TR35 Exerts Anti-tumor Effects by Modulating Mitogen-Activated Protein Kinase and STAT3 Signaling in Lung Cancer Cells
 Zhiyong Shi1†
Zhiyong Shi1†  Yang Gao1†
Yang Gao1†  Lifeng Feng1
Lifeng Feng1  Wencong Tian1
Wencong Tian1  Zhihua Dou2 Chen Liu2
Zhihua Dou2 Chen Liu2  Jie Liu1
Jie Liu1  Yang Xu1
Yang Xu1  Yachen Wang1
Yachen Wang1  Jie Yan1 Qiang Wu3
Jie Yan1 Qiang Wu3  Jing Li1
Jing Li1  Liang Yang1
Liang Yang1  Zhaocai Zhang4* Jie Yang2*
Zhaocai Zhang4* Jie Yang2*  Zhi Qi1*
Zhi Qi1*- 1Department of Molecular Pharmacology, School of Medicine, Nankai University, Tianjin, China
- 2Department of Bioengineering, College of Life Science and Technology, Xinjiang University, Ürümqi, China
- 3Key Laboratory of Emergency and Trauma of Ministry of Education, Research Unit of Island Emergency Medicine, School of Tropical Medicine and Laboratory Medicine, Chinese Academy of Medical Sciences (No. 2019RU013), Hainan Medical University, Haikou, China
- 4Department of Critical Care Medicine, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Cancer is a complex disease extremely dependent on its microenvironment and is highly regulated by a variety of stimuli inside and outside the cell. Evidence suggests that active camel whey fraction (TR35) confer anti-tumor effects in non-small cell lung cancer (NSCLC). However, its exact mechanisms remain elusive. Here, we investigated the mechanisms underlying suppression of NSCLC cell growth and proliferation by TR35. Treatment of A549 and H1299 cells with TR35 suppressed their growth and enhanced apoptosis, as revealed by CCK-8, colony formation and flow cytometric analyses. We find that TR35 suppresses tumor growth in a xenograft nude mouse model without losses in body weight. RNA-seq and KEGG pathway analyses showed that the DEGs were enriched in mitogen-activated protein kinase (MAPK) and Jak-STAT signaling pathways. After test the key factors’ activity associated with these pathways by Immunohistochemical (IHC) staining and western blotting, the activation of JNK phosphorylation and inhibition of p38 and STAT3 phosphorylation was observed both in TR35 treated lung cancer cell and tumor tissue. Taken together, these results showed that TR35 play a significant role in the NSCLC progression in the tumor microenvironment via MAPK and Jak-STAT signaling, highlighting TR35 as a potential therapeutic agent against lung cancer.
Introduction
Lung cancer is a most common malignancy that is associated with high morbidity and mortality worldwide. Non-small cell lung cancer (NSCLC) accounts for 80–85% of lung cancers (No authors listed, 2020). Although treatment advances have significantly improved lung cancer prognosis, its 5-year survival remains low, at <15% (Miller et al., 2016), underscoring the need for effective preventive and therapeutic strategies against NSCLC.
In recent years, camel milk has been shown to be one of the most important special dairy products. Some dietary ingredients exhibit effectiveness against diabetes (Agrawal et al., 2005), hepatitis (El Miniawy et al., 2014), allergies (Shabo et al., 2005), autism, lactose intolerance (Shabo and Yagil, 2005), as well as cancer (Badawy et al., 2018; Kamal et al., 2018). In the traditional medicine of ethnic minorities in Xinjiang, camel milk has long been applied as an adjuvant anti-cancer treatment, including in lung cancer. Our previous study showed that, TR35, an active component of camel milk have inhibitory effects against esophageal cancer (Yang J. et al., 2019), but its effects against lung cancer are unclear.
The mitogen-activated protein kinase (MAPK) signaling cascade is made up of P38, JNK and ERK, and influences various processes, including cell proliferation, differentiation, apoptosis, and autophagy (Li et al., 2020). Numerous small molecule drugs and natural products inhibit cancer cell proliferation through MAPK signaling activation (Sui et al., 2017), highlighting its potential as an anti-cancer therapeutic target.
Signal transducer and activator of transcription 3 (STAT3) is overexpressed in various cancer types and modulates various cellular processes like proliferation, apoptosis, and differentiation, which makes it an attractive anticancer target (Subramaniam et al., 2013). Persistent STAT3 inhibition suppresses expression of its downstream targets, such as c-Myc, Bcl-XL, blocking cell cycle progression, and promoting apoptosis (Yu et al., 2007; Laudisi et al., 2018). STAT3 signaling is vital in lung cancer treatment (Bousquet Mur et al., 2020).
Here, we find that TR35 inhibits the growth and proliferation of lung cancer cells in vitro and in vivo. RNA-seq and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses implied that TR35 may mediate its anti-cancer effects via MAPK and Jak-STAT signaling. Our data indicate that p-JNK, p-p38, and p-STAT3 might be key TR35 targets. Together, our findings highlight TR35 as a potential anti-NSCLC therapeutic factor.
Materials and Methods
Materials
TR35 was purified from Xinjiang Bactrian camel milk as previously described (Yang J. et al., 2019), dissolved in RPMI 1640 complete medium, and then filtered to remove bacteria.
Cells Culture
H1299 and A549 (human lung cancer cell lines) were obtained from the Cell Bank of Shanghai. They were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS (BI, China) and 1% penicillin/streptomycin (Gibco, 10378016), in an incubator at 37°C, 5% CO2.
Cell Cytotoxic Assay
Cell seeding was in a 96-well microplate at a density of 2,000 cells/well. Then, to allow attachment, they were cultured overnight. They were then treated with TR35 at 0, 1, 2, and 4 mg/ml for 24 and 48 h. To evaluate proliferation, 10 μL of CCK-8 (Dojindo, CK04-500) was added and cells incubated for 4 h. Next, absorbance was read at 450 nm after which we calculated the number of living cells per well. Each experiment was repeated at least three times.
Colony Formation Assay
Cell seeding at a density of 500 cells/well was done in six-well plates, in triplicate. Then, they were cultured until visible colonies formed, with media change every 3 days. After 12 days, colonies were fixed in anhydrous methanol for a duration of 15 min, stained using 0.2% crystal violet (Sigma-Aldrich, C0775-25G), and the number of cells determined using a light microscope. The clone formation rate was given by the formula: .
Analysis of Cell Cycle and Apoptosis
Cell seeding was done on six-well plate at 1 × 106 cells/well and grown to 70% confluence before treatment with TR35 at 0, 1, 2, and 4 mg/ml for 24 and 48 h. Cell cycle analysis was done using propidium iodide (PI) following manufacturer instructions (cell cycle detection kit, BD, 550825). Apoptosis analysis was done using a Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, 556547) and Annexin V-APC/PI Apoptosis Kit (Sungene Biotech, AO2001-11A-H) using manufacturer protocol and analyzed by flow cytometry on FACSCalibur (BD Biosciences). The flow cytometry data were analyzed using FlowJo X.
RNA-seq Analysis
Seeding of A549 cells was done at a density of 5 × 106 cells/flask in two 75 cm2 flasks. Treatment with TR35 (4 mg/ml) for 48 h was done in one flask while the other was used as the untreated (control). Then, a High Pure RNA Isolation Kit (Roche, 11828665001) was used to extract Total RNA. cDNA was synthesized using RNA-Seq Sample Prep Kit (Illumina) as per the manufacturers’ instructions. Quality control analysis of the sample library was done on an Agilent 2100 Bioanalyzer and ABI StepOnePlus RT-PCR. The cDNA libraries were sequenced at the Beijing Genomics Institute (BGI, Shenzhen, China) using a HiSeq 2000 platform (Illumina).
