Novel compound heterozygous variants in the USH2A gene associated with autosomal recessive retinitis pigmentosa without hearing loss
 Yanxia Huang
Yanxia Huang Lamei Yuan
Lamei Yuan Guiyun He
Guiyun He Yanna Cao
Yanna Cao Xiong Deng
Xiong Deng Hao Deng
Hao Deng- 1Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Center for Experimental Medicine, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Disease Genome Research Center, Central South University, Changsha, China
- 4Department of Ophthalmology, Hunan Provincial People’s Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, China
- 5Department of Ophthalmology, The Third Xiangya Hospital, Central South University, Changsha, China
- 6Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
Background: Retinitis pigmentosa (RP) is a group of progressive inherited retinal dystrophies characterized by the primary degeneration of rod photoreceptors and the subsequent loss of cone photoreceptors because of cell death. It is caused by different mechanisms, including inflammation, apoptosis, necroptosis, pyroptosis, and autophagy. Variants in the usherin gene (USH2A) have been reported in autosomal recessive RP with or without hearing loss. In the present study, we aimed to identify causative variants in a Han-Chinese pedigree with autosomal recessive RP.
Methods: A six-member, three-generation Han-Chinese family with autosomal recessive RP was recruited. A full clinical examination, whole exome sequencing, and Sanger sequencing, as well as co-segregation analysis were performed.
Results: Three heterozygous variants in the USH2A gene, c.3304C>T (p.Q1102*), c.4745T>C (p.L1582P), and c.14740G>A (p.E4914K), were identified in the proband, which were inherited from parents and transmitted to the daughters. Bioinformatics analysis supported the pathogenicity of the c.3304C>T (p.Q1102*) and c.4745T>C (p.L1582P) variants.
Conclusions: Novel compound heterozygous variants in the USH2A gene, c.3304C>T (p.Q1102*) and c.4745T>C (p.L1582P), were identified as the genetic causes of autosomal recessive RP. The findings may enhance the current knowledge of the pathogenesis of USH2A-associated phenotypes, expand the spectrum of the USH2A gene variants, and contribute to improved genetic counseling, prenatal diagnosis, and disease management.
Introduction
Retinitis pigmentosa (RP, MIM #268000) is one of the most severe hereditary retinal disorders and a major cause of visual disability and blindness (Taghipour et al., 2019; González-Del Pozo et al., 2020). It has wide phenotypic and genetic heterogeneity with numerous gene defects (Ávila-Fernández et al., 2010; Gao et al., 2019; Karali et al., 2019). The disorder is typically characterized by progressive degeneration of rod photoreceptors, cone photoreceptors, and retinal pigment epithelium, leading to night blindness, restricted peripheral vision (tunnel vision), and reduction of central vision (Hartong et al., 2006; Verbakel et al., 2018; Koyanagi et al., 2020). The estimated prevalence of the disorder is about 0.025%, and there are more than 1.5 million reported patients worldwide (de Castro-Miró et al., 2014; Menghini et al., 2020; Sharon et al., 2020). Affected individuals usually present difficulty with dark adaptation in adolescence, tunnel vision in young adulthood, and central vision loss in elder age, with a varying onset age (Jinda et al., 2014; Verbakel et al., 2018). RP may be inherited in three patterns: autosomal recessive RP (ARRP), autosomal dominant RP (ADRP), and X-linked RP (XLRP), which account for 50%–60%, 30%–40%, and 5%–15% of patients, as well as some non-Mendelian inheritance traits (Hartong et al., 2006; Ferrari et al., 2011; Chen et al., 2020; Dan et al., 2020). To date, at least 44 genes causing ARRP have been recorded in the Retinal Information Network (https://web.sph.uth.edu/RetNet/, updated on 10 January 2023) (Méndez-Vidal et al., 2013; Caruso et al., 2020; Koyanagi et al., 2020). Among them, the usherin gene (USH2A) was reported to be responsible for 10%–15% of ARRP. Variants in at least 23 different genes have been identified in ADRP, and there are two causative genes for XLRP (Dias et al., 2018). In the recent decade, high-throughput DNA sequencing such as the whole exome sequencing (WES) has increased the identification of the causative genes in RP patients.
In RP, early studies considered that different mechanisms were involved in photoreceptor cell death (Chang et al., 1993). Inflammation is proposed to be a result of the photoreceptor degeneration induced by the genetic defects, and meanwhile, inflammation can promote the cell death (Zhao et al., 2015). Apoptosis is known as a main mechanism for rod degeneration, and necroptosis mediates cone degeneration at later stages, which remain controversial (Murakami et al., 2012; Olivares-González et al., 2020). The nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation and pyroptosis were reported in RP models (Olivares-González et al., 2020; Olivares-González et al., 2021). Excessive autophagy may induce cone cell death (Murakami et al., 2012). Although it is clear that gene variants can disrupt photoreceptor function, the exact mechanism leading to photoreceptor cell death is still poorly understood.
The present study was aimed at identifying the pathogenic variants accounting for ARRP in a Han-Chinese pedigree by WES and Sanger sequencing. The clinical characteristics and potential genetic variants were evaluated, and the novel USH2A compound heterozygous variants, c.3304C>T (p.Q1102*) and c.4745T>C (p.L1582P), may be responsible for ARRP phenotype in this family. Our results expand the gene variant spectrum and will serve as an efficient reference for genetic diagnostic testing for patients with suspected ARRP, which will be useful for clinical management and prognosis.
Materials and methods
Subjects and clinical evaluation
A three-generation Han-Chinese family with a suspected autosomal recessive inherited retinal disease was recruited for genetic analysis from Changsha, Hunan province, China. Peripheral blood samples of the family were collected from six members. Written informed consents were signed by all the enrolled individuals or guardians before their participation, and the possible consequences were explained. The study was conducted in accordance with the Declaration of the Helsinki and approved by the Institutional Review Board of the Third Xiangya Hospital, Central South University (Changsha, China). Ophthalmic evaluations of patient were comprehensively performed, including best corrected visual acuity (BCVA), intraocular pressure, slit-lamp biomicroscopy, fundus photograph, visual field, optical coherence tomography (OCT), electroretinography (ERG), and fundus fluorescein angiography (FFA). Pure-tone audiometry was carried out to determine hearing thresholds at different frequencies, and the normal hearing can be defined when the loudness is lower than 20 dB. The videonystagmography (VNG) was employed to evaluate vestibular function.
Whole exome sequencing and bioinformatics analysis
Genomic DNA was obtained from peripheral blood samples by QIAamp DNA Mini Kit (QIAGEN, Venlo, Netherlands). Three samples of members (the patient and his parents) were submitted to KingMed Diagnostics (Changsha, China) for exome capture and sequencing. Library preparation was performed using QIAseq FX DNA Library Kit (QIAGEN), according to manufacturer’s protocols, and the targeted regions were captured by xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc., Coralville, Iowa, United States). The sequencing was performed on an Illumina NovaSeq platform. The primary data were obtained using Bcl2fastq (v2.0.1) analysis. Trimmomatic (version 0.36) was processed to obtain the clean data by removing low quality reads, bases, adaptors, etc. The clean data were aligned with the human genome reference sequence (GRCh37/hg19) using the Burrows-Wheeler Aligner software, and variants were called by Genome Analysis Toolkit. The ANNOVAR tool was used to annotate and identify genetic variants. The candidate variants were filtered by the following databases, including the 1000 Genomes Project, Single Nucleotide Polymorphism database (version 154), National Heart, Lung, and Blood Institute-Exome Sequencing Project 6500, Exome Aggregation Consortium, Genome Aggregation Database, China Metabolic Analytics Project, the Human Gene Mutation Database (HGMD), and ClinVar database. Variant pathogenicity was predicted by MutationTaster2021 (https://www.genecascade.org/MutationTaster2021/), Protein Variation Effect Analyzer (PROVEAN, https://provean.jcvi.org/), the Sorting Intolerant from Tolerant (SIFT), the Polymorphism Phenotyping v2 (Polyphen-2, http://genetics.bwh.harvard.edu/pph2), and MutationAssessor (http://mutationassessor.org/r3/) (Xiang et al., 2019a; Wu et al., 2021; Xiong et al., 2021; Yu et al., 2022).
