolactam bond is highlighted with blue shading. The stereochemistry and MIC values of the isolated malacidin A (1) (Hover et al., 2018) and synthesized diastereomers [this work 1a, the Li group 1b–1e (Sun et al., 2020)] are shown above; X in 1 represents undefined stereochemistry.
During the course of this work, the first total synthesis of malacidin A (1c) and three diastereomers (1b, 1d and 1e) was reported by Sun et al., establishing the exact stereoconfiguration of the natural product (Sun et al., 2020). This involved Fmoc-SPPS of a branched precursor followed by solution-phase β-hydroxy-mediated cyclization between 3-HyAsp5 and a salicylaldehyde ester of Lys4, employing the Ser/Thr ligation approach developed by the Li group (Li et al., 2010). Peptides 1b-1e were prepared in 2–6% overall yields with variation at residue positions 2 and 5, employing appropriately protected derivatives of (2S,3S)- or (2S,3R)-3-MeDap, and (2S,3S)- or (2S,3R)-3-HyAsp, respectively. A close match between the NMR spectra of synthetic 1c, bearing (2S,3S)-3-MeAsp1, (2S,3R)-3-MeDap2, (2S,3R)-3-HyAsp5 and (2R,3S)-3-MeAsp8 residues, and isolated malacidin A (1), established the exact stereochemistry of the natural product (Sun et al., 2020). Along with the observed bioactivity, advanced Marfey’s analysis of the synthetic amino acids used to prepare 1c, compared to those obtained by hydrolysis of isolated malacidin A (1) further confirmed the stereochemical assignment. The MIC values were not reported for the other three diastereomers of malacidin A (1b, 1d and 1e), presumably due to their inactivity; however, this information would be required to establish an SAR. As the synthesis of the diastereomer 1a described herein commenced before the total synthesis and determination of the actual configuration of malacidin A (1) was published, our original synthetic strategy was still pursued to assess the significance of the opposite β-stereocenters of the three non-canonical residues (3-MeDap2, 3-HyAsp5 and d-3-MeAsp8) on the activity of the antibiotic.
Results and Discussion
Synthesis of Unnatural Amino Acid Building Blocks
Before synthesis of the diastereomer of malacidin A (1a) could commence, appropriately protected building blocks of (2S,3S)-3-MeAsp1, (2S,3S)-3-MeDap2, (2S,3S)-3-HyAsp5, (2R,3R)-3-MeAsp8 and (2S,4R)-4-MePro10 and (2E,4Z)-8-methylnona-2,4-dienoic acid were required to facilitate incorporation using solid-phase synthesis.
Synthesis of (2S,3S)-Fmoc-3-MeAsp(OtBu)-OH (2) and (2R,3R)-Fmoc-3-MeAsp(OtBu)-OH (7)
The initial synthetic strategy of (2S,3S)-Fmoc-3-MeAsp(OtBu)-OH (2) from H-l-Asp(OtBu)-OH took inspiration from similar work reported by Giltrap et al. (2017), with the preparation of methylated l-Asp 4 adapted from procedures outlined by Xue et al. (2002) (Scheme 1A). Tri-benzylation, methylation, and global benzyl deprotection of H-l-Asp(OtBu)-OH afforded the (2S,3S) and (2S,3R) diastereomers (5 and 6, respectively) as a 1:1 mixture, which were separated by silica gel flash chromatography. Fmoc protection of 5 delivered the desired (2S,3S) building block 2. The enantiomeric (2R,3R) analogue 7 was prepared in an analogous fashion from H-d-Asp(OtBu)-OH (Scheme 1B).
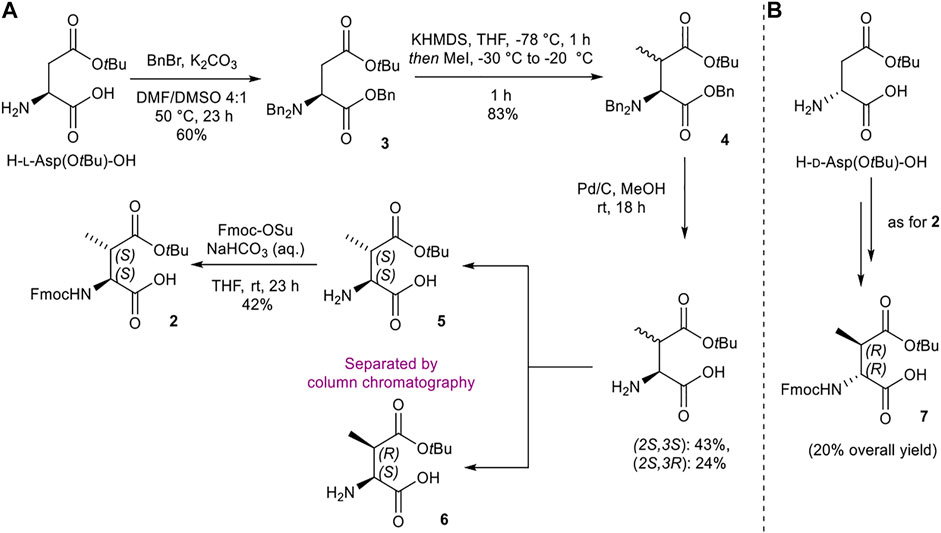
SCHEME 1. Synthesis of (2S,3S)-Fmoc-3-MeAsp(OtBu)-OH (2) and (2R,3R)-Fmoc-3-MeAsp(OtBu)-OH (7) building blocks.