Protein Extraction and Western Blot
The procedures of Western blot analysis were as described previously (Tian et al., 2020). Briefly, cells were lysed using RIPA lysis buffer (Santa, sc-24948, Germany). The BCA Protein Assay Kit (Thermo Fisher Scientific, 23227) was used to determine protein concentrations, according to the manufacturer’s instructions. Protein samples were resolved by SDS-PAGE and detected by immunoblotting with antibodies against c-Myc (CST, 13987), Bcl-XL (CST, 2764), p-STAT3 (CST, 9145), STAT3 (CST, 9139S), p-JNK (CST, 4668), JNK (CST, 9252T), p-p38 (CST, 4551), and p38 (CST, 8690T). Tubulin was used as the loading control and band intensities evaluated on Image J.
Animals and Treatment
A total of twenty 4-week old BALB/c nude mice (18–22 g) were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice were kept in specific pathogen-free (SPF) conditions, at 23 ± 2°C, 12 h light/dark cycles at the animal facility, School of Life Sciences, Nankai University. The mice were randomly split into four groups (five mice each). Two treated groups and two control groups were given TR35 (4 mg/ml, in Milli-Q H2O) and an equal volume of H2O, respectively, by oral gavage, twice a day. After 1 week, one treated group and one control group were subcutaneously injected with 2 × 106 A549 cells on the back side by axillary. The other treated and control groups were subcutaneously injected with 2 × 106 H1299 cells on the back side by axillary. The body weights of mice were determined after every 3 days. Thirty days after the appearance of subcutaneous scleroma, the nude mice were sacrificed, after which tumors were harvested and weighed. The tumors were divided into two pieces for western blot and Immunohistochemical (IHC) staining analysis.
Immunohistochemical Staining
The paraformaldehyde fixed tumor tissue were subjected to IHC staining according to standard procedures recommended by CST. Primary antibodies against p-JNK (1:200, CST, 4668), p-p38 (1:150, CST, 4551), p-STAT3 (1:200, CST 9145) diluted in 5% goat serum in 0.1% TBST were incubated with different tissues at 4°C overnight. Second and third antibody labeling was done using the rabbit anti-histochemical kit (Zhongshan Jinqiao, China), according to the manufacturer’s instructions. The IHC positive rate was determined using the H-Score method by two independent pathologists at our center.
Statistical Analysis
Statistical testing was divided into three independent experiments, each in triplicate and data were presented as mean ± SEM. The differences between the two groups were analyzed with Student’s t test. The significant differences among the three groups or more than three groups were analyzed by single factor analysis of variance (ANOVA) and then compared many times by LSD test (SPSS ver. 17). P ≤ 0.05 was set as the threshold for statistical significance. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001.
Results
TR35 Inhibits Lung Cancer Cells Proliferation
To evaluate the effects of TR35 on lung cancer cells, A549 and H1299 cells were administered with TR35 at 1, 2, and 4 mg/ml for 24 and 48 h followed by clonogenic and CCK-8 assays to assess growth and proliferation. This analysis revealed that TR35 suppressed lung cancer cells growth and proliferation in a dose-dependent manner, and these effects were most pronounced using TR35 at 4 mg/ml for 48 h (Figure 1A; inhibition rate = 76.4% in A549 and 72.5% in H1299). Examination TR35 long-term effects using clonogenic assays showed that treatment with TR35 for 12 days suppressed the colony-forming capacity of lung cancer cells (Figure 1B).
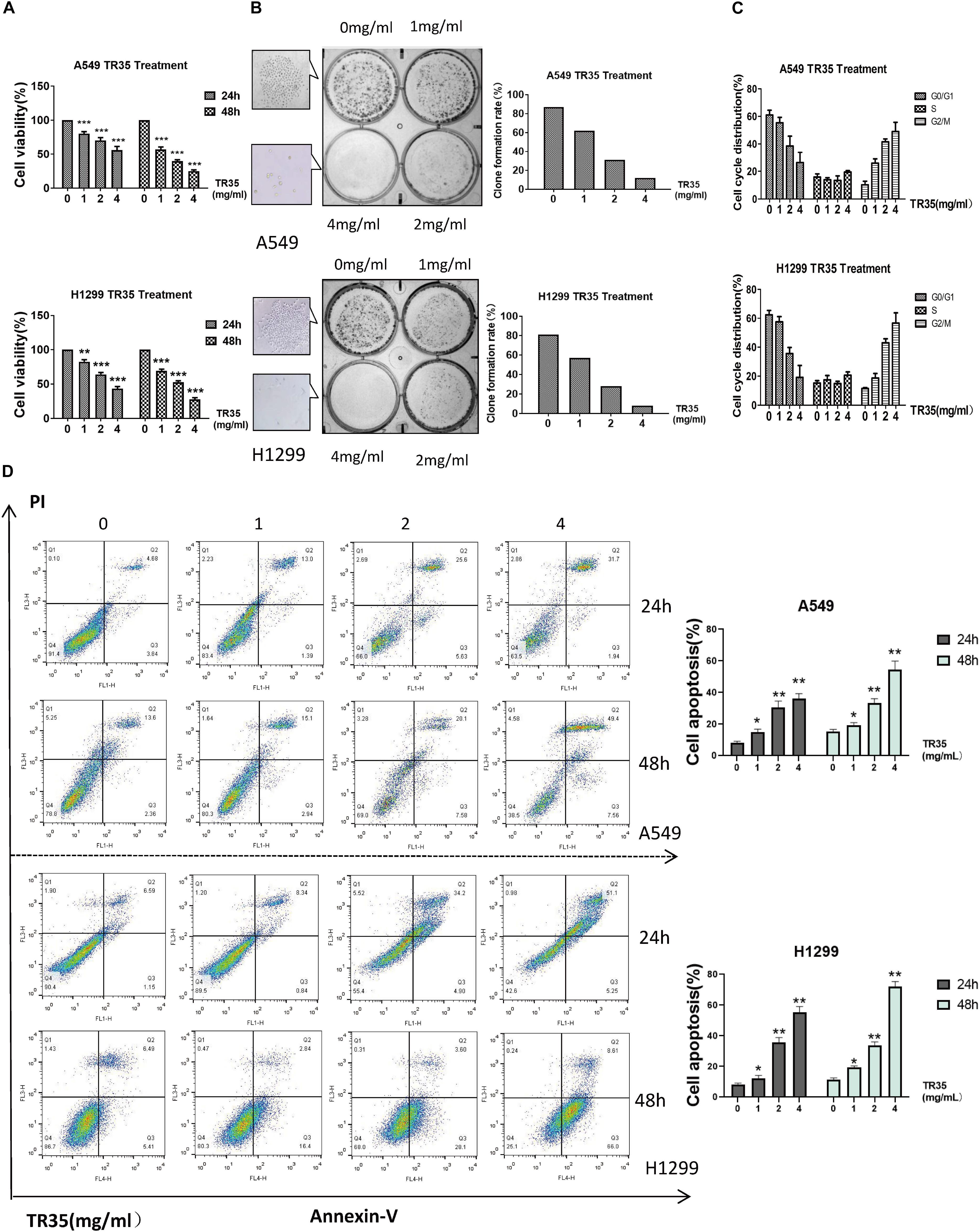
Figure 1. TR35 induced inhibition of A549 and H1299 cell growth. (A) CCK-8 assay evaluation of the inhibition of proliferation of A549 and H1299 cells incubated with TR35 at different concentrations and different times. (B) Colony-formation of A549 and H1299 cells inhibited by TR35. Cells were exposed to different concentrations of TR35 for 11 days in the colony formation assay. (C) TR35-induced G2/M cell cycle arrest. G2/M cell cycle arrest increased in the TR35 treated A549 and H1299 cells. (D) Flow cytometry of Annexin V and PI double staining of A549 and H1299 cells treated with TR35 at different concentrations for 24 and 48 h. Columns show the percentages of apoptotic cancer cells incubated with TR35. The data are representative of three independent experiments. Data are expressed as the mean ± SEM of three determinations (*p < 0.05, **p < 0.01, ***p < 0.001 vs. CON).
