Characterization of N6-Methyladenosine in Domesticated Yak Testes Before and After Sexual Maturity
 Xingdong Wang
Xingdong Wang  Jie Pei Shaoke Guo Mengli Cao
Jie Pei Shaoke Guo Mengli Cao  Pengjia Bao
Pengjia Bao  Lin Xiong
Lin Xiong  Xiaoyun Wu Min Chu Chunnian Liang Ping Yan*
Xiaoyun Wu Min Chu Chunnian Liang Ping Yan*  Xian Guo*
Xian Guo*- Key Laboratory of Yak Breeding Engineering of Gansu Province, Lanzhou Institute of Husbandry and Pharmaceutical Sciences, Chinese Academy of Agricultural Sciences, Lanzhou, China
The potential regulatory role of N6-methyladenosine (m6A), the most prominent mRNA modification in eukaryotes, has recently been identified in mammals, plants, and yeast. However, whether and how m6A methylation is involved in sexual maturation in mammals remains largely unexplored. In this study, testicular tissue was obtained from yaks before and after sexual maturation, and m6A maps were generated via preliminary experiments and methylated RNA immunoprecipitation sequencing. Only spermatogonial cells and a few primary spermatocytes were observed in the testicular tissue of yaks before sexual maturation, while spermatogenic cells at different stages of maturity could observed after sexual maturation. Experiments examining the expression of methylation-related enzymes and overall methylation levels showed that the methylation levels in yak testes increased after sexual maturation. Overall, 1,438 methylation peaks were differentially expressed before and after sexual maturation; 1,226 showed significant up-regulation and 212 showed significant down-regulation after sexual maturation. Annotation analysis showed that the differential methylation peaks were most commonly concentrated in the exon region, followed by the 3′UTR and finally the 5′UTR region. KEGG pathway analysis demonstrated that homologous recombination, the Notch signaling pathway, growth hormone synthesis, and other signaling pathways may be involved in testicular development and maturation in yaks. Levels of most m6A modifications were positively correlated with mRNA abundance, suggesting that m6A plays a regulatory role in mammalian sexual maturation. To our knowledge, this is the first report of an m6A transcriptional map of the yak testes, and our study lays the foundation for elucidating the function of m6A in the development of yak testes.
Introduction
Yaks are a type of semi-domesticated, multifunctional cattle that became separate from other cattle about 2.2 million years ago. They are primarily found in the Qinghai-Tibet Plateau region at an altitude of 2,500–6,000 m, where there is no absolute frost-free season (Das et al., 2019a; Hui et al., 2019). The annual average temperature (−3–5°C) in plateau areas is relatively low. Animals that wish to survive in high-altitude areas need to adapt to the environments with low temperatures and oxygen levels, which is a major challenge (Hui et al., 2019). Yaks have a compact body structure, no functional sweat glands, and a relatively low skin surface area per unit body weight (0.016 m2/kg) (Krishnan et al., 2018). They also have a higher breathing rate, and their heart and chest cavity are more developed than those of other cattle. Furthermore, yaks have higher red blood cell counts and hemoglobin levels than other cattle, which allow better adaptation to low atmospheric oxygen levels. While these characteristics enable yaks to better adapt to freezing temperatures and hypoxic conditions, they also make them more susceptible to heat stress (Krishnan et al., 2016).
With a yak population of over 16 million (Wang et al., 2021), China houses more than 95% of all yaks in the world (Zhao et al., 2019). Yaks are multi-purpose high-altitude bovids (Das et al., 2019b) that not only provide important products such as milk, meat, fur, and fuel but are also used as local modes of transport (Wang et al., 2014; Das et al., 2019a). Yaks, which the local herdsmen call “the boat of the plateau and an all-powerful domestic animal,” thus play important social and economic roles in the plateau area, while also maintaining the pasture ecosystem and agricultural biodiversity (Wiener et al., 2003; Shah et al., 2018).
Interestingly, yaks show later sexual maturity and lower fertilization rates than other cattle (Das et al., 2019b). This low reproductive efficiency (Zhou et al., 2020), including delayed onset of puberty, seasonal reproduction, low conception rates, and long calving intervals, limits reproductive performance in yaks. Given the important role of the testes in male reproduction, research on these gonads—especially from an epigenetics perspective—could help improve the reproductive efficiency of the yak.
Epigenetic changes are potentially heritable and environmentally modifiable changes in gene expression mediated by non-DNA-encoded mechanisms (Puigoriol-Illamola et al., 2020) and mainly include DNA methylation and histone modification (Cai et al., 2021). RNA also undergoes several post-transcriptional modifications, more than 150 of which have been identified across organisms (Sun et al., 2019; Liu et al., 2020), including N1-methyladenosine (m1A), N6-methyladenosine (m6A), and 5-methylcytosine (m5C). These modifications are extensively found across several types of RNA, including ribosomal RNA (rRNA) and messenger RNA (mRNA) (Zheng et al., 2013b). Among these, the most common internal modification in higher eukaryotes is m6A, which is important for mRNA metabolism and a variety of biological processes (Liu et al., 2018).
m6A, discovered in the early 1970s, is a reversible and dynamic modification involving the methyltransferase complex (writer), demethylases (erasers), and binding proteins (readers) (Zhao Y. et al., 2021). Methyltransferase complexes, including methyltransferase-like 3 (METTL3), METTL14, and Wilms’ tumor 1-associated protein (WTAP) (Lin et al., 2017), induce methylation, which can be reversed by fat mass and obesity-associated factor (FTO) and AlkB homologue 5 (ALKBH5) (Jia et al., 2011; Zheng et al., 2013a). The function of m6A is dependent on its recognition by specific reader proteins bound to methylated mRNA (Wang et al., 2020). m6A modifications regulate almost every stage of mRNA metabolism at different levels, including RNA folding and mRNA maturation, processing, stability, output, and translation (Meyer and Jaffrey, 2014; Zhao et al., 2017). In addition, m6A plays a role in cell fate determination, cell cycle regulation, cell differentiation, and circadian rhythm maintenance (Wu et al., 2017).
Androgens are key for the postnatal masculinization of the fetus, especially in mammals (Rey, 2021). Hence, the process of sexual maturation is a critical period for the development of the male reproductive system. Epigenetics may influence several aspects of male reproductive tract development (Mcgowan et al., 2018), including spermatogenesis—the production of haploid sperm from diploid spermatogonial stem cells (SSCs)—which is regulated at the transcriptional, post-transcriptional, and translational levels (Lin et al., 2017). Studies in knockout mouse models have shown that METTL3 regulates spermatogonial differentiation and meiosis initiation in mice (Xu et al., 2017). In early germ cells, the conditional knockout of METTL3 or METTL14 can cause the abnormal self-renewal and differentiation of SSCs (Lin and Tong, 2018). ALKBH5 knockout mice also show impaired fertility (Wojtas et al., 2017). FTO mutations are positively associated with decreased semen quality, and FTO dysfunction may reduce male fertility (Landfors et al., 2016). Further, the deletions of METTL3, METTL14, FTO, ALKBH5, YTHDF2, and YTHDC2 lead to impaired fertility or defective gametogenesis (Frye et al., 2018; Huang and Ping, 2018). Single-cell sequencing data from the studies on human testes have shown that the RNA m6A regulators are expressed in almost all cell types in the testes (Wang et al., 2018). Multiple studies have shown that m6A modification is critical for the development of the male reproductive system and especially mammalian spermatogenesis (Xu et al., 2017; Ming-Han et al., 2018). However, the regulation of m6A during sexual maturation in yaks, and especially its effect on spermatogenesis, remains unclear.
In recent years, new technology has allowed us to better understand RNA methylation, including m6A. The novel technique methylated RNA immunoprecipitation with next-generation sequencing (MeRIP-seq) was established in 2012 by two independent research groups. Subsequently, the first N6-methyladenosine modification map for the methylome of m6A RNA was generated and had a resolution of 100 nucleotides (Dominissini et al., 2012; Meyer et al., 2012). Such generation of a whole-transcriptome m6A methylation map via the detection of m6A sites at the transcriptome level could allow the further exploration of biological modifications across new areas of research and uncover the varying roles of RNA m6A modification.
To examine the regulatory mechanism of m6A modifications during the sexual maturation of yak testes, we collected healthy yak testes before and after sexual maturation and obtained the m6A profile of the whole transcriptome using MeRIP-seq. Differential methylation peaks were obtained by comparing maps before and after sexual maturation to elucidate the regulatory role of m6A methylation in yak sexual maturation.
