Toluene Dioxygenase-Catalyzed cis-Dihydroxylation of Quinolines: A Molecular Docking Study and Chemoenzymatic Synthesis of Quinoline Arene Oxides
 Derek R. Boyd1* Narain D. Sharma1 Pui L. Loke1 Jonathan G. Carroll1 Paul J. Stevenson1
Derek R. Boyd1* Narain D. Sharma1 Pui L. Loke1 Jonathan G. Carroll1 Paul J. Stevenson1  Patrick Hoering2
Patrick Hoering2  Christopher C. R. Allen2*
Christopher C. R. Allen2*- 1School of Chemistry and Chemical Engineering, Queen's University of Belfast, Belfast, United Kingdom
- 2School of Biological Sciences, Queen's University of Belfast, Belfast, United Kingdom
Molecular docking studies of quinoline and 2-chloroquinoline substrates at the active site of toluene dioxygenase (TDO), were conducted using Autodock Vina, to identify novel edge-to-face interactions and to rationalize the observed stereoselective cis-dihydroxylation of carbocyclic rings and formation of isolable cis-dihydrodiol metabolites. These in silico docking results of quinoline and pyridine substrates, with TDO, also provided support for the postulated cis-dihydroxylation of electron-deficient pyridyl rings, to give transient cis-dihydrodiol intermediates and the derived hydroxyquinolines. 2-Chloroquinoline cis-dihydrodiol metabolites were used as precursors in the chemoenzymatic synthesis of enantiopure arene oxide and arene dioxide derivatives of quinoline, in the context of its possible mammalian metabolism and carcinogenicity.
Introduction
Quinoline and substituted quinolines are widely distributed in the environment as urban particulates, resulting from partial combustion of fossil fuels and tobacco. Quinoline 1 is a mammalian hepatocarcinogen and a bacterial mutagen, with its metabolites binding covalently to DNA. Possible pathways responsible for these biological effects continue to be of interest (Hollstein et al., 1978; Tada et al., 1980, 1982; LaVoie et al., 1983; Agarwal et al., 1986, 1990; Willems et al., 1992; Saeki et al., 1993; Reigh et al., 1996; Suzuki et al., 2000; Dowers et al., 2004; Hakura et al., 2007; Diaz Duran et al., 2015; Matsumoto et al., 2018). Cytochrome P-450 (CYP-450) monooxygenases have been identified as responsible for catalyzing epoxidation and dearomatization of quinolines, during mammalian liver metabolism.
Bacterial cell metabolism can also involve an initial dearomatization step of quinolines, via arene dioxygenase-catalyzed cis-dihydroxylation, resulting in the isolation of stable, and postulation of transient, azaarene metabolites. This is exemplified by the metabolism of quinoline 1 (X = H), and substituted quinolines, which were studied earlier using different bacterial strains and arene dioxygenase enzyme types (Boyd et al., 1987, 1993, 1998, 2002; Bott et al., 1990; Fetzner et al., 1993; Kaiser et al., 1996; Zia et al., 2016).
Toluene dioxygenase (TDO), naphthalene dioxygenase (NDO), and biphenyl dioxygenase (BPDO) were among those bacterial enzymes found to catalyze the formation of cis-dihydrodiol metabolites and monohydroxylated derivatives of quinoline substrates (Boyd et al., 2002). The UV4 mutant strain of Pseudomonas putida, expressing TDO, was used in the biotransformation of quinoline 1 (X = H) to give cis-dihydrodiols 2 (33% relative yield) and the less stable cis-dihydrodiol 3 (1% relative yield, Figure 1) (Boyd et al., 1987, 1993). As the latter metabolite readily decomposed to 8-hydroxyquinoline 4 (27% relative yield), similar proportions of the cis-diols 2 and 3 were assumed to be initially formed from dihydroxylation of the carbocyclic ring of quinoline 1.