Sanger sequencing
For the potential pathogenic variants identified by WES, Sanger sequencing was applied to test the variants in the available family members, and the segregation of candidate variants with the phenotype was analyzed in the view of inheritance pattern. Primer3 (https://primer3.ut.ee) was used to design PCR amplification primers and sequencing primers for detecting the candidate variants according to the human gene reference sequences (http://genome.ucsc.edu). Three pairs of primer sequences are as follows: 5′-TAGCTCCATTCCAGCAACCT-3′ and 5′-CAGAGGAAACCACAACAGCA-3′, 5′-TTCGAACAAAAGTGCCTGAA-3′ and 5′-AGCTGAGGGCAAGTCACATT-3′, 5′-ACTCAGCCCTCCCCTGTACT-3′ and 5′-AGTGGCTTCTCCGAGTTTCA-3′. The Chromas software (v2.01, Technelysium Pty Ltd., South Brisbane, Australia) was applied to analyze the sequencing results.
Variant evaluation, conservative analysis, and structure modeling
The American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation were utilized following the terms, “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign”, and “benign” (Richards et al., 2015). Multiple sequence alignments for eleven species were conducted using Basic Local Alignment Search Tool of National Center for Biotechnology Information (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The detailed information of protein was accessed from the Universal Protein Resource (UniProt, https://www.uniprot.org/). The wild-type and variant protein structures were predicted by the online CPHmodels-3.2 (https://services.healthtech.dtu.dk/service.php?CPHmodels-3.2). The three-dimensional structures were shown by PyMOL software (v2.3, Schrödinger, Inc., New York, United States) (Xiang et al., 2019b; Wu et al., 2019; Huang et al., 2021; Yuan et al., 2021).
Results
Clinical data
The proband, a 34-year-old male, complained of night blindness, constricted vision field, and reduced vision. The BCVA test results were 0.6 in both eyes. The intraocular pressure and anterior segment examination were normal. The fundus photographs revealed waxy-pale appearance of the discs, attenuated retinal arteries, and pigment deposits (Figures 1A,B). The visual field test indicated peripheral vision loss in both eyes (Figures 1C,D). The OCT revealed that the retinal nerve fiber layer was markedly thinned (Figures 2A–F). Full-field ERG showed significantly decreased scotopic and photopic responses, with extinguished decrease of a and b waves (Figures 2G,H). FFA showed hyperfluorescence and hypofluorescence areas and attenuated vessels in both eyes (Figures 3A–F). Hearing examination was normal by pure-tone audiometry test (Figures 3G,H). Vestibular assessments showed intact vestibular function according to the VNG test (Supplementary Table S1). The patient matched the clinical diagnostic criteria of RP. Parental consanguineous marriage was denied.
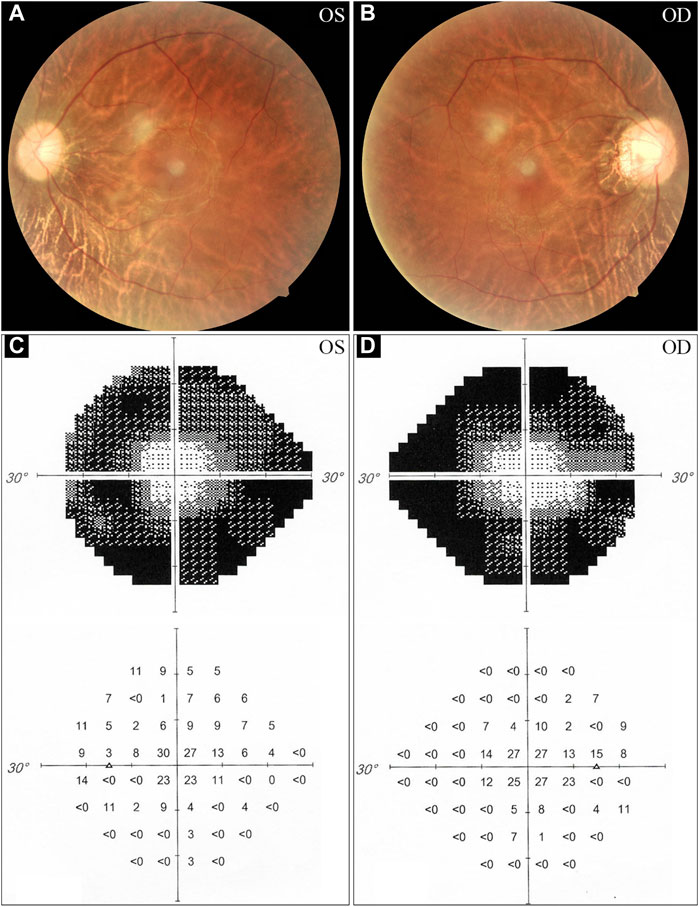
FIGURE 1. Fundus photographs and visual field tests from the proband. (A, B) The fundus examinations demonstrate pigmentary deposits, attenuated vessels, and waxy optic discs. (C, D) Visual field assessments reveal peripheral vision loss in the proband. OS, oculus sinister (left eye); OD, oculus dexter (right eye).
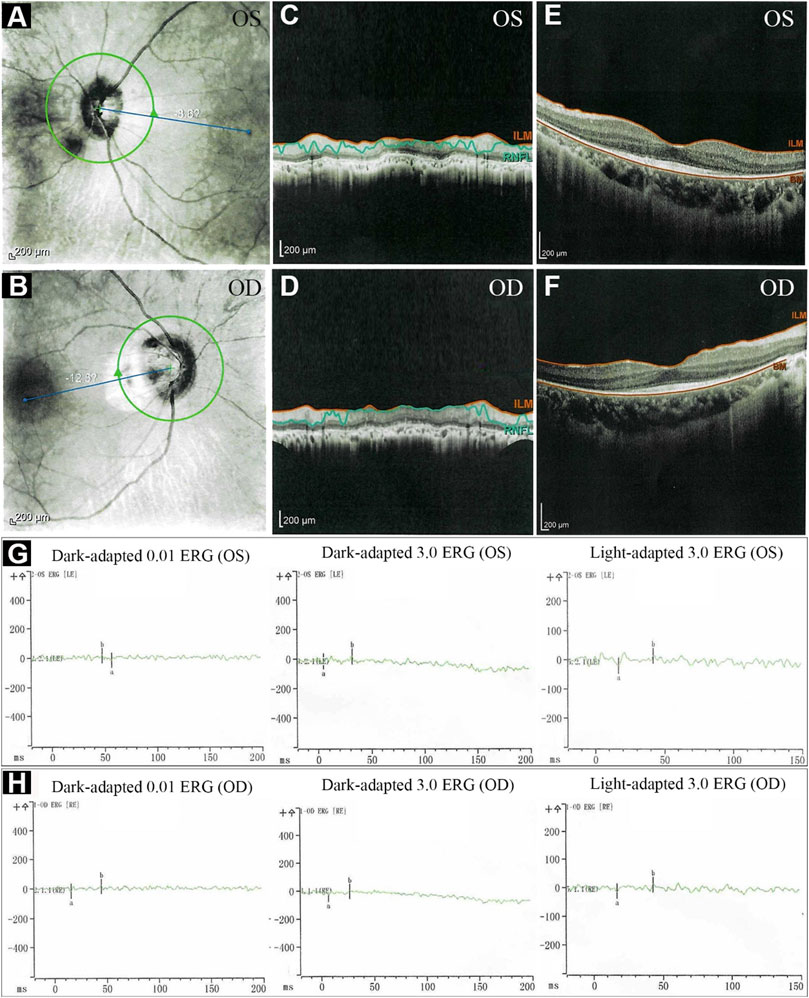
FIGURE 2. The optical coherence tomography (OCT) and electroretinography (ERG) of the proband. (A–F) OCT showed thinning of retinal nerve fiber layer. (G, H) ERG showed significantly reduced scotopic and photopic responses, with extinguished decrease of a and b waves. ILM, internal limiting membrane; RNFL, retinal nerve fiber layer; BM, Bruch’s membrane; OS, oculus sinister (left eye); OD, oculus dexter (right eye).