TR35 Induced G2/M Cell Cycle Arrest and Apoptosis
Since treatment with TR35 reduced lung cancer cells viability, we assessed its effects on the cell cycle. Flow cytometry analysis showed that treatment with TR35 enhanced cell numbers in the G2/M phase and inhibited cell numbers in the G0/G1 phase (Figure 1C). Suggesting that treatment with TR35 may suppress cell growth by promoting cell cycle arrest. Additionally, treating lung cancer cells with various TR35 concentrations for 48 h significantly increased the number of cells in early or late apoptosis, while reducing the number of viable cells (Figure 1D). Together, these data show that TR35 suppresses lung cancer cell growth by inducing cycle arrest and apoptosis.
RNA-seq
Next, RNA-seq was used to better evaluate the mechanisms driving TR35 effects on lung cancer cell growth and proliferation. To this end, a cDNA library from A549 cells treated with 4 mg/ml TR35 for 48 h and a mock-treated control library were sequenced. A total of 433 differential expression genes (DEGs) were identified (TR35-v-control), including 180 downregulated genes and 253 upregulated genes (twofold as the cut off value). The heatmap used for hierarchical clustering analysis (HCA) exhibited distinct gene expression between control and treated group (Figure 2A). KEGG pathway analysis indicated that the DEGs were enriched for eight pathways, especially MAPK and Jak-STAT signaling pathway, which were significantly different in TR35-treated cells vs. controls as determined by hypergeometric distribution (p ≤ 0.05, Figure 2B).
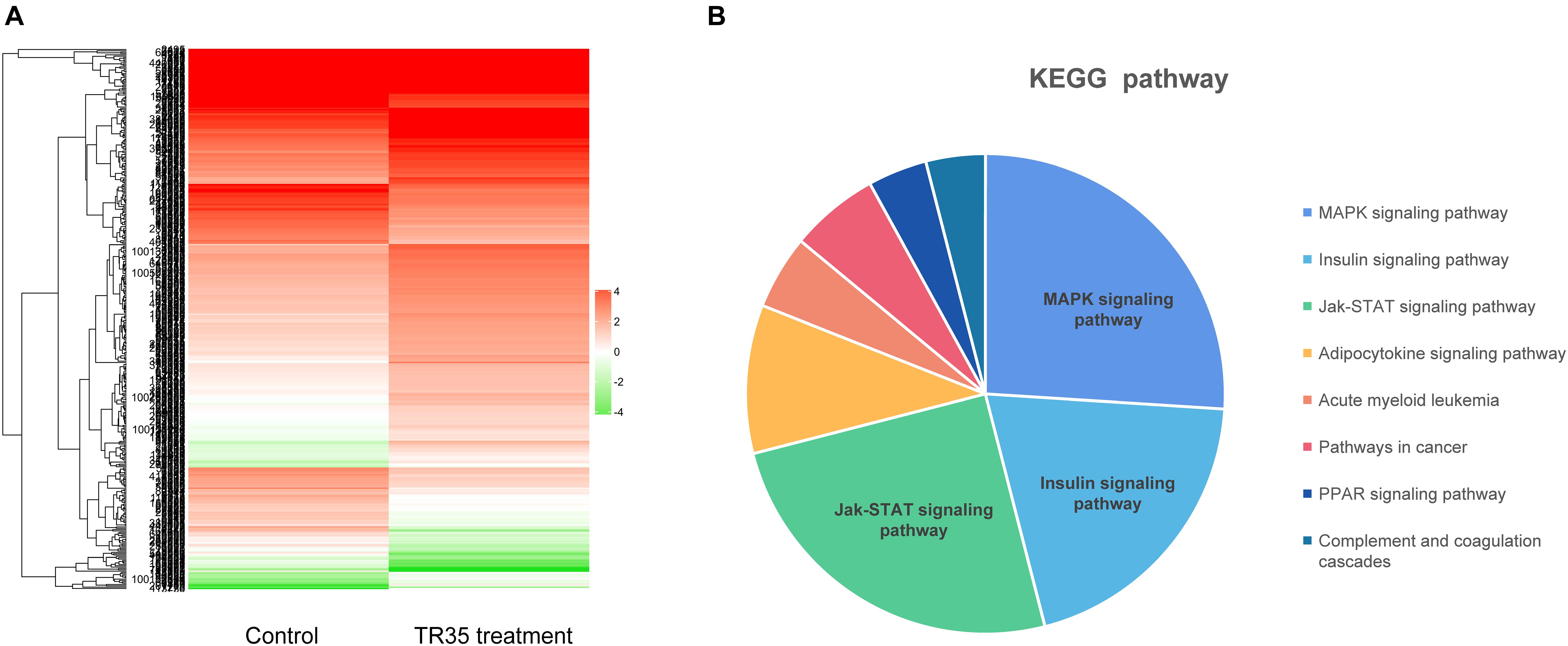
Figure 2. The Heatmap and KEGG pathway enrichment analysis of DEGs in A549 cells after TR35 treatment. (A) Heatmap of expression profile for the 433 DEGs. (B) KEGG pathway enrichment analysis of DEGs.
TR35 Activates JNK and Suppresses STAT3 Signaling in Lung Cancer Cells
Mitogen-activated protein kinase and Jak-STAT signaling are crucial mediators of extracellular stimuli to the nucleus, modulating gene expression and thus, cell proliferation and apoptosis. RNA-seq and KEGG pathway suggested that TR35 affects lung cancer cell proliferation and apoptosis through these pathways. To test this possibility, we used western blotting to evaluate the levels of total and phosphorylated p38, JNK, and STAT3 proteins in A549 and H1299 cells treated with TR35 at 1, 2, and 4 mg/ml for 24 and 48 h. This analysis showed that TR35 dose-dependently enhanced p-JNK level while reducing p-p38 and p-STAT3 levels without significantly affecting total JNK, p38, and STAT3 protein levels. At the same time, the expression level of c-Myc and Bcl-XL, which are downstream molecules of STAT3, was also downregulated (Figures 3A–D). These results indicated that TR35 affects lung cancer cell growth, proliferation and apoptosis via MAPK and Jak-STAT signaling.