Synthesis of (2S,3S)-Fmoc-3-MeDap(Dde)-OH (8)
(2S,3S)-Fmoc-3-MeDap(Dde)-OH (8) was prepared from procedures adapted from Martín et al., utilizing a N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl) (Dde) protecting group (Scheme 2) (Martín et al., 2014). tert-Butyloxycarbonyl (Boc) and benzyl protections of l-threonine afforded 9. Sequential tosylation and nucleophilic substitution with sodium azide yielded azide 11 with the correct (3S) stereochemistry, along with a considerable amount of undesired elimination byproduct 12. Hydrogenation and Dde protection of the resultant amine with 13 (prepared from dimedone) (Armaly et al., 2017), followed by exchange of the Nα-Boc protecting group for Fmoc protecting group afforded the desired building block 8.
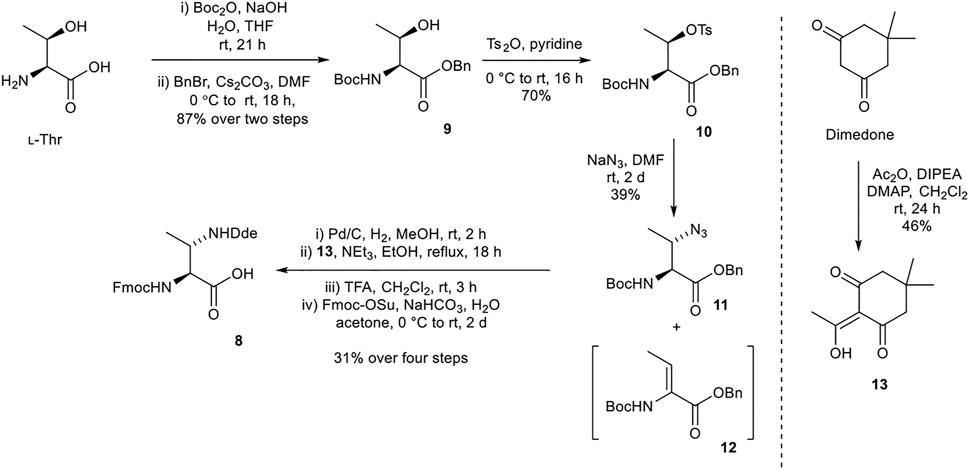
SCHEME 2. Synthesis of (2S,3S)-Fmoc-3-MeDap(Dde)-OH (8).
Synthesis of (2S,3S)- Fmoc-3-Hy(TBS)Asp(OtBu)-OH (14)
(2S,3S)-Fmoc-3-Hy(TBS)Asp(OtBu)-OH (14) was prepared following literature precedent (Scheme 3) (Moreira and Taylor, 2018). α,β-Unsaturated ester 15 was synthesized from 2-(benzyloxy)acetaldehyde via a Horner–Wadsworth–Emmons reaction under Masamune–Roush conditions. Sharpless asymmetric aminohydroxylation using FmocNHCl (19, readily available from 9-fluorenylmethyl carbamate) (Gwon et al., 2015) as a nitrogen source afforded 16. tert-Butyldimethylsilyl (TBS) protection of the hydroxyl group using TBSOTf yielded 17. Attempted benzyl deprotection using Bobbit’s salt (4-acetamido-2,2,6,6-tetramethylpiperidine·BF4) returned only starting material, while deprotection using BCl3 and pentamethylbenzene resulted in concomitant loss of the tBu ester. Hydrogenation of 17 removed the benzyl and Fmoc protecting groups concurrently, and the free amine was reprotected using Fmoc-OSu to afford 18. Finally, oxidation of alcohol 18 using TEMPO/NaClO2/NaOCl afforded the desired building block 14.
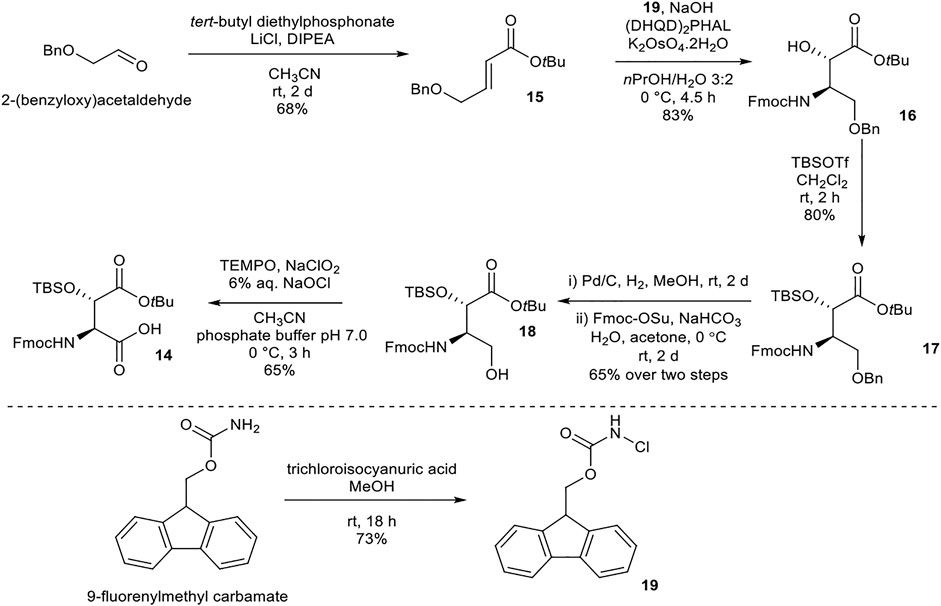
SCHEME 3. Synthesis of (2S,3S)-Fmoc-3-Hy(TBS)Asp(OtBu)-OH (14) building block.