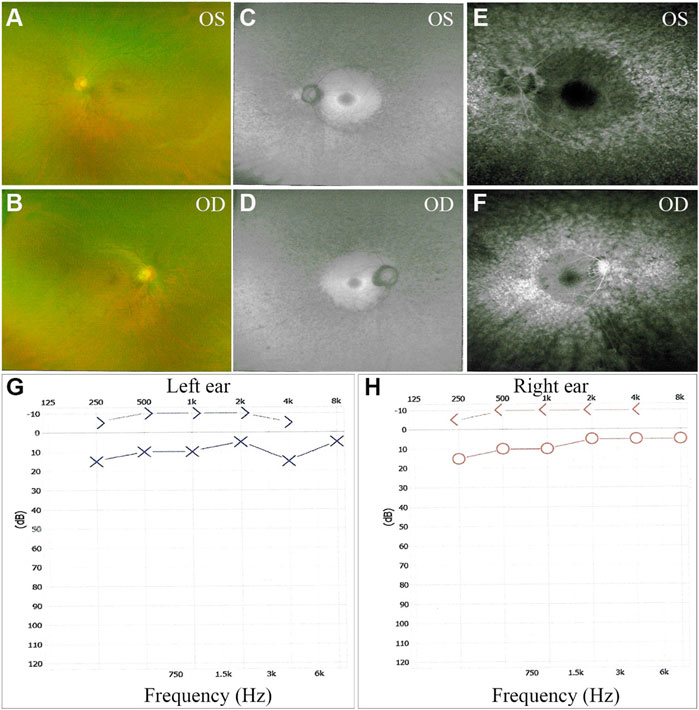
FIGURE 3. Fundus fluorescein angiography (FFA) and pure-tone audiograms of the proband. (A–F) Bilateral fluorescein leakage on FFA. (G, H) Audiograms showed no hearing loss in both ears. OS, oculus sinister (left eye); OD, oculus dexter (right eye).
Whole exome sequencing and identification of pathogenic variants
WES of three individuals (I:1, I:2, and II:1, Figure 4A) generated a total of 239.41 million (84.24 million, 73.67 million, and 81.50 million) clean reads, and the target region with a mean coverage of 99.86% (99.91%, 99.74%, and 99.92%) was mapped to the human reference genome. The mean depth of target region was 162.72-fold (174.23-fold, 149.84-fold, and 164.08-fold). A total of 51,803 single nucleotide polymorphisms (SNPs) and 8,129 insertions-deletions (indels) were found in the father (I:1), and a total of 52,281 SNPs and 8,028 indels were found in the mother (I:2). There were a total of 51,919 SNPs and 8,042 indels in the proband (II:1). Three heterozygous variants (c.3304C>T, p.Q1102*; c.4745T>C, p.L1582P; c.14740G>A, p.E4914K) in the USH2A gene (NM_206933.4), a causative gene for ARRP, were identified in the proband. The two variants c.3304C>T and c.14740G>A were inherited from his father, and the c.4745T>C variant was originated from his mother. The nonsense variant, c.3304C>T (p.Q1102*), was predicted to be deleterious by MutationTaster2021. The missense variant, c.4745T>C (p.L1582P), was predicted to be deleterious by MutationTaster2021, PROVEAN, SIFT, and PolyPhen-2. The c.14740G>A (p.E4914K) variant was predicted to be benign (Table 1). Sanger sequencing confirmed the heterozygous variants in the five family members (Figures 4A–D). Co-segregation analysis supported that compound heterozygous variants (c.3304C>T and c.4745T>C) were responsible for the disease phenotype. The c.3304C>T variant was classified as “likely pathogenic” variant, and the c.4745T>C and c.14740G>A variants were classified as “uncertain significance” variants according to the comprehensive analysis of ACMG guidelines. Conservative analysis of the USH2A protein from zebrafish to human showed that leucine at variant site (p.Leu1582) was highly conserved and glutamic acid at variant site (p.Glu4914) was conserved from house mouse to human (Figure 4E). Structural modeling showed the conformational alteration of the protein (UniProt Knowledgebase: O75445, Figure 5).
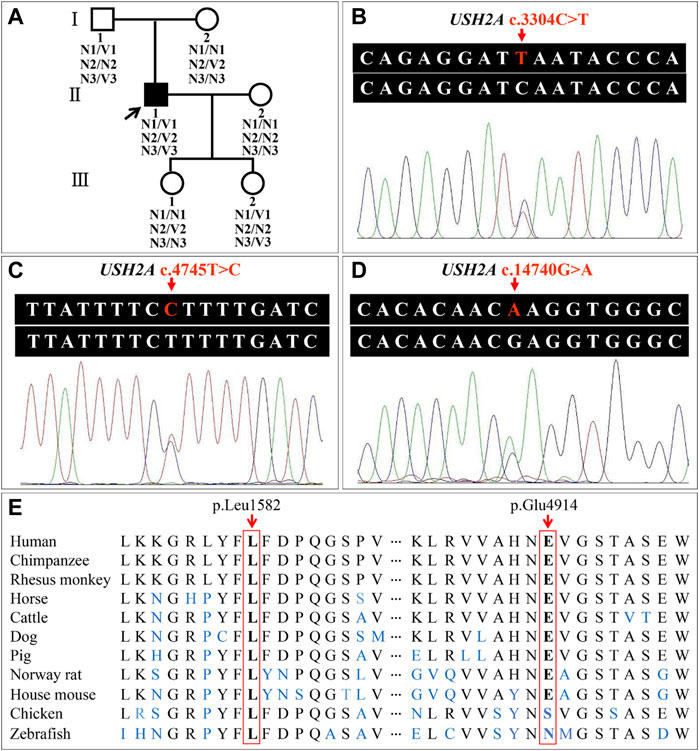
FIGURE 4. Pedigree of the family with autosomal recessive retinitis pigmentosa and analysis of the usherin gene (USH2A) variants. (A) Pedigree figure. The squares and the circles symbolize males and females, respectively. Filled symbol represents the patient, and empty symbols represent unaffected members. The arrow indicates the proband. N1, allele with USH2A c.3304C; V1, allele with USH2A c.3304T; N2, allele with USH2A c.4745T; V2, allele with USH2A c.4745C; N3, allele with USH2A c.14740G; V3, allele with USH2A c.14740A. (B–D) Patient (II:1) with variants c.3304C>T, c.4745T>C, and c.14740G>A in the USH2A gene. (E) Conservation analysis of the USH2A p.Leu1582 and p.Glu4914 residues.