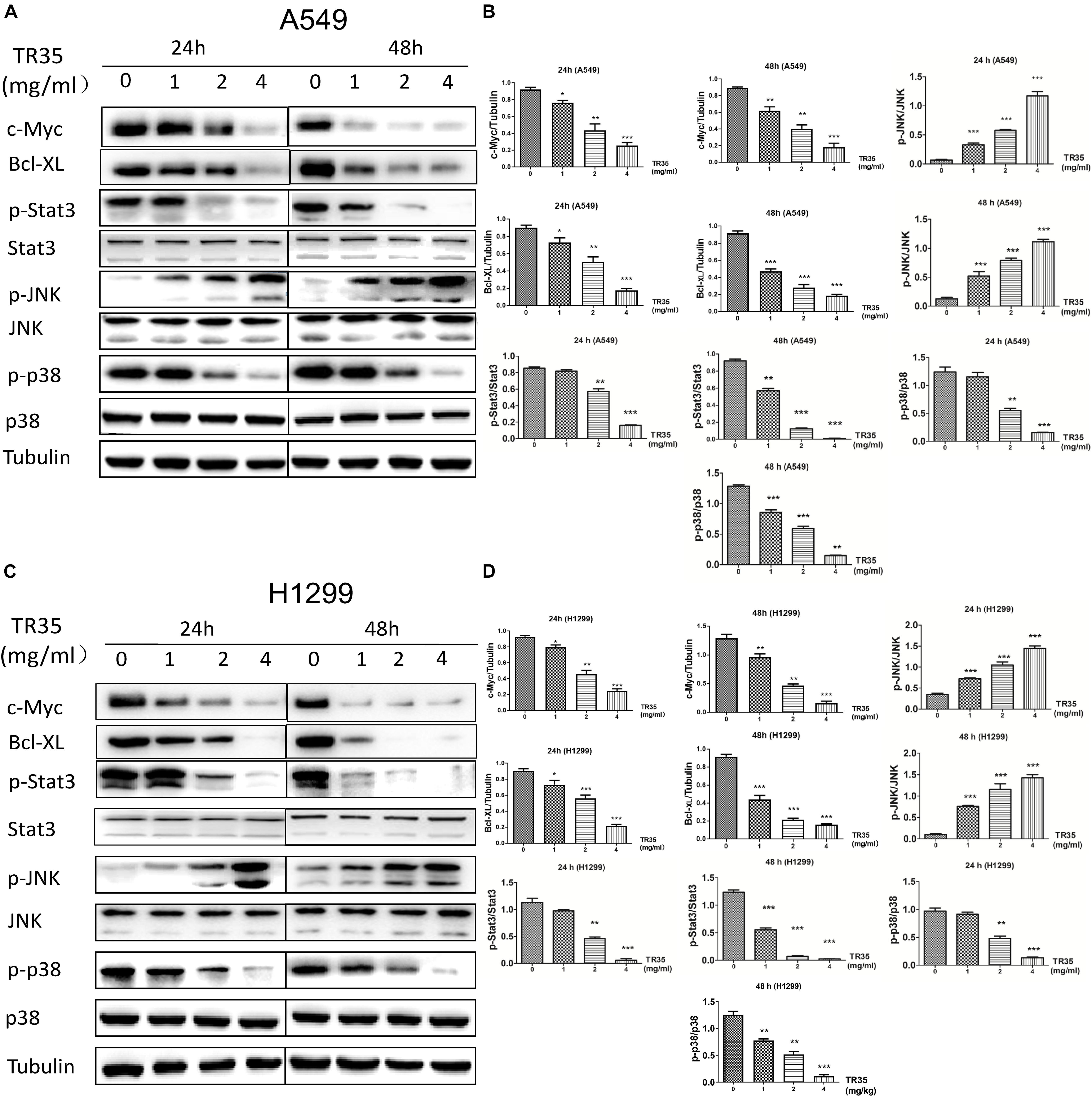
Figure 3. Activation of JNK and reduction of p38 and STAT3 activities by TR35 treatment in H1299 and A549 cells. (A,C) A549 and H1299 cells were treated with TR35 (0–4 mg/mL) for 24 and 48 h. STAT3, p38, and JNK steady state and phosphorylated levels were analyzed by western blotting. Tubulin was used as loading controls. (B,D) Quantification of the band density was analyzed by ImageJ software. Data are presented as the mean ± SEM, n = 3 (*p < 0.05, **p < 0.01, ***p < 0.001 vs. CON).
TR35 Suppresses Non-small Cell Lung Cancer Xenograft Tumor Growth in vivo
To evaluate the in vivo effects of TR35 on NSCLC cell growth and proliferation, a xenograft mouse model was established through the subcutaneous injection of A549 and H1299 cells into SPF nude mice. Two treated groups and two control groups were treated with TR35 or a corresponding volume of H2O (mock), respectively. Subcutaneous tumor formation was observed by naked eye in all nude mice 7–10 days after injection. Thirty days later, tumors were harvested and their final weights taken to assess TR35 anti-tumor effects in vivo. This analysis found that the size and weight of tumors from TR35-treated mice were significantly lower than control tumors. However, mouse weight did not differ significantly between the groups (Figures 4A–F).
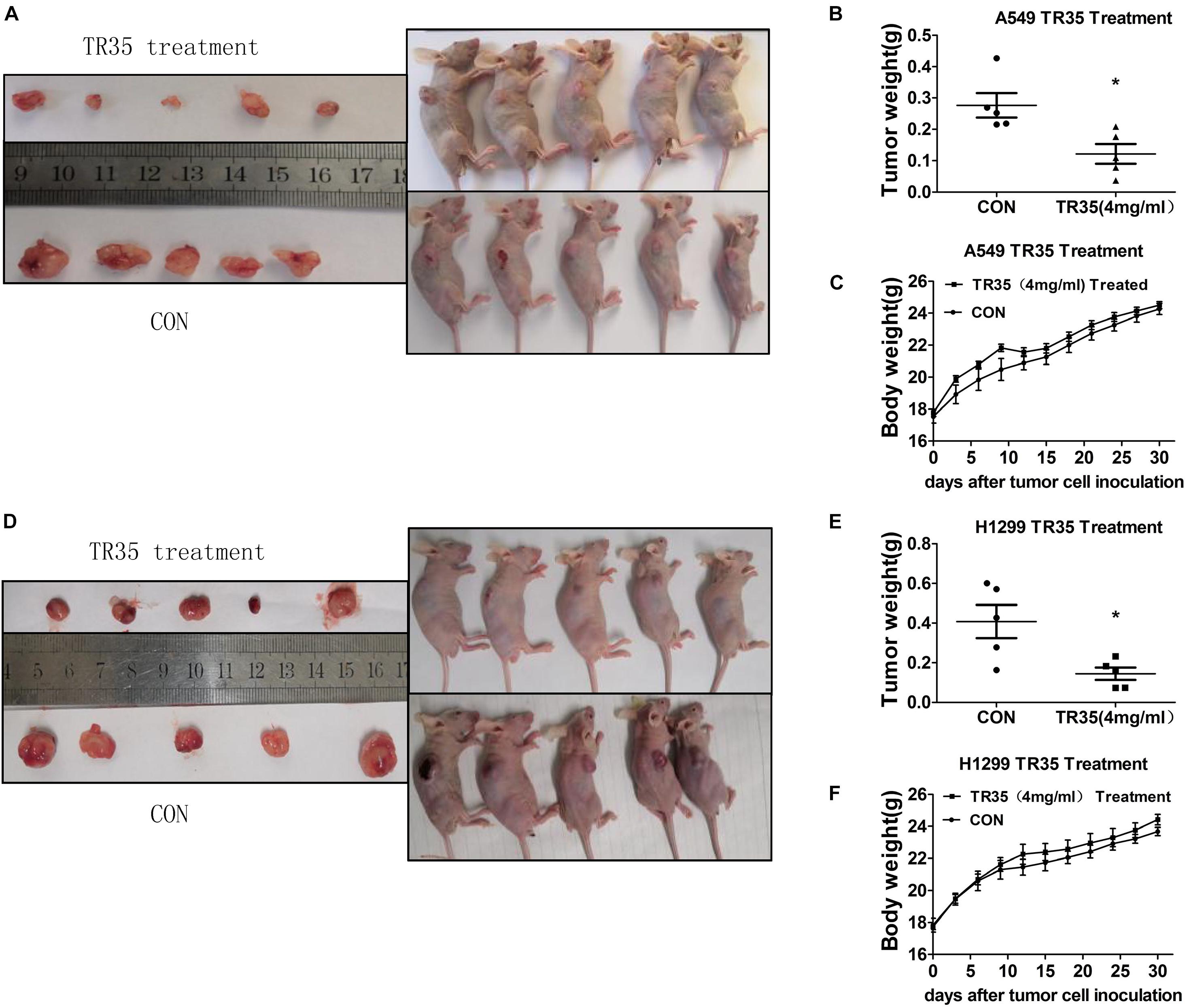
Figure 4. The inhibitory effect of TR35 intragastric feeding on the growth of lung cancer xenograft in nude mice. Equal amounts of A549 and H1299 cells were inoculated under the skin of the back of nude mice. (A,D) After TR35 treatment, the animal images of unresectable tumors and tumor images of resected tumors were obtained in animals injected with different cell lines. (B,E) The comparison of tumor weights of two groups in animals injected with different cell lines. (C,F) Animal weight was recorded once every 3 days in animals injected with different cell lines. Data are presented as the mean ± SEM, n = 5 (*p < 0.05 vs. CON).