Materials and Methods
Ethics Statement
All animal-related procedures conformed to the China Council on Animal Care and the Ministry of Agriculture of the People’s Republic of China guidelines. All yak-handling procedures were approved by The Animal Care and Use Committee of the Lanzhou Institute of Husbandry and Pharmaceutical Sciences Chinese Academy of Agricultural Sciences (Permit No: SYXK-2014-0002).
Animals and Sample Collection
Testicular tissue was collected from six healthy yaks in Maqu County, Gannan Tibetan Autonomous Prefecture (33.99°N 102.07°E). Of these, three were 1.5 years old (Y group sexual maturity not reached) and three were 5 years old (M group sexual maturity reached).
Y Group
Testicular tissue was obtained after surgical castration. Before tissue collection, the testes were locally disinfected. After collection, the wound was sutured with a surgical needle, and penicillin and streptomycin were administered to prevent wound infection.
M Group
Yaks were euthanized to obtain testicular tissue. Two sets of testicles were collected from six yaks; The entire testicular tissue was cut up and placed in a cryotube that was immediately immersed in liquid nitrogen, A small portion of the testicular tissue was fixed in Bouin’s Fluid (SolarBio, Beijing, China).
Immediately after collection, samples were sent to the Lanzhou Institute of Husbandry and Pharmaceutical Sciences, Chinese Academy of Agricultural Sciences for further experiments.
Hematoxylin–Eosin Staining
Fixed tissues were dehydrated with 75% ethanol, paraffin embedded, and then sectioned (6 mm). The sections were stained using the improved HE staining kit (SolarBio, Beijing, China) according to manufacturer’s instructions. Post-staining, the sections were sealed with neutral gum, and images were obtained on a Pannoramic 250 digital section scanner (Drnjier, Jinan, China).
RNA Extraction and cDNA Synthesis
First, total RNA was extracted from the testicular tissue of each yak using the TRIzol reagent (Invitrogen, CA, United States). RNA concentration and purity (OD260/280 ratio) were assessed using the NanoDrop 2000 spectrophotometer (ThermoFisher Scientific, Waltham, MA, United States) and were found to be 500–5,000 ng/ml and 1.9–2.1, respectively. Furthermore, the samples were subjected to 1% agarose gel electrophoresis, and 28S and 18S rRNA bands were observed. For cDNA synthesis, each RNA sample (diluted to 500 ng/ml) was reverse transcribed using the Transcriptor First Strand cDNA Synthesis Kit (Takara Bio Inc., Dalian, China). The cDNA was subsequently stored at −80°C until further use.
Quantitative Real-Time PCR
To study the m6A status in yak testicular tissue before and after sexual maturation, the levels of RNA methylation-related genes such as METTL3, METTL14, WTAP, FTO, ALKBH5, YTHDF1/2/3, YTHDC1/2, RBM15, VIRMA, and ZC3H13 were detected using qRT-PCR. The National Center for Biotechnology Information website was used for primer designing (Supplementary Table S1). RT-PCR was performed using a CFX Link Real-Time PCR Detection System. The reaction volume was 20 μl, including 10 µl of 2× PrecisionPLUS Master Mix (Primerdesign), 1 µl of diluted cDNA (25 ng), 1 µl (300 nmol) of gene-specific forward and reverse primers each, and 7 µl of RNase/DNase free water. The standard PCR reaction conditions for all transcripts were as follows: 95°C (3 min), followed by 39 cycles of 95°C (10 s) and 55°C (30 s). GAPDH was used as the reference gene, and relative gene expression was examined using the 2-ΔΔCT method (Schmittgen, 2001). Each reaction was repeated in triplicate to obtain Ct values. Analysis of variance (ANOVA) was used to analyze differences in the expression of methylation-related enzymes.
Evaluation of m6A Content
The total mRNA m6A levels in the yak testes were estimated using the EpiQuik RNA Methylation Quantitative Kit (Epigentek, P-9005, NY, United States). An m6A standard curve (0.01–0.5 ng/μl) was constructed based on the manufacturer’s instructions for the kit. Absorbance was measured using a microplate (Thermo Scientific., Shanghai, China) reader at 450 nm.
MeRIP-Seq and mRNA Sequencing
First, the quality and quantity of total RNA were estimated using Bioanalyzer 2,100 (Agilent, CA, United Stets) and NanoDrop 2000 (Thermo Scientific), both with an RIN number > 7.0. More than 100 μg of total RNA was used for mRNA isolation via an mRNA Purification Kit (Ambion Dynabeads mRNA Purification Kit). Subsequently, poly (A) mRNA fractions were created using divalent cations and a thermocycler (RNA solution kept at 94°C for exactly 5), incubated with m6A-Dynabeads (Anti-m6A, Synaptic Systems, Cat. No 202003) in an m6A-binding buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl2, 1% NP-40, 2 mM EDTA), and allowed to bind to the beads. Following this, the m6A-Dynabeads were washed and m6A-positive RNA was eluted. The RIP was extracted and cleaned, and finally, m6A “enriched RNA” was collected. We used 100 ng of RNA (100 ng of input and 100 ng of post m6A-IP positive fraction) for library construction with the Illumina TrueSeq Stranded mRNA platform. Finally, paired-end sequencing was performed on an Illumina HiSeq X10 system at OE Biotech Co., Ltd. (Shanghai, China) using manufacturer’s instructions. The data was submitted to the GENE EXPRESSION OMNIBUS (GEO) database (accession number GSE181266).
Analysis of Sequencing Data
Raw reads obtained from RNA sequences were subjected to statistical analysis and quality control (Supplementary Table S2). Raw data (raw reads) in the fastq format were processed using the Trimmomatic (Bolger et al., 2014) software, and reads with adapter sequences or poly-N sequences as well as low-quality reads were removed to obtain clean data. Following this, 250,000 paired reads were randomly extracted from the clean data, and blastn was used with the NT database (ftp://fp.ncbi.nih.gov/blast/db) to examine sequence alignment with the reads. The most high-quality results with an e value < 1e-10 and coverage > 80% were selected. Meanwhile, the SortMeRNA (Evguenia et al., 2012) software was used for removing ribosomal RNA reads. The remaining clean reads were mapped to the reference genome, LU_Bosgru_v3.0 (ftp://ftp.ensembl.org/pub/release-99/fasta/bos_grunniens/dna/Bos_grunniens.LU_Bosgru_v3.0.dna_sm.toplevel. Fa. gz) for sequence alignment using HISAT2 (Kim et al., 2015) to obtain position information on the reference genome as well as specific sequence characteristic information from the sequenced samples. Default parameters were used, and unique reads showing high mapping quality were retained. The Guitar (Cui et al., 2016) R package and deeptools (Fidel et al., 2014) software were used to evaluate m6A-seq data quality in order to assess the quality of MeRIP-seq data.
m6A-enriched peaks were detected in each ample using the MeTDiff peak calling software (screening criteria: p <= 0.05; fc >= 1.5) and the options FRAGMENT_LENGTH = 200, PEAK_CUTOFF_PVALUE = 0.01, and PEAK_CUTOFF_FDR = 0.05) (Cui et al., 2018). The number, width, and distribution of peaks were statistically analyzed, and the corresponding input sample was used as the control. The identified peaks were annotated based on their intersection with the gene architecture using ChIPseeker (Yu et al., 2015). Finally, differential analysis of m6A-seq data was performed by comparing data from yaks who had yet to reach sexual maturation with that from yaks who had already reached sexual maturation. This was done using MeTDiff (screening criteria: diff. p <= 0.05; diff. fc >= 1.5) with the following parameters: FRAGMENT_LENGTH = 200, PEAK_CUTOFF_PVALUE = 0.01, DIFF_PEAK_CUTOFF_FDR = 0.05, and PEAK_CUTOFF_FDR = 0.05). The differential peaks were again annotated using ChIPseeker.
Gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the identified peaks and differential peaks were performed using R based on hypergeometric distribution. Sequence motifs were identified using MEME (Bailey et al., 2009) and DREME (Schulz et al., 2012) and annotated using Tomtom software.
Statistical Analysis
All statistical analyses were performed using SPSS version 21.0 (Qiu et al., 2007). One-way ANOVA was used to analyze between-group differences. p < 0.05 or p < 0.01 was considered statistically significant (Lan et al., 2013).