Synthesis of (2S,4R)-Fmoc-4-MePro-OH (20)
(2S,4R)-Fmoc-4-MePro-OH (20) was prepared according to procedures adapted from Murphy et al. (Murphy et al., 2008) (Scheme 4). Nα-Boc protection and tBu esterification of (4R)-4-hydroxy-l-proline afforded alcohol 21. Dess-Martin periodinane oxidation of 21 to ketone 22, followed by Wittig reaction with Ph3PMeBr afforded alkene 23. Selective reduction of 23 using Crabtree’s catalyst installed the desired (4R) stereochemistry. Global deprotection and Nα-Fmoc protection afforded the desired building block 20.
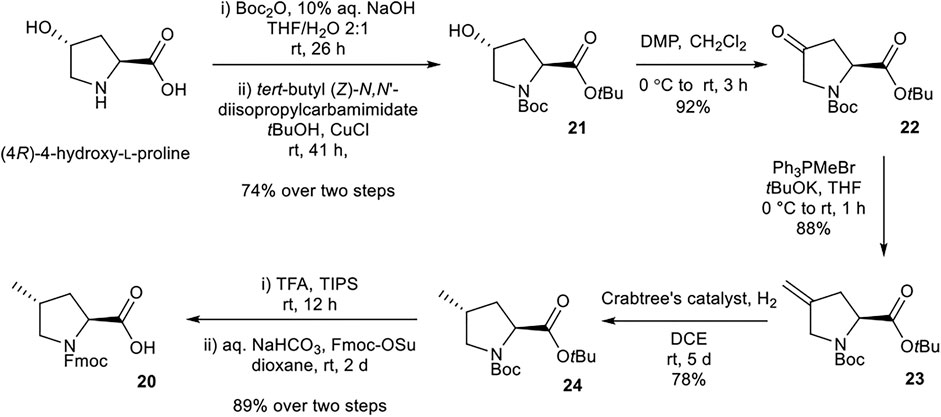
SCHEME 4. Synthesis of (2S,4R)-Fmoc-4-MePro-OH (20) building block.
Synthesis of (2E,4Z)-8-methylnona-2,4-dienoic acid (25)
The lipid building block 25 was prepared from readily available 5-methylhexyne (Scheme 5). Sequential bromination, boron-mediated reduction, and Sonogashira cross-coupling with propargyl alcohol provided enyne 28. Alkyne reduction and two-step oxidation afforded desired (2E,4Z) polyunsaturated carboxylic acid 25. The acidic nature of silica gel resulted in partial isomerization of the (4Z)-alkene upon attempted purification of 25. Instead, purification and resulting undesired isomerization could be avoided by assuring complete purity of precursor 30 which upon oxidation yielded (2E,4Z)-25 in excellent purity.
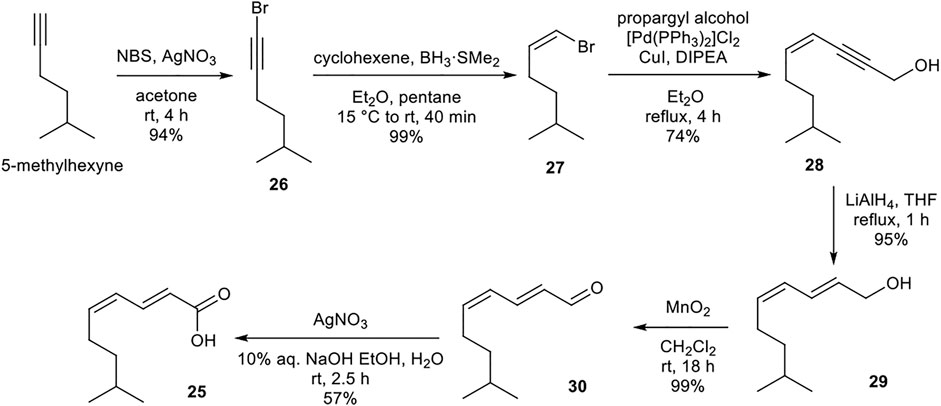
SCHEME 5. Synthesis of (2E,4Z)-8-methylnona-2,4-dienoic acid (25) building block.
Synthesis of Analogues of Malacidin A
A robust synthetic strategy toward diastereomer 1a was established by first preparing a simplified analogue (31), wherein all non-canonical amino acids and the unsaturated lipid tail were substituted for the corresponding canonical/commercially available amino acids and decanoic acid, respectively (Figure 2). The synthetic route was inspired by the synthesis of glycinocins A-C reported by the Payne group (Corcilius et al., 2017). It was envisioned that macrocyclization would take place at the same site as the native peptide’s macrolactam bond, i.e. between the β-NH2 of the Dap2 derivative and α-CO2H of Pro10. The protected linear precursor was assembled via Fmoc-SPPS on a hyper acid-labile 2-chlorotrityl chloride (2-CTC) polystyrene (PS) resin (Scheme 6). The use of 2-CTC PS resin was also required to prevent diketopiperazine formation upon incorporation of Pro as the C-terminal residue. This synthetic approach required an orthogonally protected Dap2 building block, thus, commercially available Fmoc-Dap(Alloc)-OH was used. As the peptide sequence contains an aspartamide-prone Asp6Gly7 region, dimethoxybenzyl (Dmb)Nα-protected Gly10 was used.