Immunohistochemical analysis revealed that relative to controls, TR35-treated tumors had significantly higher p-JNK levels and significantly lower levels of p-p38 and p-STAT3 (Figures 5A,B). Similar results were obtained by western blotting (Figure 5C).
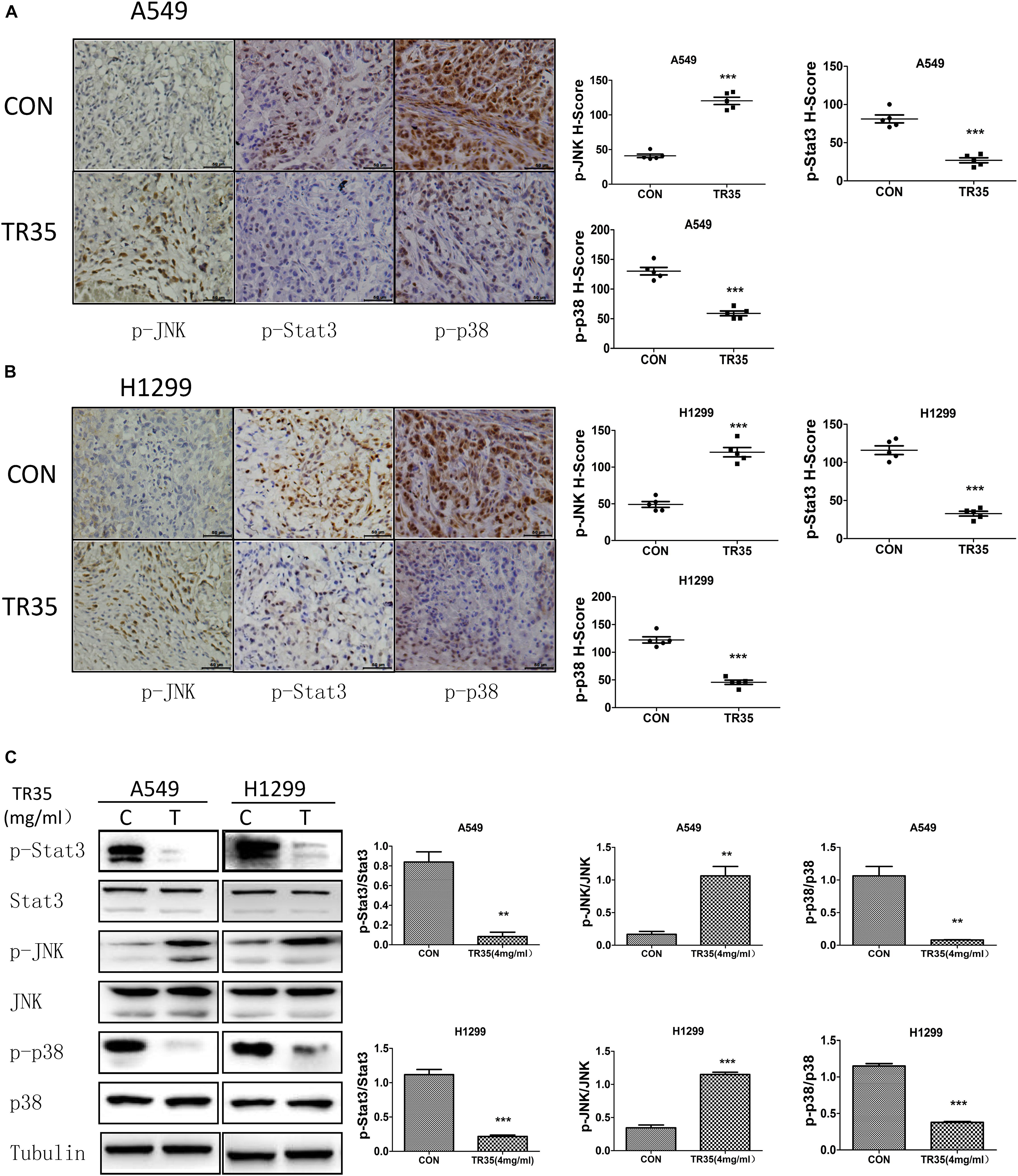
Figure 5. Western blotting and Immunohistochemistry were used to evaluate the effect of TR35 on the expression of p-JNK, p-p38, p-STAT3 protein in dissected tumor tissue. (A,B) The expressions of p-JNK, p-p38, and p-STAT3 in dissected tumor samples formed by different cell lines were measured by IHC under a light microscope. Scale bar 50 μm. (C) STAT3, p38, and JNK steady state and phosphorylated levels were analyzed in dissected tumor samples formed by different cell lines were measured by western blotting (**p < 0.01, ***p < 0.001 vs. CON).
Discussion
Some dietary constituents are known to be chemoprevention and multiple studies have reported dietary adjuvant therapy for cancer, including camel milk. However, few studies have examined molecular mechanisms underlying the anti-tumor effect of camel milk, especially in lung cancer. Here, we show that TR35, an active fraction obtained from Xinjiang Bactrian camel milk, effectively suppresses lung cancer cell growth by inducing apoptosis and G2/M cycle arrest. Mechanistically, we find that TR35 exerts anti-tumor effects by activating p-JNK/MAPK signaling and suppressing STAT3 signaling (including its downstream molecules such as c-Myc and Bcl-XL). However, p-p38 expression was reduced. These observations were suggested by RNA-seq data and confirmed using in vivo and in vitro assays (Figure 6).