Results
HE Staining of Testicular Tissue
Testicular tissue sections (Figure 1A) of yaks at the pre- (Y) and post (M)-sexual maturation stage were examined. Only spermatogonia and a few primary spermatocytes could be observed in yak testes before sexual maturation. However, after sexual maturation, spermatogenic cells at all stages of development were found to be distributed in the yak testes. Moreover, sperm cells were detected in the middle of the seminiferous tubules.
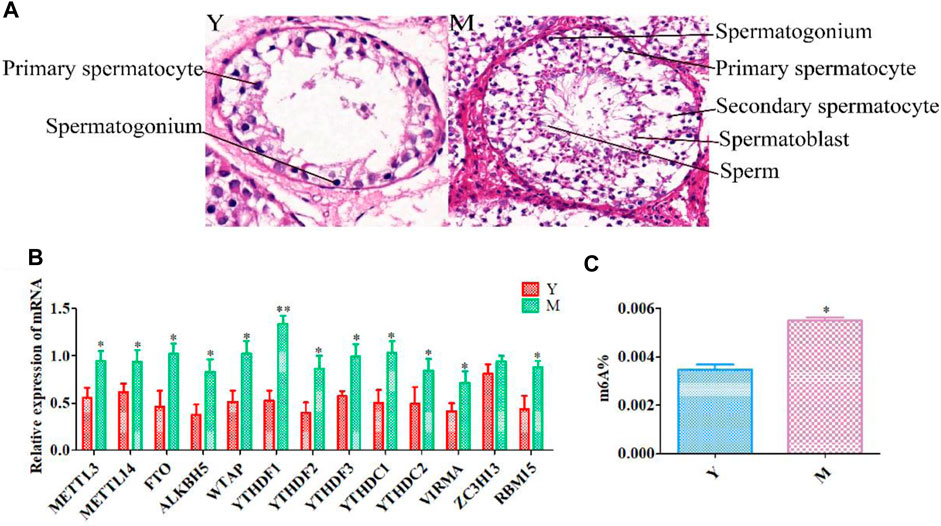
FIGURE 1. Histological and qRT-PCR analysis. (A) HE staining of yak testes before (Y) and after (M) sexual maturation. (B) Quantitative expression of methylation-related genes. (C) Overall RNA methylation levels in testicular tissue. **p < 0.01, *p < 0.05.
Quantitative RT-PCR Findings and Global Methylation Levels
After sexual maturation, demethylases (FTO and ALKBH5), methyltransferases (METTL3, METTL14, WTAP, VIRMA, and RBM15), and m6A-binding proteins (YTHDC1, YTHDC2, YTHDF2, and YTHDF3) were significantly up-regulated. Moreover, while YTHDF1 showed very significant up-regulation after sexual maturation, ZC3H13 expression did not change significantly (Figure 1B). Such changes in the expression of methylation-related enzymes could result in dynamic variations in m6A methylation in testicular tissue after sexual maturation in yaks. Therefore, we compared the methylation status of yak testicular tissue before and after sexual maturation and observed a significant increase in m6A levels after sexual maturation (Figure 1C).
Sequencing Quality Control and Reference Genome Alignment
The available data volume of each sample ranged from 6.07G to 6.83G, the Q30 base distribution ranged from 92.63 to 93.68%, and the average GC content was 48.61%. The genome alignment rate of each sample with respect to the reference genome was 88.62–94.21% (Supplementary Table S3). A high multiple comparison rate was observed in the comparison analysis, and multiple alignment reads were therefore excluded. Only single reads were retained for subsequent analysis. Due to some differences in the matching rate of the samples, the results of matching were divided into three parts: unaligned reads, uniquely mapped reads, and multiple mapped reads (Supplementary Figure S1A).
Methylation Peak Detection and Annotation
We obtained an m6A map of the complete yak transcriptome using high-throughput sequencing and analyzed the detected peaks (Supplementary Table S4; Supplementary Figure S1B). In group Y (1.5-year-old yaks), the number of peaks was 10,042, and genome percentage was 1.54%. In group M (5-year-old yaks), the number of peaks was 11,763, and the genome percentage was 1.87%. Due to the differences in the predicted methylation sites across different samples, the predicted methylation sites were divided into five categories based on reliability: none, low, moderate, high, and very high (Supplementary Figure S1C).
Annotation results from ChIPseeker were counted. We observed 1–6 m6A peaks on each gene, and 85% of genes had only one peak. (Supplementary Table S5). The gene FREM2 located on chromosome 15 had the most methylation peaks (6 m6A peaks). Statistical analyses of peak width showed that peaks of 0–1,000 bp were the most common, and those sized more than 100,000 bp were the least common (Table 1). The majority of peaks were concentrated in the exon region, followed by the 3′UTR and 5′UTR region (Figure 2A). We have chosen two genes to show the m6A methylation pattern. The peak of ROBO1 was located at the CDS and 3′ UTR regions, and the peak of ADAMTS1 was located at the 3′ UTR, the CDS, and the 5′ UTR regions (Figure 2B). A significant enrichment was observed in peaks at the 3′UTR (near the stop codon) (Figure 3A).
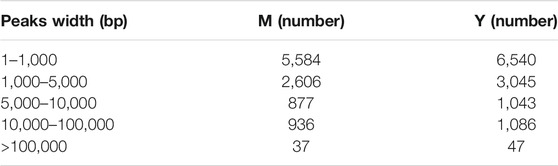
TABLE 1. Statistical analyses of peak widths.
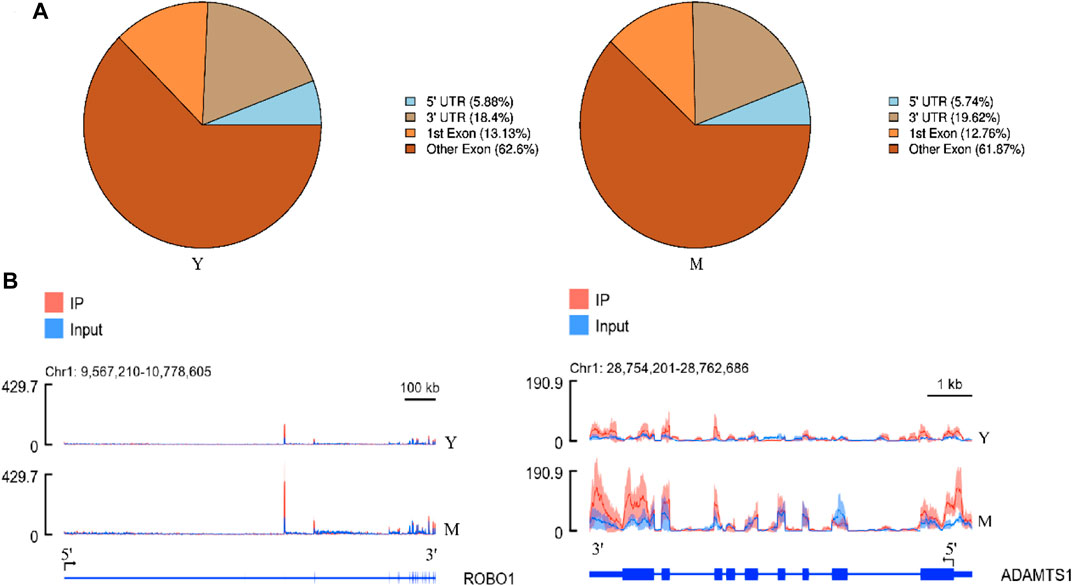
FIGURE 2. Topological distribution of m6A peaks. (A), Pie chart showing the peak in gene functional element region annotation. (B), IGV plot shows directly the peaks in the genes of ROBO1 and ADAMTS1. The peak of ROBO1 was located at the CDS and 3′ UTR regions, and the peak of ADAMTS1 was located at the 3′ UTR, the CDS, and the 5′ UTR regions.