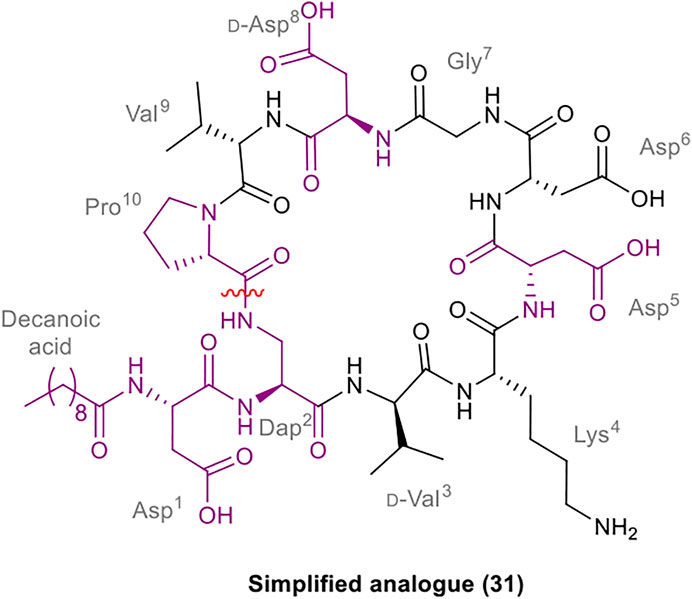
FIGURE 2. Structure of the simplified analogue of malacidin A (31); The substituted residues are highlighted in purple; the disconnection site is shown with a red wavy line.
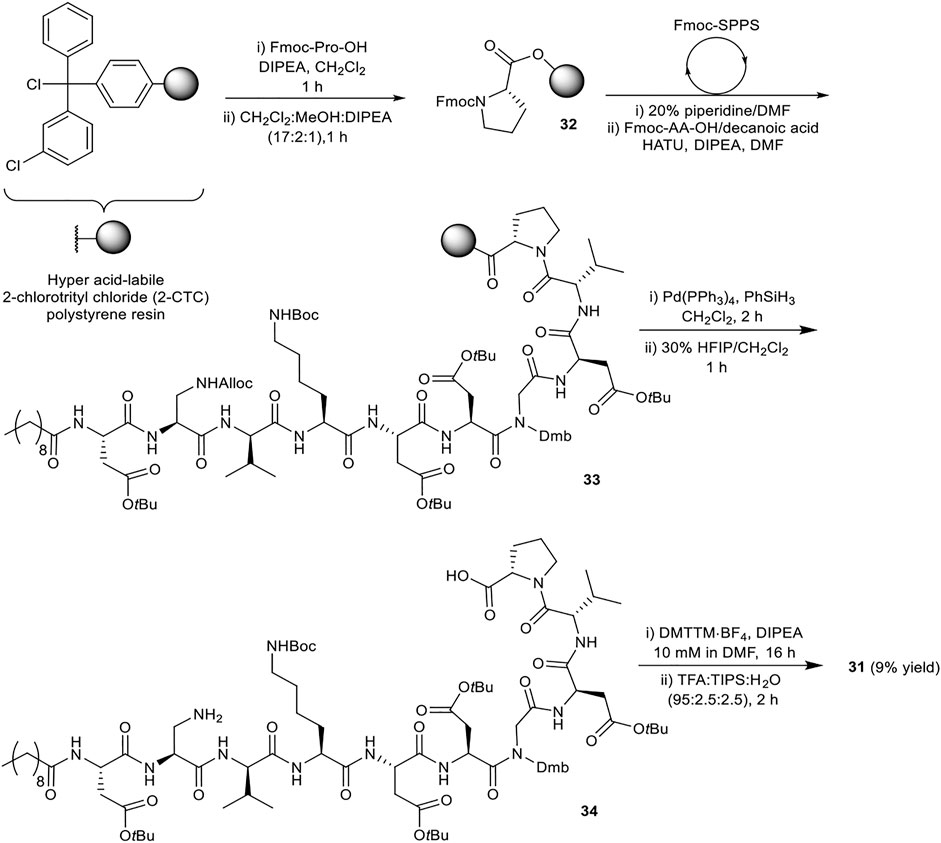
SCHEME 6. Synthesis of simplified analogue 31.
The synthesis of 31 commenced with attachment of Fmoc-Pro-OH to 2-CTC resin to give resin-bound 32 (Scheme 6). After capping of unreacted chloride sites with methanol and N,N-diisopropylethylamine (DIPEA), the assembly of orthogonally protected linear precursor 33 was undertaken. Fmoc removal was achieved by treatment with 20% piperidine in N,N-dimethyl formamide (DMF) (v/v). l-Amino acids were coupled using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) and DIPEA in DMF for 30 min, and for 2 h when using d-amino acids, Dap2, Gly7 and decanoic acid. Double coupling was found to be necessary for Fmoc-Dap(Alloc)-OH. Next, Alloc removal from Dap2 was carried out using tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) and phenylsilane (PhSiH3). Next the assembled fully protected linear peptide was cleaved from the resin with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) in CH2Cl2 (3:7, v/v) for 1 h. Following solvent evaporation the peptidic residue was dissolved in H2O:CH3CN (1:4, v/v) and lyophilized. The obtained side chain protected linear precursor 34 was subjected to macrocyclization conditions (peptide concentration 10 mM) using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate (DMTTM·BF4) and DIPEA in DMF overnight. No difficulties were observed during the handling and dissolution of the maximally protected peptide. The final side chain deprotection was performed using trifluoroacetic acid (TFA), triisopropylsilane (TIPS), and H2O (95:2.5:2.5, v/v/v) for 2 h, followed by purification by reverse-phase high pressure liquid chromatography (RP-HPLC) to give simplified analogue 31 in 9% overall yield.