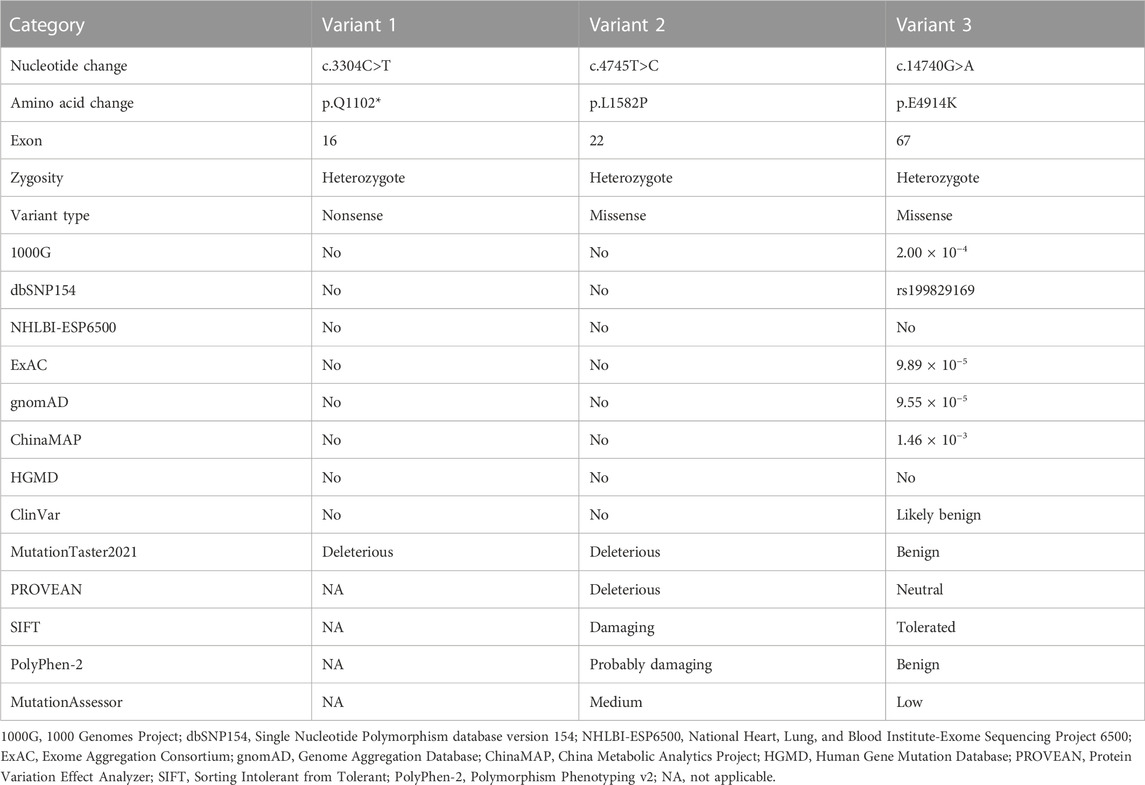
TABLE 1. Identification of USH2A variants in the proband.
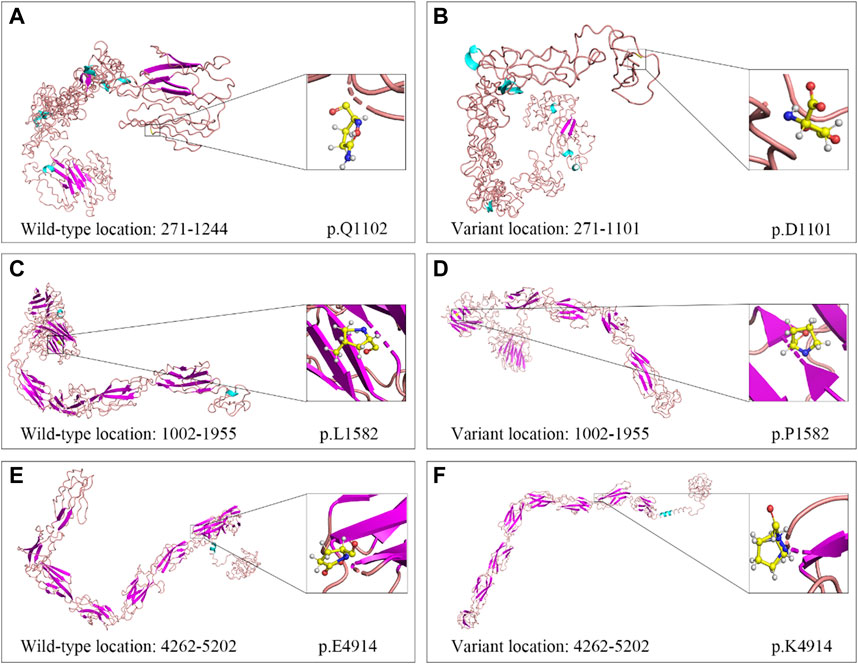
FIGURE 5. Cartoon models of wild-type and variant USH2A protein in three regions. (A, B) Segment 1 (amino acid 271–1,244): c.3304C>T (p.Q1102*). (C, D) Segment 2 (amino acid 1,002–1,955): c.4745T>C (p.L1582P). (E, F) Segment 3 (amino acid 4,262–5,202): c.14740G>A (p.E4914K).
Discussion
In 1998, three biologically important variants (c.2299del, c.2898del, and c.4336_4337del) in the USH2A gene were reported to be responsible for the autosomal recessive disorder, Usher syndrome type IIA (USH2A, MIM #276901), featured by sensorineural hearing impairment and progressive RP (Eudy et al., 1998; McGee et al., 2010). Subsequently, the USH2A missense variant p.C759F was firstly reported to be associated with ARRP without hearing impairment in 2000, which was presumed to affect the disulfide bridge in the fifth laminin epidermal growth factor-like domain of the USH2A protein (Rivolta et al., 2000; Lenassi et al., 2015). Until now, over 1,155 likely pathogenic variants in the USH2A gene have been recorded in HGMD, responsible for non-syndromic and syndromic RP (i.e., ARRP and USH2A). The USH2A gene variants were reported to be the most common cause (29%) of Usher syndrome and one of the most common causes (19%–23%) of ARRP (Gao et al., 2019; Zhu et al., 2020).
In this study, novel compound heterozygous variants in the USH2A gene, c.3304C>T (p.Q1102*) and c.4745T>C (p.L1582P), were prosecuted as disease causative factors for a Han-Chinese pedigree with RP by a full ophthalmological examination, WES, bioinformatics analysis, Sanger sequencing, and co-segregation analysis. These two variants were predicted to be harmful by several prediction softwares.