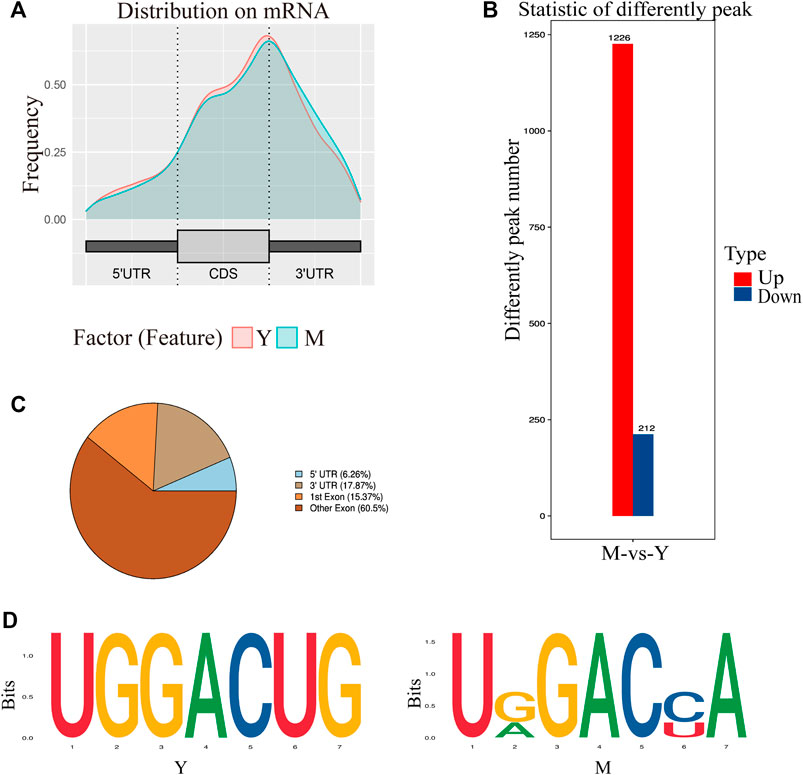
FIGURE 3. Characteristics of the m6A peak. (A) Distribution of m6A peak on mRNA. The peaks were mostly distributed on the exon region and there was a distinct enrichment peak at the 3′ terminate (near the stop codon). Y: Before sexual maturity, M: After sexual maturity. (B) Statistical analysis of differential peaks. (C) Distribution of differential methylation peaks on mRNA. (D) Top motifs with m6A peaks in the M and Y groups. This is based on the respective high-trust peak analysis in each group.
Differential peak analysis revealed 1,438 differential methylation peaks between the M and Y groups. The total length of these peaks was 5,352,512 bp and their average length was 3,722.19 bp, accounting for 0.19% of the genome. The number of significantly up-regulated and down-regulated methylation peaks in Y-VS-M was 1,226 and 212, respectively (Figure 3B). Annotation analysis revealed that the differential methylation peaks were also mainly concentrated in the exon region, followed by the 3′UTR and finally the 5′UTR region (Figure 3C).
Next, we determined whether the identified m6A peaks shared any common motifs. We found that m6A modifications were typically present in 5-RRACH-3 (R = A or G; H = A, C, or U) sequences (Figure 3D), as reported previously (Yi et al., 2021).
GO and KEGG Pathway Analysis
To elucidate the function of m6A RNA methylation in testicular sexual maturation, we performed functional enrichment analyses of genes with significantly altered m6A methylation levels based on the GO and KEGG databases. GO analysis showed enrichment for m6A methylation in the following processes across the three biological modules of GO: 1) Molecular function: protein kinase binding, phosphatidylinositol 3-kinase binding, and cadherin binding; 2) Cellular component: nucleoplasm, cytoskeleton, and cell cortex; 3) Biological process: transcription by RNA polymerase III, regulation of cholesterol biosynthesis, and neural tube closure (Figure 4A). KEGG analysis showed that genes with significant changes in m6A methylation levels were mainly enriched in processes such as homologous recombination, the Notch signaling pathway, and Growth hormone synthesis, secretion, and action (Figure 4B).
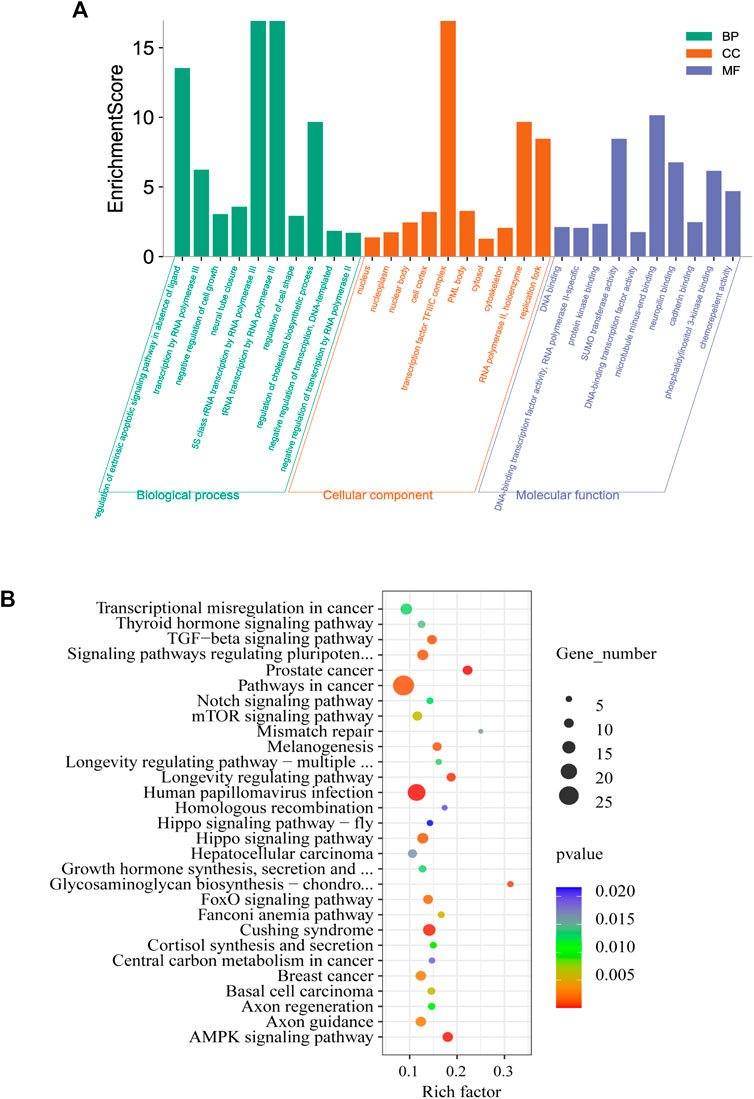
FIGURE 4. Enrichment analysis. (A,B), GO (A) and KEGG (B) analysis of genes associated with differential methylation peaks before and after sexual maturation.
We examined mRNA-level changes in the testicular tissue of yaks before and after sexual maturation based on RNA-seq data (Supplementary Table S6). Compared with the Y group, 895 and 760 genes were up- and down-regulated, respectively, in the M group (p < 0.5, log2FC > 1), indicating that mRNA expression levels changed during sexual maturation. Further analysis of these differentially expressed genes using GO analysis showed enrichment in the following processes: 1) Molecular function: transmembrane signaling receptor activity, semaphorin receptor binding, and phosphatidylinositol 3-kinase binding; 2) Cellular component: sperm flagellum, motile cilium, and cytoskeleton; 3) Biological process: spermatogenesis, cell differentiation, and actin crosslink formation (Figure 5A). KEGG analysis revealed that the differentially expressed genes had significant enrichment in pathways such as Axon guidance, the PI3K-Akt signaling pathway, the Relaxin signaling pathway, and Focal adhesion (Figure 5B).