Late-stage incorporation of the lipid enabled the preparation of five additional lipid tail analogues based on the simplified peptidic core of 31, namely hexanoyl, tetradecanoyl, hexadecanoyl, 4-pentylbenzoyl and 4-phenylbenzoyl analogues 36–40 (Scheme 7). The previously detailed SPPS protocol was used to prepare the common Fmoc protected linear sequence 35, followed by division of the resin for coupling to the various lipid tails. The remaining steps of the synthesis for each analogue were carried out separately using the same conditions as for 31. Analogues 36–40 were thus obtained in 5–17% yield in >96% purity after RP-HPLC purification.
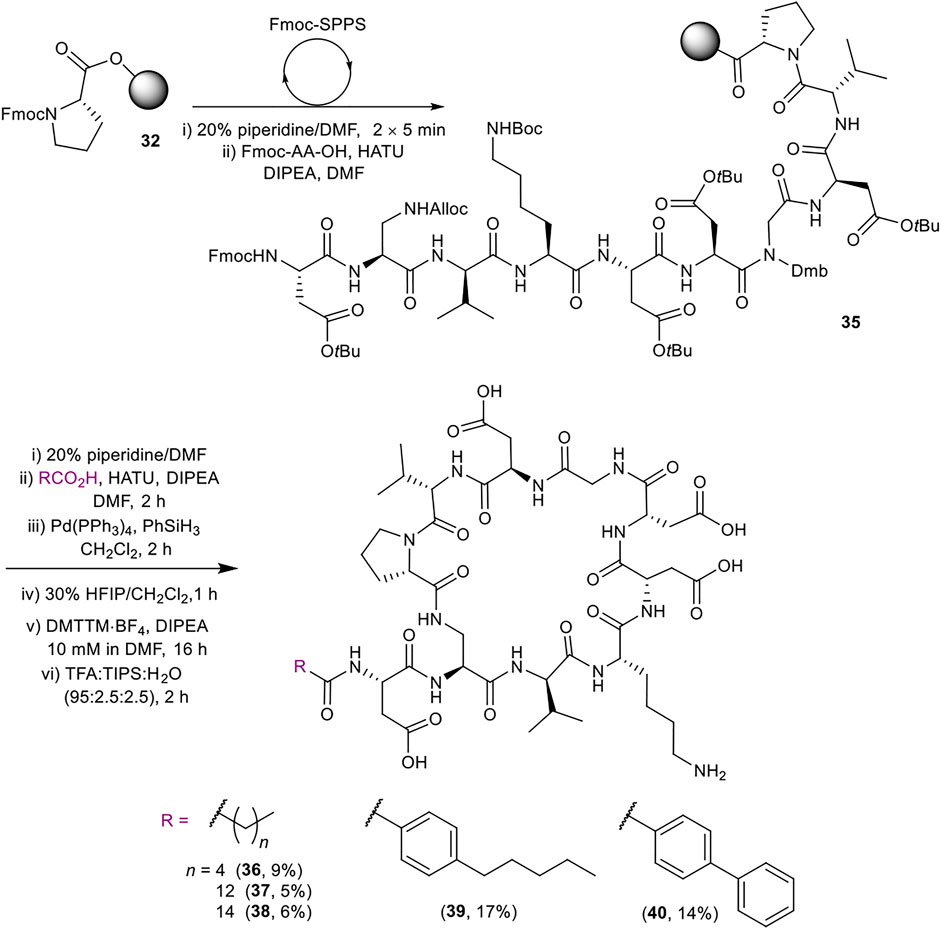
SCHEME 7. Synthesis of simplified analogues (36–40) with variation in the lipid moiety. Overall yields are given in brackets.
Attention next focused on synthesis of the selected diastereomer of malacidin A 1a. While the same overall strategy could be used, some modification of the synthesis was required to incorporate the (2S,3S)-Fmoc-3-MeDap(Dde)-OH (8), (2S,3S)-Fmoc-3-Hy(TBS)Asp(OtBu)-OH (14) and (2E,4Z)-8-methylnona-2,4-dienoic acid (25) building blocks. The unsaturated nature of the lipid tail precluded use of Alloc as a protecting group on the 3-MeDap2 residue sidechain (Keinan and Greenspoon, 1986). (2S,3S)-Fmoc-3-MeDap(Dde)-OH (8) was therefore prepared, as the Dde group could be removed from the fully assembled linear precursor under mild conditions using hydroxylamine hydrochloride/imidazole without affecting the unsaturation of the lipid tail (Díaz-Mochón et al., 2004). Lipid stability investigations (see Supplementary Material Section 5) revealed that partial cis-trans isomerization of the (4Z)-π-bond of the lipid could be minimized by performing the final side chain deprotection in 50% TFA for 30 min. However, removal of the TBS protecting group was incomplete under these conditions and a separate deprotection step was required.