The USH2A gene, located on chromosome 1q41, has two transcripts due to alternatively splicing (21 exons and 72 exons), encoding the secreted and transmembrane proteins, respectively (Inaba et al., 2020; Meng et al., 2021). The longer isoform is a transmembrane protein of 5,202 amino acids (∼600 kDa), which is a cilial protein and mainly expressed in the connecting cilia of the photoreceptors and developing inner ear hair cells (Méndez-Vidal et al., 2013; Huang et al., 2018; Castiglione and Möller, 2022). The USH2A plays an essential role in the homeostasis, development, and function of visual and auditory sensory (Molina-Ramírez et al., 2020; Toms et al., 2020). The protein includes three regions: i) a large extracellular region with a signal peptide, a laminin N-terminal, 10 laminin epidermal growth factor-like domains, and 34 fibronectin type-III domains separated by 2 laminin G-like domains; ii) a transmembrane region; iii) a short intracellular region with a PDZ-binding motif at its C-terminal end (Liu et al., 2007; Chen et al., 2014; Fu et al., 2020; Zhu et al., 2021). The USH2A variants, including missense, nonsense, duplications/insertions, deletions, indels, and splicing variants, spread throughout the 72 exons and their flanking intronic regions (Aller et al., 2006; Koyanagi et al., 2020; Gao et al., 2021). Genotype-phenotype correlation has yet to be elucidated, while analyses based on some studies showed that patients with truncating variants may have more severe phenotypes, involving visual and auditory dysfunction (Inaba et al., 2020; Meng et al., 2021). Non-syndromic RP (typical or isolated RP) may be caused by variants in genes related to retina-specific expressions or functions, whereas syndromic RP (RP with extra-ocular defects) may be aroused by variants in genes functioning in various cells or tissues (Dias et al., 2018; Verbakel et al., 2018). However, the precise mechanisms of those variants underlying RP and Usher syndrome are still unclear.
At least 189 homozygous USH2A pathogenic variants were observed in 563 patients (RP and USH2A) from different populations in the worldwide (Figures 6, 7, and Supplementary Table S2), including European (47.60%, 268/563), Asian (30.91%, 174/563), American (5.86%, 33/563), African (1.78%, 10/563), and Oceanian (0.35%, 2/563). The most frequent homozygous variants in RP patients are missense variants, in which p.C759F and p.C934W account for 46.77% (58/124). Five most frequent variants in USH2A patients are p.E767Sfs*21 (24.37%, 107/439), p.W3955* (6.38%, 28/439), p.Q81Yfs*28 (3.64%, 16/439), c.8559–2A>G (3.19%, 14/439), and c.12067–2A>G (3.19%, 14/439). The common homozygous missense variant p.C759F is liable to lead to RP, while the homozygous c.2299del (p.E767Sfs*21) variant tends to cause USH2A. The extra-, even intra-familial clinical differences including penetrance and manifestations related to the same variant suggest that genetic background, epigenetics, and the environmental factors might influence the phenotypes.
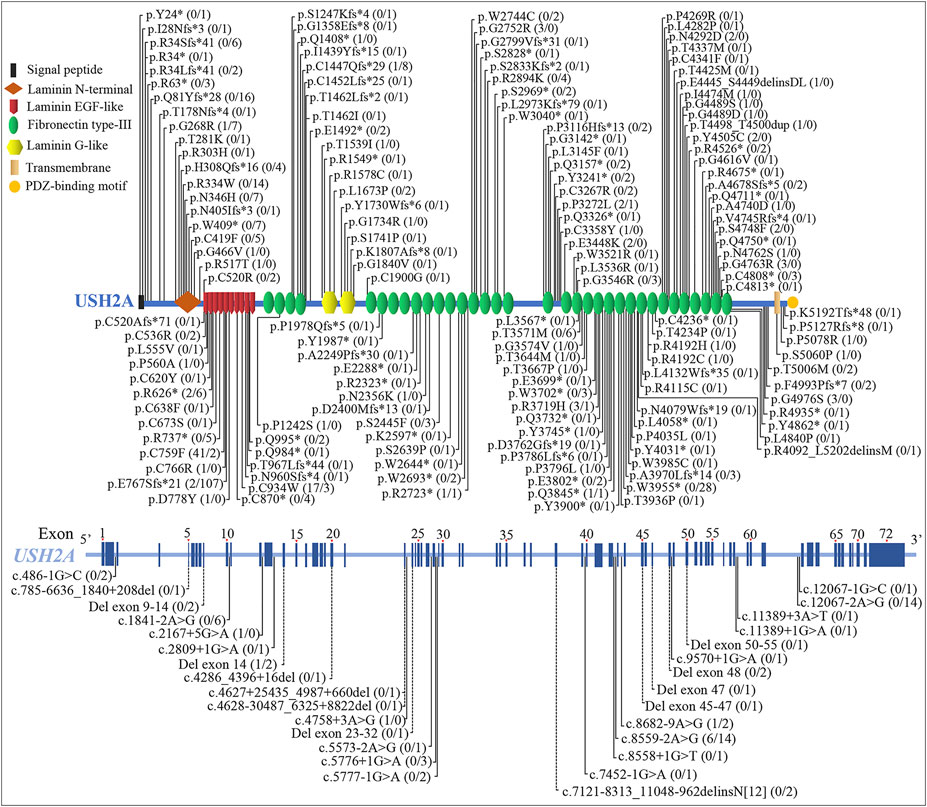
FIGURE 6. Schematic distribution of the usherin gene (USH2A) homozygous variants associated with retinitis pigmentosa (RP) and Usher syndrome type IIA (USH2A). Numbers in parentheses after the variants represent the number of patients with RP and USH2A, separated by the slash. The dotted line indicates deletion or deletion-insertion with changes over 20 bp. EGF, epidermal growth factor.