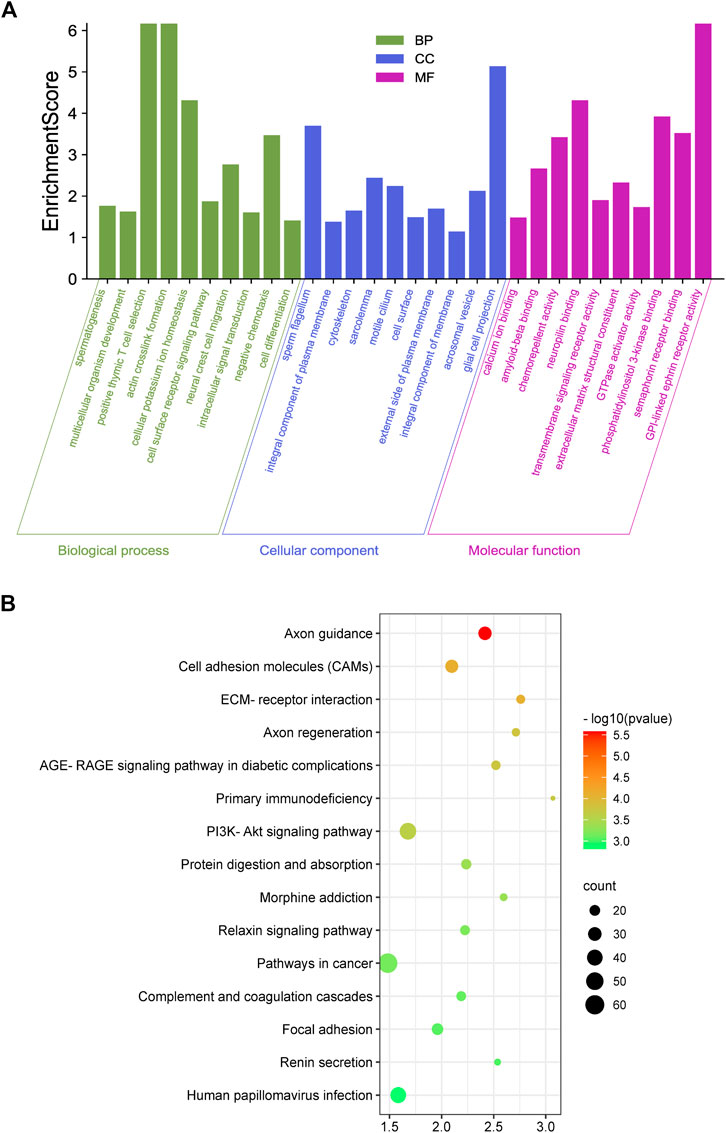
FIGURE 5. Enrichment analysis. (A,B), GO (A) and KEGG (B) enrichment analysis of genes differentially expressed before and after sexual maturation.
By comparing the GO and KEGG analysis results of differentially expressed genes and differential methylation of corresponding genes, we found enrichment in the GO terms for phosphatidylinositol 3-kinase binding and other kinase binding, cytoskeleton, and spermatogenesis. KEGG analysis showed that all of the genes were enriched in spermatogenesis-related signaling pathways. These results indicated that the sexual maturity-related changes in gene expression and methylation levels in yaks were associated with spermatogenesis.
Combined Comparison of m6A Methylation and Gene Expression Before and After Sexual Maturation
To further elucidate the relationship between m6A methylation and RNA expression levels, we used peaks with a Log2 Fold Change >0.5, p < 0.01 and mRNAs showing a log2 foldchange >0.5, p < 0.01. The difference in the m6A methylation level was less than 0.05. Both m6A peaks and mRNA levels were significantly different for 396 mRNAs. Of these, 286 showed an up-regulation of m6A peaks and mRNA expression, 38 showed an up-regulation of m6A peaks and down-regulation of mRNA expression, 5 showed a down-regulation of m6A peaks and up-regulation of mRNA expression, and 67 showed a down-regulation of both m6A peaks and mRNA expression (Supplementary Table S7). The association of m6A methylation with mRNA expression is depicted in Figure 6A.
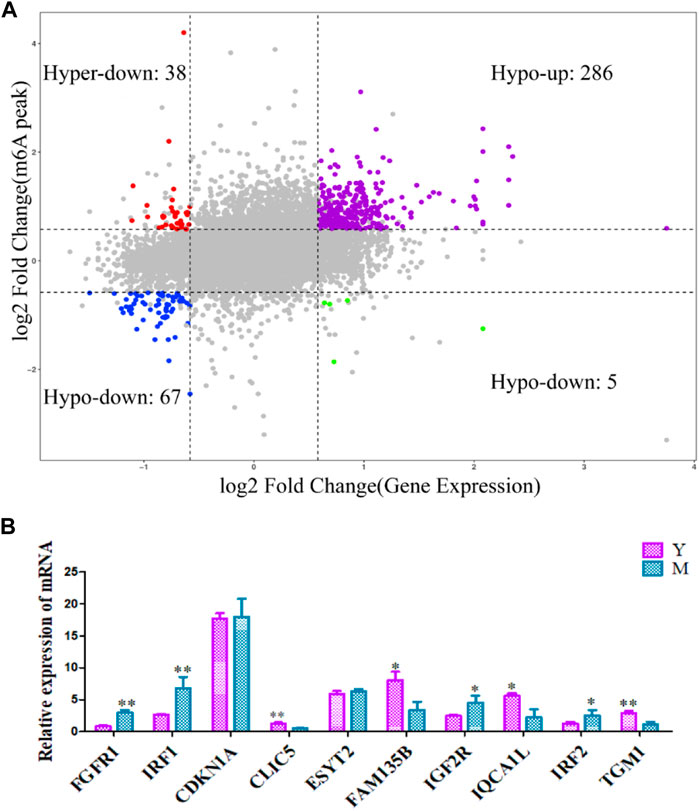
FIGURE 6. Differentially methylated and expressed genes examined using conjoint analysis and verified using qRT-PCR. (A) Combined four-quadrant scatter plot of genes showing differential methylation and differential expression (gray dots, genes with no significant differences; colored dots, genes with significant differences). (B) The differentially expressed genes were verified using qRT-PCR. Y: before sexual maturation. M: after sexual maturation. **p < 0.01, *p < 0.05.
Verification of m6A-Modified Differentially Expressed Genes
qRT-PCR was used to verify the levels of differentially methylated genes in yak testes before and after sexual maturation. The observed expression patterns corroborated the RNA-seq data (Figure 6B), confirming the validity of our transcriptome analysis.
Discussion
Sexual maturation is a continuous process that allows males to produce mature sperm and maintain fertility. Spermatogenesis, a complex developmental process, is meticulously controlled by complex regulatory processes at several levels (Lin and Tong, 2018). m6A RNA modification is critical for the development of the male reproductive system (Zhao T. et al., 2021). The development of testicular tissue is a hot-spot in reproductive biology research. In our study, HE staining was performed on testicular tissue sections. This staining revealed significant differences in testicular tissue before and after sexual maturity. This was consistent with experimental findings from Qiu et al., who observed no sperm in smears prepared using testicular tissue from 1.5-year-old Datong yak testicles but observed normal sperm in those prepared using testicles obtained from yaks aged 2 years or older (Qiu et al., 2010). These results indicated that the stages at which we obtained yak testicular tissue in the present study were suitable for the purpose of our research.
qRT-PCR confirmed that methylation-related enzymes in the yak testes showed different levels before and after sexual maturation. ALKBH5, YTHDC2, FTO, METTL3, and METTL14 showed significantly higher levels after sexual maturation. Murine studies have shown that ALKBH5 knockout results in impaired spermatogenesis and male sterility (Zheng et al., 2013a). In YTHDC2 knockout mice, germ cells do not mature beyond the zygotic period (Hsu et al., 2017). METTL3 and METTL14 double-knockout down-regulates the translation of the key spermatogenesis-related m6A modification transcripts, leading to anomalies in spermatogenesis during anaphase (Lin et al., 2017). FTO mutations are positively associated with decreased semen quality and may reduce male fertility (Landfors et al., 2016). Therefore, it appears thar m6A methylation-related enzymes are vital for maintaining the normal function of the male reproductive system. We also observed that the overall methylation levels in yak testicular tissue increased after sexual maturation. This was in line with results from Sun et al.’s (2020) study , in which methylation levels were observed to increase with age in mice, peaking in adult males.
In mammals, m6A enrichment is observed near the stop codon and the 3′UTR. Ke et al. (2015) found that most m6A residues are located in the last exon, allowing for 3′UTR regulation. In contrast, in amphibians such as Xenopus laevis, m6A enrichment is observed near the start and stop codons (Sai et al., 2020). By analyzing the distribution of methylation peaks in the yak testes, we found m6A peaks to be primarily enrichmented around the 3′UTR near the stop codon, consistent with the results of human and mouse studies (Dominissini et al., 2012; Meyer et al., 2012; Schwartz et al., 2014). Different genes of last Exon are different and the statistical difficulty is high, so the experimental statistics are only divided into two parts, namely the first exon and other exons. Hence, the overall distribution of m6A sites appears to be comparable across mammals. m6A has been found to contain a conserved RRACH motif (R = a purine, A = m6A, and H = non-guanine base) (Dominissini et al., 2012; Meyer et al., 2012). By analyzing the enrichment motifs around the m6A peak, we also found a large number of RRACH motifs, consistent with the results observed in plants, mammals, yeast, and amphibians (Sai et al., 2020). This strongly suggests that RNA adenosine methylation is conserved across mammals.
m6A modifications are known to generate mRNA instability (Geula et al., 2015). To elucidate the role of m6A methylation in sexual maturation, we probed the different expression peaks across differentially expressed mRNAs. A total of 396 mRNAs showed differential expression peaks, 353 of which showed patterns consistent with the trends of peak expression, accounting for 89% of all the identified mRNAs. m6A peaks showed a positive correlation with the expression of related genes.