The synthesis of malacidin analogue 1a began with loading of (2S,4R)-Fmoc-4-MePro-OH (20) onto 2-CTC resin followed by iterative Fmoc-SPPS (Scheme 8). Commercially available amino acids were incorporated using the previously established conditions. Custom building blocks (2, 7, 8, 14, 25) were coupled using (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU)/ethyl cyano(hydroxyimino)acetate (Oxyma Pure)/DIPEA as the coupling reagents to maximize the coupling efficiency and reduce the number of molecular equivalents required for complete coupling (1.2 equiv.). Next, TBS removal from the side chain hydroxy group of 3-HyAsp5 residue using tetrabutylammonium fluoride (TBAF) buffered with AcOH (1:1, 15 eq.) was performed on peptidyl-resin 42 followed by Fmoc removal and coupling of the polyunsaturated lipid 25. Nβ-Dde removal from the 3-MeDap2 residue was carried out under mild conditions using 3.6 M NH2OH·HCl and 2.7 M imidazole in NMP/CH2Cl2 (5:1, v/v) for 4 h. Following cleavage of the resulting peptide sequence from the resin with HFIP:CH2Cl2 (3:7, v/v) and solvent evaporation the crude peptide was dissolved in H2O:CH3CN (1:4, v/v) and lyophilized. The obtained protected linear peptide 43 was subjected to macrocyclization with DMTTM·BF4 and DIPEA at 10 mM dilution. As above, no difficulties were experienced during manipulations of the protected peptide. Gratifyingly, the reaction proceeded smoothly with complete consumption of the starting material in 4.5 h. Finally, side chain removal was carried out using the optimized deprotection cocktail of TFA:CH2Cl2:H2O:TIPS (50:45:2.5:2.5, v/v/v/v) for 30 min with no detectable isomerization of the polyunsaturated lipid. To reduce exposure of the acid-sensitive polyunsaturated lipid to highly acidic TFA during HPLC purification, an eluent system of H2O and CH3CN containing 0.1% formic acid was employed, providing 1a in 7% overall yield.
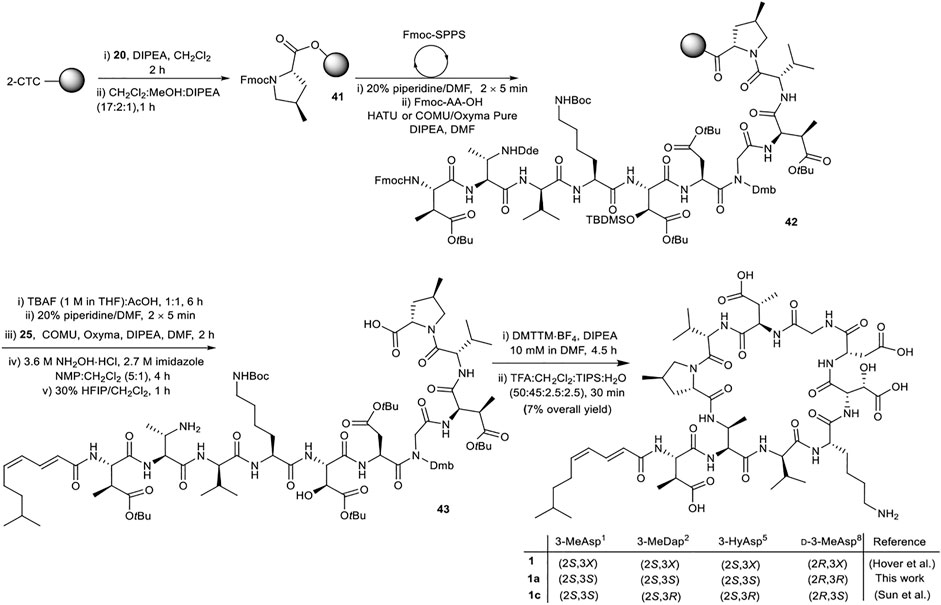
SCHEME 8. Synthesis of diastereomer of malacidin A, 1a, employing chemically synthesized building blocks. X represents undefined β-carbon-stereochemistry in the isolated malacidin A (Hover et al., 2018), 1c represents the correct stereochemistry of malacidin A (Sun et al., 2020).
Evaluation of the antibacterial activity of the synthesized analogues 1a, 31 and 36–40 was then undertaken. Unfortunately, no activity toward S. aureus was observed for these analogues using media supplemented with 1.25–1.5 mM CaCl2, as recommended for biotesting of daptomycin (Wiegand et al., 2008) (Supplementary Tables S2, S3). As native malacidin A (1) showed maximum activity at 15 mM CaCl2 (Hover et al., 2018), diastereomer 1a was also tested at this concentration but no activity was observed (Supplementary Table S2). This observation indicates that both the presence and absolute configuration of the β-substituents of the non-canonical amino acids are important for the activity of the antibiotic, most likely through their interaction with Ca2+ ions to form the active antibiotic-Ca2+ complex. This observation is not unusual, as previous reports of SAR studies of A54145 D and daptomycin CDLAs showed that removal or reversal of configuration at even one stereocenter may result in a significant reduction or complete loss of bioactivity (Kralt et al., 2019; Xu et al., 2019). It is likely that orientation of the β-OH of 3-HyAsp5, that forms part of the calcium binding motif, is highly important to provide efficient coordination to Ca2+ ions. Further SAR studies are required to assess the contribution of the individual non-canonical residues to the antimicrobial activity of malacidin A.
Comparison of the 1H and 13C NMR spectra of diastereomer 1a to the reported spectra of malacidin A (under similar conditions, see SI), showed significant differences in the α-proton region (Hover et al., 2018) (Supplementary Figure S12). The 1H spectrum of daptomycin is known to change upon Ca2+ binding, demonstrating significant line broadening that is characteristic of aggregation (Ball et al., 2004). To investigate the Ca2+ binding capability of 1a, the 1H NMR spectra of 1a was recorded in the presence of CaCl2 at 1.5 mM and 15 mM (Supplementary Figure S16), concentrations similar to those used in MIC assays for daptomycin (typically 1.25 mM) and malacidin (1) respectively. Disappointingly, no detectable signal shifts or line broadening were observed (Supplementary Figure S14), indicating that 1a fails to interact with Ca2+ ions, hence the lack of antibacterial activity. Despite these differences, it was observed that the lipid sp2 proton signals closely matched that of the natural product and no peaks arising from cis-trans isomerization were observed, indicating the tolerance of acid-sensitive lipid 25 to the optimized TFA side chain deprotection conditions and HPLC purification protocols.