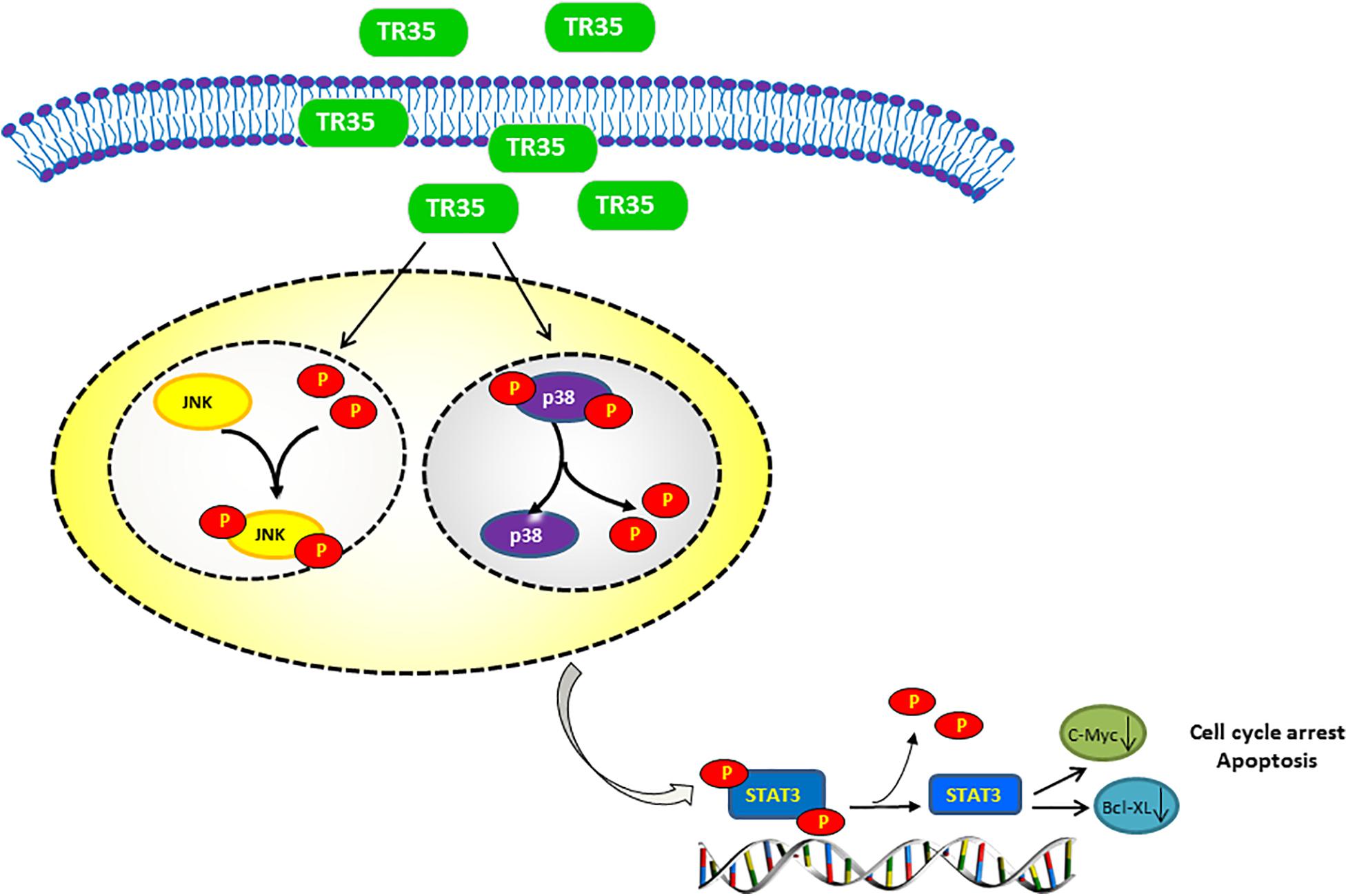
Figure 6. Proposed model of the mechanism underlying the TR35 inhibit NSCLC cell growth and proliferation via MAPK and Jak-STAT signaling pathway. Mechanistically, In TR35 treated NSCLC cells, JNK activation in the JNK-STAT3 signaling axis suppresses STAT3 phosphorylation accompanying the p-p38 downregulation, and thereby suppressing the growth and proliferation of NSCLC cells.
Natural compounds are reported to induce apoptosis via MAPK signaling (Park et al., 2017; Chen and Chen, 2018). Apoptosis is influenced by a variety of intracellular proteins and complex signaling pathways. Mounting evidence implicates STAT3 and MAPK signaling in cancer cell proliferation and apoptosis (Zou et al., 2016; Meng et al., 2018). MAPK signaling pathways fall into 3 main classes: ERK, JNK and p38 (Haagenson and Wu, 2010). ERK regulates cancer cell differentiation, proliferation and apoptosis (Zhai et al., 2016). Toxic and environmental stresses activate JNK, which modulates inflammation by controlling cell differentiation, proliferation, survival, as well as migration (Park et al., 2016). Cell-stress induced signaling activates P38 in response to oxidative stress and toxic chemicals (Tsai et al., 2017). STAT3 activation promotes cell proliferation, angiogenesis, multidrug resistance, and suppresses apoptosis (Miao and Zhang, 2015). Our findings show that TR35 activates JNK signaling, while inhibiting p38 and STAT3 signaling. JNK signaling is associated with the development of numerous disorders, including cancers (Johnson and Nakamura, 2007; Weston and Davis, 2007; Wang et al., 2016). STAT3 is constitutively activated in many cancers and may be oncogenic (Kim et al., 2007). Various studies suggest STAT3 expression is elevated in tumors relative to normal tissues and its long-term activation correlates with development of various cancers (Inoue et al., 2007). Elevated STAT3 function is reported to prevent tumor cell apoptosis, while its inhibition suppresses proliferation and induces apoptosis in cancer cells (Thorburn et al., 2008; Tournier, 2013; Tan et al., 2017). This may be due to many proteins downstream of STAT3, which are crucial for tumor cell proliferation and survival were also downregulated in this process, such as c-Myc and Bcl-XL (Yu et al., 2007). JNK has been suggested as an upstream STAT3 kinase (Sun et al., 2014), and its activation in the JNK-STAT3 signaling axis suppresses STAT3 phosphorylation. JNK activation and STAT3 inhibition are reported to induce M1 macrophage polarization in lung cancer, which may have anti-tumor effects (Cui et al., 2020). These reports are consistent with our data from NSCLC cells and tumor tissues (Figures 3, 5).
Numerous studies have shown that JNK/MAPK and P38/MAPK signaling inactivate STAT3, inducing apoptosis. In this process, p-JNK and p-p38 levels are elevated. However, we observed that TR35 suppressed p-p38 levels in NSCLC cells and tumor tissues. Previous studies have reported the persistent phosphorylation of p38 and STAT3 in NSCLC. The constitutive activation of p38 and STAT3 is related to increase in cell proliferation and metastasis in NSCLC (Greenberg et al., 2002; Dutta et al., 2014; Harada et al., 2014). Greenberg et al. (2002) analyzed tissues from 20 NSCLC cases and found that the activity level of p38 was twice higher than in adjacent tissues, suggesting that p38 modulates malignant growth and transformation of cells. Additionally, Zhou et al. confirmed that reducing p-p38 levels and increasing p-JNK level inhibits cell proliferation (Zhou et al., 2020). These reports are consistent with our findings, indicating that the anti-tumor effects of p-p38 downregulation may occur via other factors, apart from p38-STAT3 signaling axis. However, the specific mechanism is unclear and further research is needed.