Subsequently, we delineated the role of m6A modifications in sexual maturation. GO annotation showed that most mRNAs containing m6A modifications were enriched in different domains across the molecular function, cellular component, and biological process modules, and their functions were mainly concentrated in aspects such as binding and catalytic activity. KEGG analysis showed that differential m6A modification was primarily associated with signal transduction and hormone synthesis. Androgenic action is key for the postnatal masculinization of the fetus, especially in humans and other mammals, and the preadolescent stage is characterized by a marked lack of gonadal steroid secretion. During pubertal development, the testes resume androgen production, and the effects of androgens become apparent during the development of male secondary sexual characteristics (Rey, 2021). Hence, m6A modifications occur not only in insects and during virus infection (Zhang et al., 2019), but also in the testes during yak sexual maturation.
We observed that m6A methylation levels of genes in testicular tissue changed significantly after sexual maturation in the yak testes. UBE3A is a ubiquitin-linked protease E3A. Ubiquitin is known to regulate many important biochemical processes in the testes, including DNA repair, structural control of meiotic chromatin (Grootegoed et al., 1998), and histone protamine transformation during spermatogenesis (Nickel et al., 1989). Previous studies have suggested that the candidate genes for male infertility are involved in ubiquitination. Wong et al. (Wong et al., 2002) confirmed the specific interaction between VCY2 and UBE3A through yeast two-hybridization and in vitro co-immunoprecipitation and also confirmed that both the VCY2 and UBE3A genes are located in the germ cell compartment. VCY2, present in the AZFC region of the YQ chromosome, is often absent in infertile men with severe oligozoospermia or azoospermia. These data suggest that UBE3A ubiquitination may be necessary for the function of VCY2 and that UBE3A and VCY2 act synergically to regulate spermatogenesis.
The presence of m6A is essential for normal animal development. KEGG analysis revealed that genes showing differential m6A mRNA methylation play a role in several biological pathways, many of which are involved in male reproduction. The evolutionarily conserved Notch pathway has been found to regulate cell fate determination in some tissues (Borggrefe and Oswald, 2009). The expression of Notch pathway components has been reported in neonatal and adult mammal testes (Dirami et al., 2001; Hayashi et al., 2001). In addition, this pathway is involved in germ cell development in Caenorhabditis elegans (Kimble and Crittenden, 2007) and Drosophila melanogaster (Song et al., 2007). Notch pathway activity in Sertoli cells is known to be critical for determining germ cell fate during the early development of testes. It is also crucial for regulating the differentiation of spermatogonia in adult testes (Garcia et al., 2017), and its interruption leads to increased apoptosis and spermatogenesis defects (Murta et al., 2014). Abnormal Notch activity is also associated with male infertility in rodents and humans (Hayashi et al., 2001; Hayashi et al., 2004). Findings from Murta et al. (Murta et al., 2013) strongly suggest that Notch signaling helps maintain the spermatogonial cell bank, initiation of spermatogenesis, regulation of spermatogenesis speed, maintenance and differentiation of germ cells, and function of mesenchymal cells.
Although methylation-related enzymes are known to play an important role in spermatogenesis and an m6A transcriptional map of sexual maturation in yaks has been developed, how changes in methylation levels affect sexual maturation in mammals remains unclear. The methylation levels in yak testes before and after sexual maturity were measured at the overall tissue level in the present study. However, testicular tissue contains different cell types at different stages, and it is currently challenging to specifically identify which cells show changes in methylation levels. Therefore, understanding the regulation of methylation level changes during the process of sexual maturation is difficult. Currently, there are technical and experience-related limitations in isolating and culturing the different types of cells from yak testes, which creates further problems in elucidating how methylation precisely controls sexual maturation in these animals. In the future, we aim to isolate different cell types from yak testicular tissue using enzymatic hydrolysis and thereby establish an in vitro culture system. By detecting methylation levels in these cell types and perturbing the key methylation regulators in the process of sexual maturation, we hope to further delineate the effect of methylation on sexual maturity.
Conclusion
In this study, the methylation levels in yak testicular tissue were compared before and after sexual maturation and gene enrichment analyses were conducted. We found that methylation levels increase in yak testes after sexual maturation and that several genes showing differential m6A expression are related to male reproduction. To our knowledge, this is the first report of an m6A transcriptional map of the yak testes, and our study lays the foundation for elucidating the function of m6A in the development of yak testes. Although the mechanism underlying post-transcriptional gene regulation and its biological significance in the process of sexual maturation in yaks are not currently clear, functional studies on methylation-related genes and the signaling pathways they are involved in should be carried out to confirm their significance in yak sexual maturation in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by The All animal-related procedures conformed to the China Council on Animal Care and the Ministry of Agriculture of the People’s Republic of China guidelines. All yak-handling procedures were approved by The Animal Care and Use Committee of the Lanzhou Institute of Husbandry and Pharmaceutical Sciences Chinese Academy of Agricultural Sciences (Permit No: SYXK-2014-0002). Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
Conceptualization, XG and XDW; Investigation, PB and MLC; Methodology and software, JP, MC, LX, CL, and XYW; Data curation, XDW and XG; Writing—original draft, XDW and SG; Writing—review and editing, XG and XDW; Funding acquisition, XG and PY. All authors contributed to the interpretation of the results and writing of the article.
Funding
This work was supported by the China Agriculture Research System of MOF and MARA (CARS-37) and Innovation Project of Chinese Academy of Agricultural Sciences (25-LZIHPS-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.755670/full#supplementary-material
References
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME Suite: Tools for Motif Discovery and Searching. Nucleic Acids Res. 37, W202–W208. doi:10.1093/nar/gkp335
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a Flexible Trimmer for Illumina Sequence Data. Bioinformatics (Oxford, England) 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Borggrefe, T., and Oswald, F. (2009). The Notch Signaling Pathway: Transcriptional Regulation at Notch Target Genes. Cell. Mol. Life Sci. 66 (10), 1631–1646. doi:10.1007/s00018-009-8668-7