In summary, seven novel analogues of malacidin A (1) were synthesized using primarily an Fmoc-SPPS-based strategy followed by late stage solution-phase macrolactamization and subsequent side chain deprotection. One diastereomeric analogue of the native sequence, 1a, and six simplified analogues containing all canonical/commercially available amino acids with variations in the lipid tail (31, 36–40) were obtained in good overall yields. Despite the lack of activity observed for these analogues, the concise and versatile synthetic strategy reported herein lays a foundation for further SAR studies of malacidin A. In contrast to the reported synthesis of malacidin A, the synthetic route described herein has improved yields, requires no additional amino acids bearing auxiliary groups to aid cyclization, and involves minimal solution-phase manipulations. The mostly solid-phase strategy also permits a single, final purification step. Additionally, a late stage incorporation of the lipid moiety on resin enables facile preparation of lipid analogues to probe the role of the lipid unsaturation and branching on antibacterial activity.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
NK and YH performed the peptide synthesis, GH and JS synthesized amino acid building blocks, AC undertook bioassays and NR analysis, GMC and SAF performed bioassays and interpreted the results, LS and MB designed and supervised the amino acids building block approaches, PH and MB planned and oversaw the peptide synthesis. All authors contributed to manuscript preparation and all authors have given approval to the final version of the manuscript.
Funding
The authors wish to acknowledge the Ministry of Business, Innovation and Employment (MBIE Endeavour grant UOAX 2010) for generous financial support and the Maurice Wilkins Centre for Molecular Biodiscovery. AC acknowledges the Lottery Health Research for a Postdoctoral Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Mel Knottenbelt is thanked for generous technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.687875/full#supplementary-material
References
Armaly, A. M., Bar, S., and Schindler, C. S. (2017). Acid Chlorides as Formal Carbon Dianion Linchpin Reagents in the Aluminum Chloride-Mediated Dieckmann Cyclization of Dicarboxylic Acids. Org. Lett. 19, 3962–3965. doi:10.1021/acs.orglett.7b01623
Ball, L.-J., Goult, C. M., Donarski, J. A., Micklefield, J., and Ramesh, V. (2004). NMR Structure Determination and Calcium Binding Effects of Lipopeptide Antibiotic Daptomycin. Org. Biomol. Chem. 2, 1872–1878. doi:10.1039/B402722A
Cassini, A., Högberg, L. D., Plachouras, D., Quattrocchi, A., Hoxha, A., Simonsen, G. S., et al. (2019). Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: a Population-Level Modelling Analysis. Lancet Infect. Dis. 19, 56–66. doi:10.1016/S1473-3099(18)30605-4
Centers for Disease Control and Prevention (U.S.) (2019). Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: Centers for Disease Control and Prevention (U.S.). doi:10.15620/cdc:82532
Corcilius, L., Elias, N. T., Ochoa, J. L., Linington, R. G., and Payne, R. J. (2017). Total Synthesis of Glycinocins A-C. J. Org. Chem. 82, 12778–12785. doi:10.1021/acs.joc.7b01959
Díaz-Mochón, J. J., Bialy, L., and Bradley, M. (2004). Full Orthogonality Between Dde and Fmoc: The Direct Synthesis of PNA--Peptide Conjugates. Org. Lett. 6 (7), 1127–1129. doi:10.1021/ol049905y
Giltrap, A. M., Haeckl, F. P. J., Kurita, K. L., Linington, R. G., and Payne, R. J. (2017). Total Synthesis of Skyllamycins A-C. Chem. Eur. J. 23, 15046–15049. doi:10.1002/chem.201704277
Gwon, D., Hwang, H., Kim, H. K., Marder, S. R., and Chang, S. (2015). Synthesis of 8-Aminoquinolines by Using Carbamate Reagents: Facile Installation and Deprotection of Practical Amidating Groups. Chem. Eur. J. 21, 17200–17204. doi:10.1002/chem.201503511
Hover, B. M., Kim, S.-H., Katz, M., Charlop-Powers, Z., Owen, J. G., Ternei, M. A., et al. (2018). Culture-independent Discovery of the Malacidins as Calcium-dependent Antibiotics with Activity against Multidrug-Resistant Gram-Positive Pathogens. Nat. Microbiol. 3, 415–422. doi:10.1038/s41564-018-0110-1
Jin, K. (2020). Developing Cyclic Peptide-Based Drug Candidates: an Overview. Future Med. Chem. 12, 1687–1690. doi:10.4155/fmc-2020-0171
Keinan, E., and Greenspoon, N. (1986). Highly Chemoselective Palladium-Catalyzed Conjugate Reduction of .alpha.,.beta.-unsaturated Carbonyl Compounds with Silicon Hydrides and Zinc Chloride Cocatalyst. J. Am. Chem. Soc. 108, 7314–7325. doi:10.1021/ja00283a029