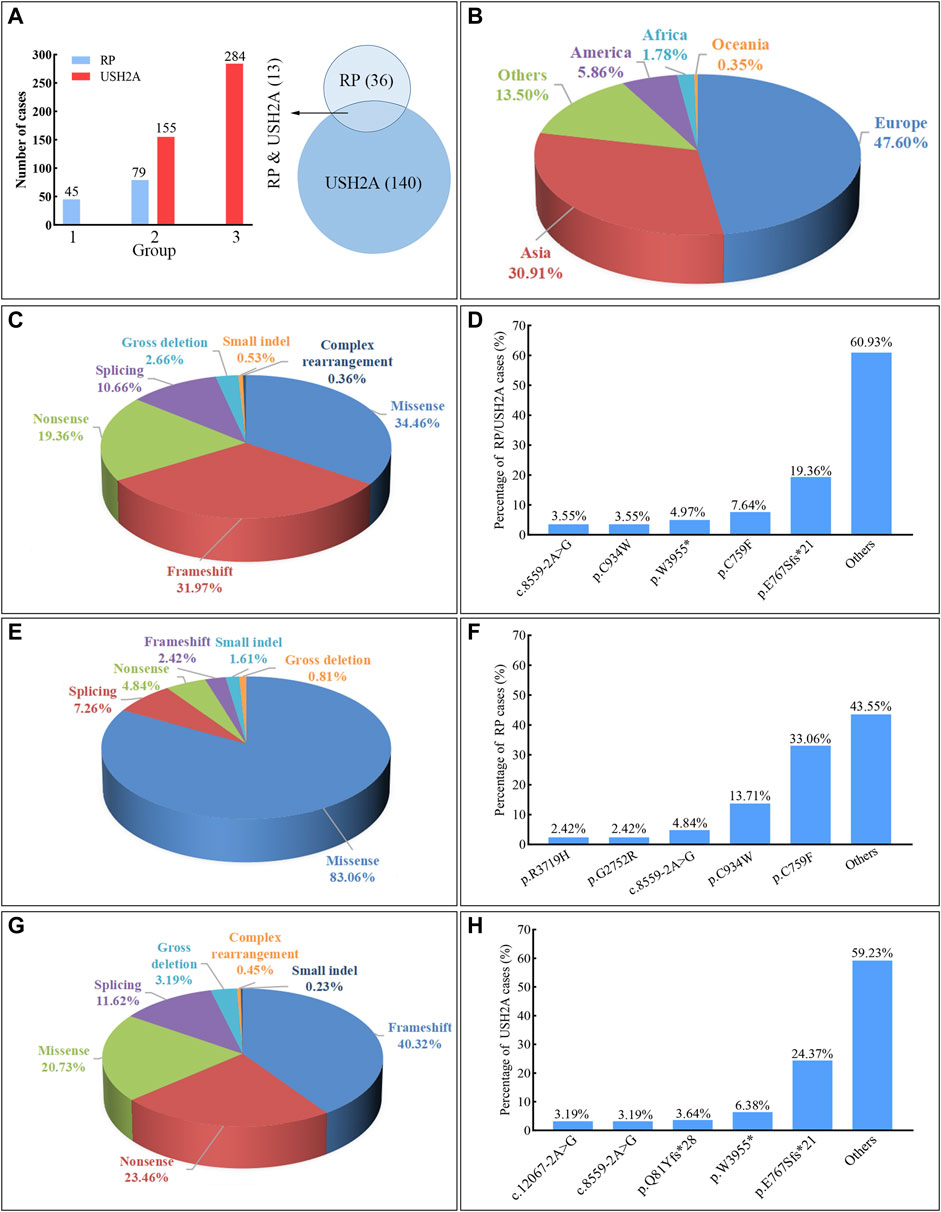
FIGURE 7. Summary of the usherin gene (USH2A) homozygous variants associated with retinitis pigmentosa (RP) and Usher syndrome type IIA (USH2A). Frameshift includes small deletion and duplication/insertion with changes involving 20 bp or less leading to reading frame shift. Small indel includes duplication and deletion-insertion with changes involving 20 bp or less leading to one or more amino acids inserted or replaced. Gross deletion or complex rearrangement (i.e., complex deletion-insertion) refers to changes over 20 bp. (A) The bar chart indicates the case number of RP or USH2A caused by various variants, in which the group “1” represents the variant causing RP, the group “2” represents the variant causing both RP and USH2A, and the group “3” represents the variant causing USH2A. The Venn diagram shows the reported numbers of variants causing RP and USH2A. (B) The proportion of RP and USH2A patients carrying the USH2A homozygous variants in different continents. (C) The proportion of cases with RP and USH2A by different variant types. (D) The percentage of cases with RP and USH2A observed at different variants. (E) The proportion of cases with RP caused by different variant types. (F) The percentage of cases with RP observed at different variants. (G) The proportion of cases with USH2A by different variant types. (H) The percentage of cases with USH2A observed at different variants.
Homozygous Ush2a knockout mice and zebrafish have progressive photoreceptor degeneration and non-progressive hearing impairment, similar to the visual and auditory deficits in human patients with USH2A variants. Two mutant ush2a zebrafish with different domains ablated had early-onset retinal dysfunction aggravated by sustained light exposure, indicating the critical role of the protein in maintenance of photoreceptors and development of hair cells, especially the intracellular domain in visual function (Dona et al., 2018; Han et al., 2018).
RP is caused by different cell death pathways and mechanisms. Some studies showed that necroptosis was implicated in both rod and cone cell death in animal models, and upregulation of receptor-interacting protein 1 and 3 kinase complexes was related with the necroptosis in photoreceptors (Murakami et al., 2012; Sato et al., 2013; Viringipurampeer et al., 2016; Viringipurampeer et al., 2019). The high expression level of tumor necrosis factor alpha and oxidative stress could induce the activation of NLRP3 inflammasome, including NLRP3, caspase-1, and interleukin 1 beta expression, and precipitate pyroptosis in rd10 mouse retinas at postnatal day 23 (Olivares-González et al., 2020). Autophagy was found to be involved in the photoreceptor cell death in some RP models, and the inhibition could exert a protective role (Rodríguez-Muela et al., 2015; Yao et al., 2018; Qiu et al., 2019). These studies demonstrate that more than one cellular mechanism is activated in retinal degeneration, which may be genotype-specific. Therefore, further research on the mechanisms of rod and cone photoreceptor cell death would be beneficial for the potential treatment of RP.
Given that patients may miss or refuse hearing examinations, the diagnosis of USH2A may be omitted in some cases, and the true frequency of USH2A-associated ARRP may be lower. In this study, pure-tone audiometry and vestibular examination were conducted, and the hearing acuity of the proband fell within the normal range, indicating the compound heterozygous USH2A variants may cause ARRP alone.
In conclusion, novel compound heterozygous variants, c.3304C>T (p.Q1102*) and c.4745T>C (p.L1582P), were identified as the genetic causes for ARRP in a Han-Chinese family by WES and Sanger sequencing. Our study expands the spectrum of the USH2A gene variants and may contribute to improved genetic counseling, prenatal diagnosis, and disease management for this family. Further construction of site-specific gene-deficient animal models and functional study may help to understand the genetic mechanism of USH2A-associated ARRP, which may further provide a clue to develop target therapy of this complex disorder.
Data availability statement
Data of this project can be accessed after an approval application by the China National GeneBank DataBase (CNGBdb). Please refer to https://db.cngb.org/, or email: CNGBdb@cngb.org for detailed application guidance. The project accession code CNP0003957 should be included in the application.
Ethics statement
Written informed consents were signed by all the enrolled individuals or guardians before their participation, and the possible consequences were explained. The study was conducted in accordance with the Declaration of the Helsinki and approved by the Institutional Review Board of the Third Xiangya Hospital, Central South University (Changsha, China, No: 2018-S400).
Author contributions
YH, LY, and HD conceptualized and designed the study. YH, LY, GH, YC, XD, and HD performed the research and analyzed the data. YH, LY, YC, and HD wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by National Natural Science Foundation of China (81873686 and 81800219), Natural Science Foundation of Hunan Province (2020JJ3057 and 2020JJ4830), Scientific Research Project of Health Commission of Hunan Province (A202303018385), Distinguished Professor of the Lotus Scholars Award Program of Hunan Province, Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University (YX202109), and Fundamental Research Funds for the Central Universities of Central South University (2021zzts218).
Acknowledgments
We would like to thank the participating members and investigators for their cooperation and efforts in collecting clinical data and genetic information.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1129862/full#supplementary-material
References
Aller, E., Jaijo, T., Beneyto, M., Nájera, C., Oltra, S., Ayuso, C., et al. (2006). Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J. Med. Genet. 43 (11), e55. doi:10.1136/jmg.2006.041764
Ávila-Fernández, A., Cantalapiedra, D., Aller, E., Vallespín, E., Aguirre-Lambán, J., Blanco-Kelly, F., et al. (2010). Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol. Vis. 16, 2550–2558.