Cai, Z., Niu, Y., and Li, H. (2021). RNA N6-Methyladenosine Modification, Spermatogenesis, and Human Male Infertility. Mol. Hum. Reprod. 27 (6), 6. doi:10.1093/molehr/gaab020
Cui, X., Wei, Z., Zhang, L., Liu, H., Sun, L., Zhang, S.-W., et al. (2016). Guitar: An R/Bioconductor Package for Gene Annotation Guided Transcriptomic Analysis of RNA-Related Genomic Features. Biomed. Research International 2016, 1–8. doi:10.1155/2016/8367534
Cui, X., Zhang, L., Meng, J., Rao, M. K., Chen, Y., and Huang, Y. (2018). MeTDiff: A Novel Differential RNA Methylation Analysis for MeRIP-Seq Data. Ieee/acm Trans. Comput. Biol. Bioinf. 15 (2), 526–534. doi:10.1109/TCBB.2015.2403355
Das, P. P., Krishnan, G., Doley, J., Bhattacharya, D., Deb, S. M., Chakravarty, P., et al. (2019a). Establishing Gene Amelogenin as Sex-specific Marker in Yak by Genomic Approach. J. Genet. 98, 7. doi:10.1007/s12041-019-1061-x
Das, P. P., Krishnan, G., Doley, J., Biswas, T. K., Paul, V., Chakravarty, P., et al. (2019b). Identification and Expression Profiling of MSY Genes of Yak for Bull Fertility. J. Genet. 98 (2), 1–10. doi:10.1007/s12041-019-1091-4
Dirami, G., Ravindranath, N., Achi, M. V., and Dym, M. (2001). Expression of Notch Pathway Components in Spermatogonia and Sertoli Cells of Neonatal Mice. J. Androl. 22 (6), 944–952. doi:10.1002/j.1939-4640.2001.tb03434.x
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the Human and Mouse m6A RNA Methylomes Revealed by m6A-Seq. Nature 485 (7397), 201–206. doi:10.1038/nature11112
Frye, M., Harada, B. T., Behm, M., and He, C. (2018). RNA Modifications Modulate Gene Expression during Development. Science 361 (6409), 1346–1349. doi:10.1126/science.aau1646
Garcia, T. X., Parekh, P., Gandhi, P., Sinha, K., and Hofmann, M.-C. (2017). The NOTCH Ligand JAG1 Regulates GDNF Expression in Sertoli Cells. Stem Cell Development 26 (8), 585–598. doi:10.1089/scd.2016.0318
Geula, S., Moshitch-Moshkovitz, S., Dominissini, D., Mansour, A. A., Kol, N., Salmon-Divon, M., et al. (2015). m 6 A mRNA Methylation Facilitates Resolution of Naïve Pluripotency toward Differentiation. Science 347 (6225), 1002–1006. doi:10.1126/science.1261417
Grootegoed, J. A., Baarends, W. M., Roest, H. P., and Hoeijmakers, J. H. (1998). Knockout Mouse Model and Gametogenic Failure. Mol. Cell Endocrinol 145 (1-2), 161–166. doi:10.1016/S0303-7207(98)00183-X
Hayashi, T., Kageyama, Y., Ishizaka, K., Xia, G., Kihara, K., and Oshima, H. (2001). Requirement of Notch 1 and its Ligand Jagged 2 Expressions for Spermatogenesis in Rat and Human Testes. J. Androl. 22 (6), 999–1011. doi:10.1002/j.1939-4640.2001.tb03441.x
Hayashi, T., Yamada, T., Kageyama, Y., Negishi, T., and Kihara, K. (2004). Expression Failure of the Notch Signaling System Is Associated with the Pathogenesis of Maturation Arrest in Male Infertility Patients. Fertil. Sterility 81 (3), 697–699. doi:10.1016/j.fertnstert.2003.08.026
Hsu, P. J., Zhu, Y., Ma, H., Guo, Y., Shi, X., Liu, Y., et al. (2017). Ythdc2 Is an N6-Methyladenosine Binding Protein that Regulates Mammalian Spermatogenesis. Cell Res 27 (9), 1115–1127. doi:10.1038/cr.2017.99
Huang, J., and Yin, P. (2018). Structural Insights into N 6 -methyladenosine (M 6 A) Modification in the Transcriptome. Genomics, Proteomics & Bioinformatics 16 (002), 85–98. doi:10.1016/j.gpb.2018.03.001
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat. Chem. Biol. 7 (12), 885–887. doi:10.1038/nchembio.687
Ke, S., Alemu, E. A., Mertens, C., Gantman, E. C., Fak, J. J., Mele, A., et al. (2015). A Majority of m6A Residues Are in the Last Exons, Allowing the Potential for 3′ UTR Regulation. Genes Dev. 29 (19), 2037–2053. doi:10.1101/gad.269415.115
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Kimble, J., and Crittenden, S. L. (2007). Controls of Germline Stem Cells, Entry into Meiosis, and the Sperm/Oocyte Decision in Caenorhabditis elegans. Annu. Rev. Cell Dev. Biol. 23 (1), 405–433. doi:10.1146/annurev.cellbio.23.090506.123326
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 28 (24), 3211–3217. doi:10.1093/bioinformatics/bts611
Krishnan, G., Paul, V., Biswas, T. K., Chouhan, V. S., Das, P. J., and Sejian, V. (2018). Adaptation Strategies of Yak to Seasonally Driven Environmental Temperatures in its Natural Habitat. Int. J. Biometeorol. 62 (8), 1497–1506. doi:10.1007/s00484-018-1549-8
Krishnan, G., Paul, V., Hanah, S. S., Bam, J., and Das, P. J. (2016). Effects of Climate Change on Yak Production at High Altitude. Indian J. Anim. Sci. 86 (6), 621–626.
Lan, X., Cretney, E. C., Kropp, J., Khateeb, K., Berg, M. A., Peñagaricano, F., et al. (2013). Maternal Diet during Pregnancy Induces Gene Expression and DNA Methylation Changes in Fetal Tissues in Sheep. Front. Genet. 4, 49. doi:10.3389/fgene.2013.00049
Landfors, M., Nakken, S., Fusser, M., Dahl, J.-A., Klungland, A., and Fedorcsak, P. (2016). Sequencing of FTO and ALKBH5 in Men Undergoing Infertility Work-Up Identifies an Infertility-Associated Variant and Two Missense Mutations. Fertil. Sterility 105 (5), 1170–1179. doi:10.1016/j.fertnstert.2016.01.002
Lin, Z., Hsu, P. J., Xing, X., Fang, J., Lu, Z., Zou, Q., et al. (2017). Mettl3-/Mettl14-mediated mRNA N6-Methyladenosine Modulates Murine Spermatogenesis. Cell Res 27 (10), 1216–1230. doi:10.1038/cr.2017.117
Lin, Z., and Tong, M.-H. (2019). m6A mRNA Modification Regulates Mammalian Spermatogenesis. Biochim. Biophys. Acta (Bba) - Gene Regul. Mech. 1862 (3), 403–411. doi:10.1016/j.bbagrm.2018.10.016
Liu, H., Xu, Y., Yao, B., Sui, T., Lai, L., and Li, Z. (2020). A Novel N6-Methyladenosine (m6A)-dependent Fate Decision for the lncRNA THOR. Cell Death Dis 11 (8), 613. doi:10.1038/s41419-020-02833-y
Liu, Z.-X., Li, L.-M., Sun, H.-L., and Liu, S.-M. (2018). Link between m6A Modification and Cancers. Front. Bioeng. Biotechnol. 6, 89. doi:10.3389/FBIOE.2018.00089
Mcgowan, M., Holland, M. K., and Boe-Hansen, G. (2018). Review: Ontology and Endocrinology of the Reproductive System of Bulls from Fetus to Maturity. Animal 12, s19–s26. doi:10.1017/S1751731118000460
Meyer, K. D., and Jaffrey, S. R. (2014). The Dynamic Epitranscriptome: N6-Methyladenosine and Gene Expression Control. Nat. Rev. Mol. Cell Biol 15 (5), 313–326. doi:10.1038/nrm3785
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and Near Stop Codons. Cell 149 (7), 1635–1646. doi:10.1016/j.cell.2012.05.003
Murta, D., Batista, M., Silva, E., Trindade, A., Henrique, D., Duarte, A., et al. (2013). Dynamics of Notch Pathway Expression during Mouse Testis Post-Natal Development and along the Spermatogenic Cycle. PLoS ONE 8 (8), e72767. doi:10.1371/journal.pone.0072767
Murta, D., Batista, M., Trindade, A., Silva, E., Henrique, D., Duarte, A., et al. (2014). In Vivo Notch Signaling Blockade Induces Abnormal Spermatogenesis in the Mouse. Plos One 9 (11), e113365. doi:10.1371/journal.pone.0113365
Nickel, B. E., Allis, C. D., and Davie, J. R. (1989). Ubiquitinated Histone H2B Is Preferentially Located in Transcriptionally Active Chromatin. Biochemistry 28 (3), 958–963. doi:10.1021/bi00429a006
Puigoriol-Illamola, D., Martínez-Damas, M., Griñán-Ferré, C., and Pallàs, M. (2020). Chronic Mild Stress Modified Epigenetic Mechanisms Leading to Accelerated Senescence and Impaired Cognitive Performance in Mice. Ijms 21 (3), 1154. doi:10.3390/ijms21031154