Cell cycle homeostasis is important in the maintenance of intracellular stability. However, cell cycle is arrested through various mechanisms, including inhibition of cyclins and expression of CDKs, when cells are damaged (Sarita Rajender et al., 2010; Bonelli et al., 2014; Cheng et al., 2017). Here, flow cytometry revealed that TR35 elevated cell numbers in the G2/M phase and suppressed cell numbers in the G0/G1 phase. Previous studies have showed that the inhibition of STAT3 signaling pathways will lead to tumor-associated G2/M phase arrest. This is because STAT3 can mediate the activity of cyclin B1/CDK protein complex (Shao et al., 2017; Yang L. et al., 2019; Nyiramana et al., 2020).
In summary, we find that TR35 inhibits NSCLC cells growth and proliferation, induces G2/M cell cycle arrest and apoptosis. This suppression process in NSCLC is via MAPK and Jak-STAT signaling. Our data highlight TR35 as a promising candidate for lung cancer therapy.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: European Nucleotide Archive, ERS7253991 and ERS7253992.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Nankai University.
Author Contributions
ZS: writing – original draft and investigation. YG: investigation and writing – review and editing. LF: software and writing – review and editing. WT: validation and writing – review and editing. ZD, CL, JLiu, YX, YW, JYan, QW, JLi, and LY: writing – review and editing. ZZ: conceptualization and writing – review and editing. JYang: resources and writing – review and editing. ZQ: writing – original draft and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 82072187, 81971887, 32071263, 81772252, 31971194, 81801949, 81802091, and 81860339), Key Technology Research and Development Program in Autonomous Region (Grant 2018B01003), Natural Science Foundation of Tianjin (Nos. 20JCYBJC01260, 20JCQNJC01850, 17JCQNJC10700, and 20YDTPJC00250), and Key Laboratory of Emergency and Trauma (Hainan Medical University), Ministry of Education (No. KLET-201906).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agrawal, R. P., Beniwal, R., Kochar, D. K., Tuteja, F. C., Ghorui, S. K., Sahani, M. S., et al. (2005). Camel milk as an adjunct to insulin therapy improves long-term glycemic control and reduction in doses of insulin in patients with type-1 diabetes A 1 year randomized controlled trial. Diabetes Res. Clin. Pract. 68, 176–177. doi: 10.1016/j.diabres.2004.12.007
Badawy, A. A., El-Magd, M. A., and AlSadrah, S. A. (2018). Therapeutic Effect of Camel Milk and Its Exosomes on MCF7 Cells In Vitro and In Vivo. Integr. Cancer Ther. 17, 1235–1246. doi: 10.1177/1534735418786000
Bonelli, P., Tuccillo, F. M., Borrelli, A., Schiattarella, A., and Buonaguro, F. M. (2014). CDK/CCN and CDKI alterations for cancer prognosis and therapeutic predictivity. Biomed Res. Int. 2014:361020. doi: 10.1155/2014/361020
Bousquet Mur, E., Bernardo, S., Papon, L., Mancini, M., Fabbrizio, E., Goussard, M., et al. (2020). Notch inhibition overcomes resistance to tyrosine kinase inhibitors in EGFR-driven lung adenocarcinoma. J. Clin. Invest. 130, 612–624. doi: 10.1172/jci126896
Chen, X., and Chen, J. (2018). miR-3188 Regulates Cell Proliferation, Apoptosis, and Migration in Breast Cancer by Targeting TUSC5 and Regulating the p38 MAPK Signaling Pathway. Oncol. Res. 26, 363–372. doi: 10.3727/096504017X14953948675421
Cheng, C. W., Leong, K. W., Ng, Y. M., Kwong, Y. L., and Tse, E. (2017). The peptidyl-prolyl isomerase PIN1 relieves cyclin-dependent kinase 2 (CDK2) inhibition by the CDK inhibitor p27. J. Biol. Chem. 292, 21431–21441. doi: 10.1074/jbc.M117.801373
Cui, L., Yang, G., Ye, J., Yao, Y., Lu, G., Chen, J., et al. (2020). Dioscin elicits anti-tumour immunity by inhibiting macrophage M2 polarization via JNK and STAT3 pathways in lung cancer. J. Cell. Mol. Med. 24, 9217–9230. doi: 10.1111/jcmm.15563
Dutta, P., Sabri, N., Li, J., and Li, W. X. (2014). Role of STAT3 in lung cancer. JAKSTAT 3:e999503. doi: 10.1080/21623996.2014.999503
El Miniawy, H. M. F., Ahmed, K. A., Tony, M. A., Mansour, S. A., and Salah Khattab, M. M. (2014). Camel milk inhibits murine hepatic carcinogenesis, initiated by diethylnitrosamine and promoted by phenobarbitone. Int. J. Vet. Sci. Med. 2, 136–141. doi: 10.1016/j.ijvsm.2014.10.004
Greenberg, A. K., Basu, S., Hu, J., Yie, T. A., Tchou-Wong, K. M., Rom, W. N., et al. (2002). Selective p38 activation in human non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 26, 558–564. doi: 10.1165/ajrcmb.26.5.4689
Haagenson, K. K., and Wu, G. S. (2010). Mitogen activated protein kinase phosphatases and cancer. Cancer Biol. Ther. 9, 337–340. doi: 10.4161/cbt.9.5.11217
Harada, D., Takigawa, N., and Kiura, K. (2014). The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 6, 708–722. doi: 10.3390/cancers6020708
Inoue, J., Gohda, J., Akiyama, T., and Semba, K. (2007). NF-kappaB activation in development and progression of cancer. Cancer Sci. 98, 268–274. doi: 10.1111/j.1349-7006.2007.00389.x
Johnson, G. L., and Nakamura, K. (2007). The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim. Biophys. Acta 1773, 1341–1348. doi: 10.1016/j.bbamcr.2006.12.009
Kamal, H., Jafar, S., Mudgil, P., Murali, C., Amin, A., and Maqsood, S. (2018). Inhibitory properties of camel whey protein hydrolysates toward liver cancer cells, dipeptidyl peptidase-IV, and inflammation. J. Dairy Sci. 101, 8711–8720. doi: 10.3168/jds.2018-14586
Kim, H. J., Hwang, N. R., and Lee, K. J. (2007). Heat shock responses for understanding diseases of protein denaturation. Mol. Cells 23, 123–131.