Caruso, S. M., Ryu, J., Quinn, P. M., and Tsang, S. H. (2020). Precision metabolome reprogramming for imprecision therapeutics in retinitis pigmentosa. J. Clin. Invest. 130 (8), 3971–3973. doi:10.1172/JCI139239
Castiglione, A., and Möller, C. (2022). Usher syndrome. Audiol. Res. 12 (1), 42–65. doi:10.3390/audiolres12010005
Chang, G. Q., Hao, Y., and Wong, F. (1993). Apoptosis: Final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron 11 (4), 595–605. doi:10.1016/0896-6273(93)90072-y
Chen, C., Sun, Q., Gu, M., Qian, T., Luo, D., Liu, K., et al. (2020). Multimodal imaging and genetic characteristics of Chinese patients with USH2A-associated nonsyndromic retinitis pigmentosa. Mol. Genet. Genomic Med. 8 (11), e1479. doi:10.1002/mgg3.1479
Chen, X., Sheng, X., Liu, X., Li, H., Liu, Y., Rong, W., et al. (2014). Targeted next-generation sequencing reveals novel USH2A mutations associated with diverse disease phenotypes: Implications for clinical and molecular diagnosis. PLoS One 9 (8), e105439. doi:10.1371/journal.pone.0105439
Dan, H., Huang, X., Xing, Y., and Shen, Y. (2020). Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol. Genet. Genomic Med. 8 (3), e1131. doi:10.1002/mgg3.1131
de Castro-Miró, M., Pomares, E., Lorés-Motta, L., Tonda, R., Dopazo, J., Marfany, G., et al. (2014). Combined genetic and high-throughput strategies for molecular diagnosis of inherited retinal dystrophies. PLoS One 9 (2), e88410. doi:10.1371/journal.pone.0088410
Dias, M. F., Joo, K., Kemp, J. A., Fialho, S. L., da Silva Cunha, A., Woo, S. J., et al. (2018). Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 63, 107–131. doi:10.1016/j.preteyeres.2017.10.004
Dona, M., Slijkerman, R., Lerner, K., Broekman, S., Wegner, J., Howat, T., et al. (2018). Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 173, 148–159. doi:10.1016/j.exer.2018.05.015
Eudy, J. D., Weston, M. D., Yao, S., Hoover, D. M., Rehm, H. L., Ma-Edmonds, M., et al. (1998). Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 280 (5370), 1753–1757. doi:10.1126/science.280.5370.1753
Ferrari, S., Di Iorio, E., Barbaro, V., Ponzin, D., Sorrentino, F. S., and Parmeggiani, F. (2011). Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genomics 12 (4), 238–249. doi:10.2174/138920211795860107
Fu, J., Cheng, J., Zhou, Q., Khan, M. A., Duan, C., Peng, J., et al. (2020). Novel compound heterozygous nonsense variants, p.L150* and p.Y3565*, of the USH2A gene in a Chinese pedigree are associated with Usher syndrome type IIA. Mol. Med. Rep. 22 (4), 3464–3472. doi:10.3892/mmr.2020.11400
Gao, F. J., Li, J. K., Chen, H., Hu, F. Y., Zhang, S. H., Qi, Y. H., et al. (2019). Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology 126 (11), 1549–1556. doi:10.1016/j.ophtha.2019.04.038
Gao, F. J., Wang, D. D., Chen, F., Sun, H. X., Hu, F. Y., Xu, P., et al. (2021). Prevalence and genetic-phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br. J. Ophthalmol. 105 (1), 87–92. doi:10.1136/bjophthalmol-2020-315878
González-Del Pozo, M., Fernández-Suárez, E., Martín-Sánchez, M., Bravo-Gil, N., Méndez-Vidal, C., Rodríguez-de la Rúa, E., et al. (2020). Unmasking retinitis pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: Hidden recessive inheritance and potential oligogenic variants. J. Transl. Med. 18 (1), 73. doi:10.1186/s12967-020-02258-3
Han, S., Liu, X., Xie, S., Gao, M., Liu, F., Yu, S., et al. (2018). Knockout of ush2a gene in zebrafish causes hearing impairment and late onset rod-cone dystrophy. Hum. Genet. 137 (10), 779–794. doi:10.1007/s00439-018-1936-6
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368 (9549), 1795–1809. doi:10.1016/S0140-6736(06)69740-7
Huang, L., Mao, Y., Yang, J., Li, Y., Li, Y., and Yang, Z. (2018). Mutation screening of the USH2A gene in retinitis pigmentosa and USHER patients in a Han Chinese population. Eye 32 (10), 1608–1614. doi:10.1038/s41433-018-0130-3
Huang, Y., Yuan, L., Cao, Y., Tang, R., Xu, H., Tang, Z., et al. (2021). Novel compound heterozygous mutations in the CHST6 gene cause macular corneal dystrophy in a Han Chinese family. Ann. Transl. Med. 9 (8), 622. doi:10.21037/atm-20-7178
Inaba, A., Maeda, A., Yoshida, A., Kawai, K., Hirami, Y., Kurimoto, Y., et al. (2020). Truncating variants contribute to hearing loss and severe retinopathy in USH2A-associated retinitis pigmentosa in Japanese patients. Int. J. Mol. Sci. 21 (21), 7817. doi:10.3390/ijms21217817
Jinda, W., Taylor, T. D., Suzuki, Y., Thongnoppakhun, W., Limwongse, C., Lertrit, P., et al. (2014). Whole exome sequencing in Thai patients with retinitis pigmentosa reveals novel mutations in six genes. Invest. Ophthalmol. Vis. Sci. 55 (4), 2259–2268. doi:10.1167/iovs.13-13567
Karali, M., Testa, F., Brunetti-Pierri, R., Di Iorio, V., Pizzo, M., Melillo, P., et al. (2019). Clinical and genetic analysis of a European cohort with pericentral retinitis pigmentosa. Int. J. Mol. Sci. 21 (1), 86. doi:10.3390/ijms21010086
Koyanagi, Y., Ueno, S., Ito, Y., Kominami, T., Komori, S., Akiyama, M., et al. (2020). Relationship between macular curvature and common causative genes of retinitis pigmentosa in Japanese patients. Invest. Ophthalmol. Vis. Sci. 61 (10), 6. doi:10.1167/iovs.61.10.6
Lenassi, E., Vincent, A., Li, Z., Saihan, Z., Coffey, A. J., Steele-Stallard, H. B., et al. (2015). A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 23 (10), 1318–1327. doi:10.1038/ejhg.2014.283
Liu, X., Bulgakov, O. V., Darrow, K. N., Pawlyk, B., Adamian, M., Liberman, M. C., et al. (2007). Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. U. S. A. 104 (11), 4413–4418. doi:10.1073/pnas.0610950104
McGee, T. L., Seyedahmadi, B. J., Sweeney, M. O., Dryja, T. P., and Berson, E. L. (2010). Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med. Genet. 47 (7), 499–506. doi:10.1136/jmg.2009.075143
Méndez-Vidal, C., González-Del Pozo, M., Vela-Boza, A., Santoyo-López, J., López-Domingo, F. J., Vázquez-Marouschek, C., et al. (2013). Whole-exome sequencing identifies novel compound heterozygous mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa. Mol. Vis. 19, 2187–2195.