Qiu, H., Jin, G. Q., and Jin, R. F. (2007). Analysis of Variance of Repeated Data Measured by Water Maze with SPSS. J. Chin. Integr. Med. 5 (1), 101–105. doi:10.3736/jcim20070121
Qiu, X., Lin, Y. Q., Wang, G. S., Ruan, W. Y., Liu, W., Wang, C. L., et al. (2010). Analysis of Energy Metabolism-Related Enzyme Activities in Yak Testis Tissue. Sichuan J. Zoolog. 29 (05), 590–592+500. doi:10.1080/00949651003724790
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A., and Manke, T. (2014). deepTools: a Flexible Platform for Exploring Deep-Sequencing Data. Nucleic Acids Res. 42 (W1), W187–W191. doi:10.1093/nar/gku365
Rey, R. A. (2021). The Role of Androgen Signaling in Male Sexual Development at Puberty. Endocrinology 162 (2), bqaa215. doi:10.1210/endocr/bqaa215
Sai, L., Li, Y., Zhang, Y., Zhang, J., Qu, B., Guo, Q., et al. (2020). Distinct m6A Methylome Profiles in Poly(A) RNA from Xenopus laevis Testis and that Treated with Atrazine. Chemosphere 245 (Apr), 125631–125638. doi:10.1016/j.chemosphere.2019.125631
Schmittgen, T. D. (2001). Real-time Quantitative PCR. Methods 25 (4), 383–385. doi:10.1006/meth.2001.1260
Schulz, M. H., Devanny, W. E., Gitter, A., Zhong, S., Ernst, J., and Bar-Joseph, Z. (2012). DREM 2.0: Improved Reconstruction of Dynamic Regulatory Networks from Time-Series Expression Data. Bmc Syst. Biol. 6 (104), 104–109. doi:10.1186/1752-0509-6-104
Schwartz, S., Mumbach, M. R., Jovanovic, M., Wang, T., Maciag, K., Bushkin, G. G., et al. (2014). Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 8 (1), 284–296. doi:10.1016/j.celrep.2014.05.048
Shah, M. A., Xu, C., Wu, S., Zhao, W., Luo, H., Yi, C., et al. (2018). Isolation and Characterization of Spermatogenic Cells from Cattle, Yak and Cattleyak. Anim. Reprod. Sci. 193, 182–190. doi:10.1016/j.anireprosci.2018.04.067
Song, X., Call, G. B., Kirilly, D., and Xie, T. (2007). Notch Signaling Controls Germline Stem Cell Niche Formation in theDrosophilaovary. Development 134 (6), 1071–1080. doi:10.1242/dev.003392
Sun, T., Wu, R., and Ming, L. (2019). The Role of m6A RNA Methylation in Cancer. Biomed. Pharmacother. 112, 108613. doi:10.1016/j.biopha.2019.108613
Sun, X., Zhang, J., Jia, Y., Shen, W., and Cao, H. (2020). Characterization of m6A in Mouse Ovary and Testis. Clin. Translational Med. 10 (4), e141. doi:10.1002/ctm2.141
Wang, H., Chai, Z., Hu, D., Ji, Q., Xin, J., Zhang, C., et al. (2019). A Global Analysis of CNVs in Diverse Yak Populations Using Whole-Genome Resequencing. BMC Genomics 20 (1), 61. doi:10.1186/s12864-019-5451-5
Wang, K., Hu, Q., Ma, H., Wang, L., Yang, Y., Luo, W., et al. (2014). Genome-wide Variation within and between Wild and Domestic Yak. Mol. Ecol. Resour. 14 (4), 794–801. doi:10.1111/1755-0998.12226
Wang, L., Wu, Z., Zou, C., Liang, S., Zou, Y., Liu, Y., et al. (2020). Sex-Dependent RNA Editing and N6-Adenosine RNA Methylation Profiling in the Gonads of a Fish, the Olive Flounder (Paralichthys olivaceus). Front. Cell Dev. Biol. 8, 751. doi:10.3389/fcell.2020.00751
Wang, M., Liu, X., Chang, G., Chen, Y., An, G., Yan, L., et al. (2018). Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell Stem Cell 23 (4), 599–614. doi:10.1016/j.stem.2018.08.007
Wang, X., Pei, J., Bao, P., Cao, M., Guo, S., Song, R., et al. (2021). Mitogenomic Diversity and Phylogeny Analysis of Yak (Bos Grunniens). BMC genomics 22 (1), 325. doi:10.1186/S12864-021-07650-X
Wojtas, M. N., Pandey, R. R., Mendel, M., Homolka, D., Sachidanandam, R., and Pillai, R. S. (2017). Regulation of m6A Transcripts by the 3ʹ→5ʹ RNA Helicase YTHDC2 Is Essential for a Successful Meiotic Program in the Mammalian Germline. Mol. Cell 68 (2), 374–387. doi:10.1016/j.molcel.2017.09.021
Wong, E. Y. M., Tse, J. Y. M., Yao, K.-M., Tam, P.-C., and Yeung, W. S. B. (2002). VCY2 Protein Interacts with the HECT Domain of Ubiquitin-Protein Ligase E3A. Biochem. Biophysical Res. Commun. 296 (5), 1104–1111. doi:10.1016/S0006-291X(02)02040-5
Wu, Y., Zhou, C., and Yuan, Q. (2017). Role of DNA and RNA N6-Adenine Methylation in Regulating Stem Cell Fate. Curr. Stem Cell Res Ther 13 (1), 31–38. doi:10.2174/1574888X12666170621125457
Xu, K., Yang, Y., Feng, G.-H., Sun, B.-F., Chen, J.-Q., Li, Y.-F., et al. (2017). Mettl3-mediated m6A Regulates Spermatogonial Differentiation and Meiosis Initiation. Cell Res 27 (9), 1100–1114. doi:10.1038/cr.2017.100
Yi, D., Wang, Q., Zhao, Y., Song, Y., You, H., Wang, J., et al. (2021). Alteration of N6 -Methyladenosine mRNA Methylation in a Rat Model of Cerebral Ischemia-Reperfusion Injury. Front. Neurosci. 15, 605654. doi:10.3389/FNINS.2021.605654
Yu, G., Wang, L.-G., and He, Q.-Y. (2015). ChIPseeker: an R/Bioconductor Package for ChIP Peak Annotation, Comparison and Visualization. Bioinformatics 31 (14), 2382–2383. doi:10.1093/bioinformatics/btv145
Zhang, X., Zhang, Y., Dai, K., Liang, Z., Zhu, M., Pan, J., et al. (2019). N6-Methyladenosine Level in Silkworm Midgut/Ovary Cell Line Is Associated with Bombyx mori Nucleopolyhedrovirus Infection. Front. Microbiol. 10, 2988. doi:10.3389/fmicb.2019.02988
Zhao, B. S., Roundtree, I. A., and He, C. (2017). Post-transcriptional Gene Regulation by mRNA Modifications. Nat. Rev. Mol. Cell Biol 18 (1), 31–42. doi:10.1038/nrm.2016.132
Zhao, J., Xu, Z., Chen, A., You, X., Zhao, Y., He, W., et al. (2019). Identification of Meat from Yak and Cattle Using SNP Markers with Integrated Allele-specific Polymerase Chain Reaction-Capillary Electrophoresis Method. Meat Sci. 148 (FEB), 120–126. doi:10.1016/j.meatsci.2018.08.019
Zhao, T., Wang, J., Wu, Y., Han, L., Chen, J., Wei, Y., et al. (2021b). Increased m6A Modification of RNA Methylation Related to the Inhibition of Demethylase FTO Contributes to MEHP-Induced Leydig Cell Injury☆. Environ. Pollut. 268 (Pt A), 115627. doi:10.1016/j.envpol.2020.115627
Zhao, Y., Hu, J., Sun, X., Yang, K., Yang, L., Kong, L., et al. (2021a). Loss of m6A Demethylase ALKBH5 Promotes post‐ischemic Angiogenesis via post‐transcriptional Stabilization of WNT5A. Clin. translational Med. 11 (5), e402. doi:10.1002/ctm2.402
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C.-M., Li, C. J., et al. (2013a). ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 49 (1), 18–29. doi:10.1016/j.molcel.2012.10.015
Zheng, G., Dahl, J. A., Niu, Y., Fu, Y., Klungland, A., Yang, Y.-G., et al. (2013b). Sprouts of RNA Epigenetics. Rna Biol. 10 (6), 915–918. doi:10.4161/rna.24711
Keywords: yak, before sexual maturity, after sexual maturity, N6-methyladenosine, testicular tissue
Citation: Wang X, Pei J, Guo S, Cao M, Bao P, Xiong L, Wu X, Chu M, Liang C, Yan P and Guo X (2021) Characterization of N6-Methyladenosine in Domesticated Yak Testes Before and After Sexual Maturity. Front. Cell Dev. Biol. 9:755670. doi: 10.3389/fcell.2021.755670
Received: 09 August 2021; Accepted: 27 October 2021;
Published: 11 November 2021.
Edited by:
Julian Valdes, National Autonomous University of Mexico, MexicoReviewed by:
Yun Bai, ShanghaiTech University, ChinaDiana Guallar, University of Santiago de Compostela, Spain
Copyright © 2021 Wang, Pei, Guo, Cao, Bao, Xiong, Wu, Chu, Liang, Yan and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Yan, yanping@caas.cn; Xian Guo, guoxian@caas.cn