Laudisi, F., Cherubini, F., Monteleone, G., and Stolfi, C. (2018). STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 19:1787. doi: 10.3390/ijms19061787
Li, Z., Zhang, L., Gao, M., Han, M., Liu, K., Zhang, Z., et al. (2020). Retraction Note: endoplasmic reticulum stress triggers Xanthoangelol-induced protective autophagy via activation of JNK/c-Jun Axis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 39:164. doi: 10.1186/s13046-020-01668-x
Meng, L. Q., Liu, C., Luo, Y. H., Piao, X. J., Wang, Y., Zhang, Y., et al. (2018). Quinalizarin exerts an anti-tumour effect on lung cancer A549 cells by modulating the Akt, MAPK, STAT3 and p53 signalling pathways. Mol. Med. Rep. 17, 2626–2634. doi: 10.3892/mmr.2017.8110
Miao, D., and Zhang, L. (2015). Leptin modulates the expression of catabolic genes in rat nucleus pulposus cells through the mitogen-activated protein kinase and Janus kinase 2/signal transducer and activator of transcription 3 pathways. Mol. Med. Rep. 12, 1761–1768. doi: 10.3892/mmr.2015.3646
Miller, K. D., Siegel, R. L., Lin, C. C., Mariotto, A. B., Kramer, J. L., Rowland, J. H., et al. (2016). Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 66, 271–289. doi: 10.3322/caac.21349
No authors listed. (2020). Erratum: global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 70:313. doi: 10.3322/caac.21609
Nyiramana, M. M., Cho, S. B., Kim, E. J., Kim, M. J., Ryu, J. H., Nam, H. J., et al. (2020). Sea Hare Hydrolysate-Induced Reduction of Human Non-Small Cell Lung Cancer Cell Growth through Regulation of Macrophage Polarization and Non-Apoptotic Regulated Cell Death Pathways. Cancers 12:726. doi: 10.3390/cancers12030726
Park, K. R., Yun, H. M., Quang, T. H., Oh, H., Lee, D. S., Auh, Q. S., et al. (2016). 4-Methoxydalbergione suppresses growth and induces apoptosis in human osteosarcoma cells in vitro and in vivo xenograft model through down-regulation of the JAK2/STAT3 pathway. Oncotarget 7, 6960–6971. doi: 10.18632/oncotarget.6873
Park, S. G., Kim, S. H., Kim, K. Y., Yu, S. N., Choi, H. D., Kim, Y. W., et al. (2017). Toyocamycin induces apoptosis via the crosstalk between reactive oxygen species and p38/ERK MAPKs signaling pathway in human prostate cancer PC-3 cells. Pharmacol. Rep. 69, 90–96. doi: 10.1016/j.pharep.2016.10.014
Sarita Rajender, P., Ramasree, D., Bhargavi, K., Vasavi, M., and Uma, V. (2010). Selective inhibition of proteins regulating CDK/cyclin complexes: strategy against cancer–a review. J. Recept. Signal Transduct. Res. 30, 206–213. doi: 10.3109/10799893.2010.488649
Shabo, Y., Barzel, R., Margoulis, M., and Yagil, R. (2005). Camel milk for food allergies in children. Isr. Med. Assoc. J. 7, 796–798.
Shabo, Y., and Yagil, R. (2005). Etiology of autism and camel milk as therapy. Int. J. Disabil. Hum. Dev. 4, 67–70. doi: 10.1515/IJDHD.2005.4.2.67
Shao, D., Ma, J., Zhou, C., Zhao, J. N., Li, L. L., Zhao, T. J., et al. (2017). STAT3 down-regulation induces mitochondria-dependent G2/M cell cycle arrest and apoptosis in oesophageal carcinoma cells. Clin. Exp. Pharmacol. Physiol. 44, 413–420. doi: 10.1111/1440-1681.12708
Subramaniam, A., Shanmugam, M. K., Perumal, E., Li, F., Nachiyappan, A., Dai, X., et al. (2013). Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim. Biophys. Acta 1835, 46–60. doi: 10.1016/j.bbcan.2012.10.002
Sui, C., Zhuang, C., Sun, D., Yang, L., Zhang, L., and Song, L. (2017). Notch1 regulates the JNK signaling pathway and increases apoptosis in hepatocellular carcinoma. Oncotarget 8, 45837–45847. doi: 10.18632/oncotarget.17434
Sun, J., Yu, M., Lu, Y., Thakur, C., Chen, B., Qiu, P., et al. (2014). Carcinogenic metalloid arsenic induces expression of mdig oncogene through JNK and STAT3 activation. Cancer Lett. 346, 257–263. doi: 10.1016/j.canlet.2014.01.002
Tan, W., Zhu, S., Cao, J., Zhang, L., Li, W., Liu, K., et al. (2017). Inhibition of MMP-2 Expression Enhances the Antitumor Effect of Sorafenib in Hepatocellular Carcinoma by Suppressing the PI3K/AKT/mTOR Pathway. Oncol. Res. 25, 1543–1553. doi: 10.3727/096504017X14886444100783
Thorburn, A., Behbakht, K., and Ford, H. (2008). TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist. Updat. 11, 17–24. doi: 10.1016/j.drup.2008.02.001
Tian, W., Yang, L., Liu, Y., He, J., Yang, L., Zhang, Q., et al. (2020). Resveratrol attenuates doxorubicin-induced cardiotoxicity in rats by up-regulation of vascular endothelial growth factor B. J. Nutr. Biochem. 79:108132. doi: 10.1016/j.jnutbio.2019.01.018
Tournier, C. (2013). The 2 Faces of JNK Signaling in Cancer. Genes Cancer 4, 397–400. doi: 10.1177/1947601913486349
Tsai, W. C., Bai, L. Y., Chen, Y. J., Chu, P. C., Hsu, Y. W., Sargeant, A. M., et al. (2017). OSU-A9 inhibits pancreatic cancer cell lines by modulating p38-JAK-STAT3 signaling. Oncotarget 8, 29233–29246. doi: 10.18632/oncotarget.16450
Wang, L., Luo, J. Y., Li, B., Tian, X. Y., Chen, L. J., Huang, Y., et al. (2016). Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582. doi: 10.1038/nature20602
Weston, C. R., and Davis, R. J. (2007). The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149. doi: 10.1016/j.ceb.2007.02.001
Yang, J., Dou, Z., Peng, X., Wang, H., Shen, T., Liu, J., et al. (2019). Transcriptomics and proteomics analyses of anti-cancer mechanisms of TR35-An active fraction from Xinjiang Bactrian camel milk in esophageal carcinoma cell. Clin. Nutr. 38, 2349–2359. doi: 10.1016/j.clnu.2018.10.013
Yang, L., Li, J., Xu, L., Lin, S., Xiang, Y., Dai, X., et al. (2019). Rhein shows potent efficacy against non-small-cell lung cancer through inhibiting the STAT3 pathway. Cancer Manag. Res. 11, 1167–1176. doi: 10.2147/CMAR.S171517
Yu, H., Kortylewski, M., and Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51.
Zhai, H., Hu, S., Liu, T., Wang, F., Wang, X., Wu, G., et al. (2016). Nitidine chloride inhibits proliferation and induces apoptosis in colorectal cancer cells by suppressing the ERK signaling pathway. Mol. Med. Rep. 13, 2536–2542. doi: 10.3892/mmr.2016.4827
Zhou, H., Liu, J., and Chen, Z. (2020). Coronarin D suppresses proliferation, invasion and migration of glioma cells via activating JNK signaling pathway. Pathol. Res. Pract. 216:152789. doi: 10.1016/j.prp.2019.152789
Keywords: anti-tumor, camel milk, lung cancer, microenvironment, MAPK, STAT3
Citation: Shi Z, Gao Y, Feng L, Tian W, Dou Z, Liu C, Liu J, Xu Y, Wang Y, Yan J, Wu Q, Li J, Yang L, Zhang Z, Yang J and Qi Z (2021) TR35 Exerts Anti-tumor Effects by Modulating Mitogen-Activated Protein Kinase and STAT3 Signaling in Lung Cancer Cells. Front. Cell Dev. Biol. 9:723346. doi: 10.3389/fcell.2021.723346
Received: 10 June 2021; Accepted: 05 October 2021;
Published: 25 October 2021.
Edited by:
Hongming Miao, Army Medical University, ChinaReviewed by:
Zhongxiao Wan, Soochow University, ChinaXiaolei Zhang, Sun Yat-sen University, China
Lihui Wang, Shenyang Pharmaceutical University, China
Copyright © 2021 Shi, Gao, Feng, Tian, Dou, Liu, Liu, Xu, Wang, Yan, Wu, Li, Yang, Zhang, Yang and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaocai Zhang, 2313003@zju.edu.cn; Jie Yang, yangjie234@xju.edu.cn; Zhi Qi, qizhi@nankai.edu.cn
†These authors have contributed equally to this work