Kralt, B., Moreira, R., Palmer, M., and Taylor, S. D. (2019). Total Synthesis of A54145 Factor D. J. Org. Chem. 84, 12021–12030. doi:10.1021/acs.joc.9b01938
Li, X., Lam, H. Y., Zhang, Y., and Chan, C. K. (2010). Salicylaldehyde Ester-Induced Chemoselective Peptide Ligations: Enabling Generation of Natural Peptidic Linkages at the Serine/Threonine Sites. Org. Lett. 12, 1724–1727. doi:10.1021/ol1003109
Liu, Y., Shi, J., Tong, Z., Jia, Y., Yang, B., and Wang, Z. (2021). The Revitalization of Antimicrobial Peptides in the Resistance Era. Pharmacol. Res. 163, 105276. doi:10.1016/j.phrs.2020.105276
Martín, M. J., Rodríguez-Acebes, R., García-Ramos, Y., Martínez, V., Murcia, C., Digón, I., et al. (2014). Stellatolides, a New Cyclodepsipeptide Family from the Sponge Ecionemia Acervus: Isolation, Solid-phase Total Synthesis, and Full Structural Assignment of Stellatolide A. J. Am. Chem. Soc. 136, 6754–6762. doi:10.1021/ja502744a
Moreira, R., and Taylor, S. D. (2018). Asymmetric Synthesis of Fmoc-Protected β-Hydroxy and β-Methoxy Amino Acids via a Sharpless Aminohydroxylation Reaction Using FmocNHCl. Org. Lett. 20, 7717–7720. doi:10.1021/acs.orglett.8b03458
Murphy, A. C., Mitova, M. I., Blunt, J. W., and Munro, M. H. G. (2008). Concise, Stereoselective Route to the Four Diastereoisomers of 4-Methylproline. J. Nat. Prod. 71, 806–809. doi:10.1021/np070673w
Müller, C., Nolden, S., Gebhardt, P., Heinzelmann, E., Lange, C., Puk, O., et al. (2007). Sequencing and Analysis of the Biosynthetic Gene Cluster of the Lipopeptide Antibiotic Friulimicin in Actinoplanes Friuliensis. Antimicrob. Agents Chemother. 51, 1028–1037. doi:10.1128/AAC.00942-06
Owen, J. G., Reddy, B. V. B., Ternei, M. A., Charlop-Powers, Z., Calle, P. Y., Kim, J. H., et al. (2013). Mapping Gene Clusters within Arrayed Metagenomic Libraries to Expand the Structural Diversity of Biomedically Relevant Natural Products. Proc. Natl. Acad. Sci. 110, 11797–11802. doi:10.1073/pnas.1222159110
Sun, Z., Shang, Z., Forelli, N., Po, K. H. L., Chen, S., Brady, S. F., et al. (2020). Total Synthesis of Malacidin A by β‐Hydroxyaspartic Acid Ligation‐Mediated Cyclization and Absolute Structure Establishment. Angew. Chem. Int. Ed. 59, 19868–19872. doi:10.1002/anie.202009092
Wiegand, I., Hilpert, K., and Hancock, R. E. W. (2008). Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc. 3, 163–175. doi:10.1038/nprot.2007.521
Wood, T. M., and Martin, N. I. (2019). The Calcium-dependent Lipopeptide Antibiotics: Structure, Mechanism, & Medicinal Chemistry. Med. Chem. Commun. 10, 634–646. doi:10.1039/C9MD00126C
World Health Organization (2019). 2019 Antibacterial Agents in Clinical Development: an Analysis of the Antibacterial Clinical Development Pipeline. Available at: https://www.who.int/publications/i/item/9789240000193 (Accessed February 28, 2021).
World Health Organization (2015). Global Action Plan on Antimicrobial Resistance. Available at: https://www.who.int/publications/i/item/9789241509763 (Accessed March 13, 2021).
Wu, C., Shang, Z., Lemetre, C., Ternei, M. A., and Brady, S. F. (2019). Cadasides, Calcium-dependent Acidic Lipopeptides from the Soil Metagenome that Are Active against Multidrug-Resistant Bacteria. J. Am. Chem. Soc. 141, 3910–3919. doi:10.1021/jacs.8b12087
Xu, B., Hermant, Y., Yang, S. H., Harris, P. W. R., and Brimble, M. A. (2019). A Versatile Boc Solid Phase Synthesis of Daptomycin and Analogues Using Site Specific, On‐Resin Ozonolysis to Install the Kynurenine Residue. Chem. Eur. J. 25, 14101–14107. doi:10.1002/chem.201903725
Keywords: calcium-dependent, lipopeptide, antibiotic, antimicrobial, solid-phase peptide synthesis
Citation: Kovalenko N, Howard GK, Swain JA, Hermant Y, Cameron AJ, Cook GM, Ferguson SA, Stubbing LA, Harris PWR and Brimble MA (2021) A Concise Synthetic Strategy Towards the Novel Calcium-dependent Lipopeptide Antibiotic, Malacidin A and Analogues. Front. Chem. 9:687875. doi: 10.3389/fchem.2021.687875
Received: 30 March 2021; Accepted: 25 May 2021;
Published: 04 August 2021.
Edited by:
Maria Luisa Mangoni, Sapienza University of Rome, ItalyReviewed by:
George Kokotos, National and Kapodistrian University of Athens, GreeceAnnemieke Madder, Ghent University, Belgium
Copyright © 2021 Kovalenko, Howard, Swain, Hermant, Cameron, Cook, Ferguson, Stubbing, Harris and Brimble. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul W. R. Harris, paul.harris@auckland.ac.nz; Margaret A. Brimble, m.brimble@auckland.ac.nz
†ORCID: Alan J. Cameron, orcid.org/0000-0003-0680-6921