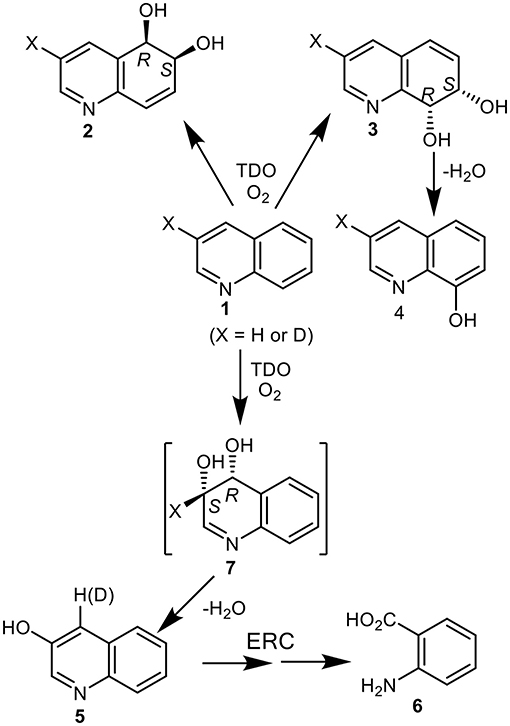
Figure 1. Isolated metabolites, 2–6, and the postulated transient cis-dihydrodiol intermediate 7, resulting from TDO-catalyzed oxidation of quinoline 1 (X = H or D).
No direct evidence for cis-dihydroxylation of the electron-poor pyridyl ring was found during analysis of the biotransformation products (Boyd et al., 1987, 1993). The combined relative yields of the isolated achiral metabolites 3-hydroxyquinoline 5 (13%) and anthranilic acid 6 (27%), were however consistent with the initial formation of the transient cis-dihydrodiol 7 as a further major initial metabolite (Figure 1). Formation of 3-hydroxyquinoline 5 could result from spontaneous dehydration of intermediate 7 and of anthranilic acid 6 from enzymatic ring-cleavage (ERC) of 3-hydroxyquinoline 5. If anthranilic acid 6 and phenol 5 were derived from intermediate cis-dihydrodiol 7, the relative ratio of initially formed cis-dihydrodiols diols 2, 3, and 7 would be estimated as ca. 33:27:40. Further in silico support for the formation of transient cis-dihydrodiol 7 - the major metabolite of quinoline 1 (TDO as biocatalyst), was obtained from a joint study with a collaborating laboratory using the GOLD molecular docking program (unpublished data).This prompted our interest in employing the Autodock Vina program for the current study.
Indirect evidence for the undetected heterocyclic cis-dihydrodiol 7 was acquired from the results of an earlier biotransformation (P. putida UV4) of 3-deuterioquinoline 1 (X = 87% D); it yielded 4-deuterioquinolin-3-ol 5 (23% D) along with other metabolites (Figure 1) (Barr et al., 1998). This could be accounted for by the migration and partial retention of deuterium, from the aromatization of the intermediate cis-dihydrodiol 7 (X = D, Figure 1) via an NIH shift mechanism, as observed during aromatization of the isolated carbocyclic cis-dihydrodiols of naphthalene and quinoline 2 and 3.
Pseudomonas strains are among the most common quinoline-degrading bacteria. Monohydroxylation at the C-2 position of quinoline often occurs during Pseudomonad biotransformations, to yield 2-hydroxyquinoline, which prefers to exist as the 2-quinolone tautomer (Kaiser et al., 1996). Substitution at the C-2 position could thus, in principle, improve the isolated yields of cis-dihydrodiol metabolites resulting from dioxygenase-catalyzed biotransformations of quinoline substrates. It may be a factor in: (i) the excellent isolated yield (80%) of the corresponding 7,8-cis-dihydrodiol obtained from NDO-catalyzed biotransformation of 2-cyanoquinoline (Zia et al., 2016) and (ii) the improved isolated yields (35–45%) of cis-dihydrodiol metabolites of 2-chloroquinoline (with TDO) and 2-methoxyquinoline (with BPDO) (Boyd et al., 2002), relative to the very low isolated yields of cis-dihydrodiols 2 and 3 obtained from quinoline 1 (X = H, <5% using TDO, Figure 1) (Boyd et al., 1993).