Meng, X., Liu, X., Li, Y., Guo, T., and Yang, L. (2021). Correlation between genotype and phenotype in 69 Chinese patients with USH2A mutations: A comparative study of the patients with Usher syndrome and nonsyndromic retinitis pigmentosa. Acta Ophthalmol. 99 (4), e447–e460. doi:10.1111/aos.14626
Menghini, M., Cehajic-Kapetanovic, J., and MacLaren, R. E. (2020). Monitoring progression of retinitis pigmentosa: Current recommendations and recent advances. Expert Opin. Orphan Drugs 8 (2-3), 67–78. doi:10.1080/21678707.2020.1735352
Molina-Ramírez, L. P., Lenassi, E., Ellingford, J. M., Sergouniotis, P. I., Ramsden, S. C., Bruce, I. A., et al. (2020). Establishing genotype-phenotype correlation in USH2A-related disorders to personalize audiological surveillance and rehabilitation. Otol. Neurotol. 41 (4), 431–437. doi:10.1097/MAO.0000000000002588
Murakami, Y., Matsumoto, H., Roh, M., Suzuki, J., Hisatomi, T., Ikeda, Y., et al. (2012). Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. U. S. A. 109 (36), 14598–14603. doi:10.1073/pnas.1206937109
Olivares-González, L., Velasco, S., Campillo, I., and Rodrigo, R. (2021). Retinal inflammation, cell death and inherited retinal dystrophies. Int. J. Mol. Sci. 22 (4), 2096. doi:10.3390/ijms22042096
Olivares-González, L., Velasco, S., Millán, J. M., and Rodrigo, R. (2020). Intravitreal administration of adalimumab delays retinal degeneration in rd10 mice. FASEB J. 34 (10), 13839–13861. doi:10.1096/fj.202000044RR
Qiu, Y., Yao, J., Jia, L., Thompson, D. A., and Zacks, D. N. (2019). Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: A novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 10 (8), 547. doi:10.1038/s41419-019-1780-1
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rivolta, C., Sweklo, E. A., Berson, E. L., and Dryja, T. P. (2000). Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 66 (6), 1975–1978. doi:10.1086/302926
Rodríguez-Muela, N., Hernández-Pinto, A. M., Serrano-Puebla, A., García-Ledo, L., Latorre, S. H., de la Rosa, E. J., et al. (2015). Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 22 (3), 476–487. doi:10.1038/cdd.2014.203
Sato, K., Li, S., Gordon, W. C., He, J., Liou, G. I., Hill, J. M., et al. (2013). Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J. Neurosci. 33 (44), 17458–17468. doi:10.1523/JNEUROSCI.1380-13.2013
Sharon, D., Ben-Yosef, T., Goldenberg-Cohen, N., Pras, E., Gradstein, L., Soudry, S., et al. (2020). A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 41 (1), 140–149. doi:10.1002/humu.23903
Taghipour, M., Derakhshan, N., Saffarian, A., and Ghanbari, M. (2019). Undetected pituitary adenoma in a patient with retinitis pigmentosa. Chin. Neurosurg. J. 5, 20. doi:10.1186/s41016-019-0168-5
Toms, M., Pagarkar, W., and Moosajee, M. (2020). Usher syndrome: Clinical features, molecular genetics and advancing therapeutics. Ther. Adv. Ophthalmol. 12, 2515841420952194. doi:10.1177/2515841420952194
Verbakel, S. K., van Huet, R. A. C., Boon, C. J. F., den Hollander, A. I., Collin, R. W. J., Klaver, C. C. W., et al. (2018). Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 66, 157–186. doi:10.1016/j.preteyeres.2018.03.005
Viringipurampeer, I. A., Gregory-Evans, C. Y., Metcalfe, A. L., Bashar, E., Moritz, O. L., and Gregory-Evans, K. (2019). Cell death pathways in mutant rhodopsin rat models identifies genotype-specific targets controlling retinal degeneration. Mol. Neurobiol. 56 (3), 1637–1652. doi:10.1007/s12035-018-1192-8
Viringipurampeer, I. A., Metcalfe, A. L., Bashar, A. E., Sivak, O., Yanai, A., Mohammadi, Z., et al. (2016). NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum. Mol. Genet. 25 (8), 1501–1516. doi:10.1093/hmg/ddw029
Wu, S., Deng, S., Song, Z., Xu, H., Yang, Z., Liu, X., et al. (2019). A disease-causing FRMD7 variant in a Chinese family with infantile nystagmus. J. Mol. Neurosci. 67 (3), 418–423. doi:10.1007/s12031-018-1245-5
Wu, S., Guo, Y., Liu, C., Liu, Q., Deng, H., and Yuan, L. (2021). Identification of a de novo TSC2 variant in a Han-Chinese family with tuberous sclerosis complex. J. Chin. Med. Assoc. 84 (1), 46–50. doi:10.1097/JCMA.0000000000000455
Xiang, Q., Cao, Y., Xu, H., Guo, Y., Yang, Z., Xu, L., et al. (2019b). Identification of novel pathogenic ABCA4 variants in a Han Chinese family with Stargardt disease. Biosci. Rep. 39 (1), BSR20180872. doi:10.1042/BSR20180872
Xiang, Q., Yuan, L., Cao, Y., Xu, H., Li, Y., and Deng, H. (2019a). Identification of a heterozygous mutation in the TGFBI gene in a Hui-Chinese family with corneal dystrophy. J. Ophthalmol. 2019, 2824179. doi:10.1155/2019/2824179
Xiong, Y., Xia, H., Yuan, L., Deng, S., Ding, Z., and Deng, H. (2021). Identification of compound heterozygous DNAH11 variants in a Han-Chinese family with primary ciliary dyskinesia. J. Cell. Mol. Med. 25 (18), 9028–9037. doi:10.1111/jcmm.16866
Yao, J., Qiu, Y., Frontera, E., Jia, L., Khan, N. W., Klionsky, D. J., et al. (2018). Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 14 (7), 1226–1238. doi:10.1080/15548627.2018.1463121
Yu, X., Yuan, L., Deng, S., Xia, H., Tu, X., Deng, X., et al. (2022). Identification of DNAH17 variants in Han-Chinese patients with left-right asymmetry disorders. Front. Genet. 13, 862292. doi:10.3389/fgene.2022.862292
Yuan, M., Guo, Y., Xia, H., Xu, H., Deng, H., and Yuan, L. (2021). Novel SCN5A and GPD1L variants identified in two unrelated Han-Chinese patients with clinically suspected Brugada syndrome. Front. Cardiovasc. Med. 8, 758903. doi:10.3389/fcvm.2021.758903
Zhao, L., Zabel, M. K., Wang, X., Ma, W., Shah, P., Fariss, R. N., et al. (2015). Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 7 (9), 1179–1197. doi:10.15252/emmm.201505298
Zhu, T., Chen, D. F., Wang, L., Wu, S., Wei, X., Li, H., et al. (2021). USH2A variants in Chinese patients with Usher syndrome type II and non-syndromic retinitis pigmentosa. Br. J. Ophthalmol. 105 (5), 694–703. doi:10.1136/bjophthalmol-2019-315786
Keywords: compound heterozygous variants, retinitis pigmentosa, usherin gene, whole exome sequencing, photoreceptor cell death
Citation: Huang Y, Yuan L, He G, Cao Y, Deng X and Deng H (2023) Novel compound heterozygous variants in the USH2A gene associated with autosomal recessive retinitis pigmentosa without hearing loss. Front. Cell Dev. Biol. 11:1129862. doi: 10.3389/fcell.2023.1129862
Received: 22 December 2022; Accepted: 25 January 2023;
Published: 15 February 2023.
Edited by:
Degui Wang, Lanzhou University, ChinaReviewed by:
Junjiang Fu, Southwest Medical University, ChinaWanqin Xie, Hunan Provincial Maternal and Child Healthcare Hospital, China
Copyright © 2023 Huang, Yuan, He, Cao, Deng and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Deng, hdeng008@163.com
†These authors have contributed equally to this work and share first authorship.