Biotransformations of 2-chloroquinoline 8, conducted using P. putida UV4 whole cells resulted in the isolation of multigram quantities of cis-diol metabolites 9, 10, and 11, possibly via intermediate 12, and traces of 2-quinolone 13 (Figure 2) (Boyd et al., 1998, 2002). Since the enantiopure cis-dihydrodiols 9, 10, and 11 are more stable, and are available in much higher yields compared with quinoline cis-dihydrodiols 2 and 3, they are herein used in the chemoenzymatic synthesis of enantiopure mammalian metabolites of quinoline 1.
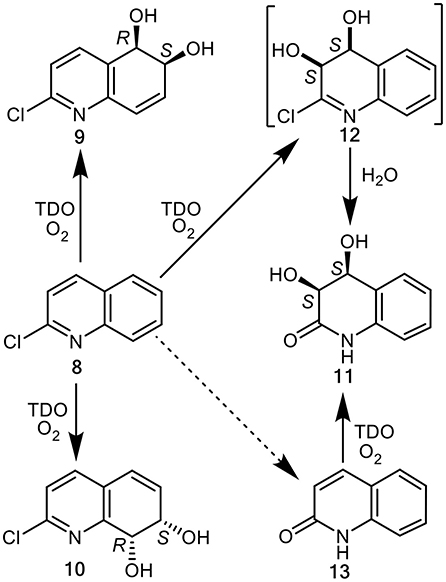
Figure 2. TDO-catalyzed cis-dihydroxylation of 2-chloroquinoline 8 and 2-quinolone 13 to yield isolated metabolites 9–11 and postulated transient intermediate 12.
While the dioxygenase-catalyzed cis-dihydroxylation of electron-rich furan and thiophene rings has been reported, to give heterocyclic dihydrodiol metabolites (Boyd et al., 2012; Lewis, 2016), little evidence is available for the dearomatization of electron-poor pyridine rings by a similar mechanism. The possibility that TDO-catalyzed cis-dihydroxylation of a pyridyl ring was proposed as a result of P. putida UV4 biotransformations of: (i) quinoline 1, to yield 3-hydroxyquinoline 5 via unstable intermediate 7 (Figure 1) (Boyd et al., 1987, 1993), (ii) 2-chloroquinoline 8, to yield cis-diol 11 via transient intermediate 12 (Figure 2) (Boyd et al., 1998, 2002), and (iii) 2-chloropyridine 14, to yield 2-chloro-3-hydroxypyridine 16 via undetected intermediate 15 (Figure 3) (Garrett et al., 2006). Since none of the cis-dihydrodiols 7, 12, and 15 could be detected directly, evidence for their intermediacy is sought through the current in silico molecular docking studies of TDO with the corresponding substrates, quinoline 1, 2-chloroquinoline 8 and 2-chloropyridine 14 (Results section).
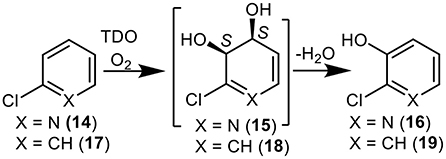
Figure 3. TDO-catalyzed oxidation of 2-chloropyridine 14 and chlorobenzene 17 to yield isolated metabolites 16, 18, and 19 and undetected cis-dihydrodiol intermediate 15.
Major objectives of this study were to: (i) review proposed metabolic pathways of quinoline 1 and 2-chloroquinoline 8 by P. putida UV 4 cells, and to compare with TDO docking results (ii) use the isolated cis-dihydrodiol metabolites 9 and 10 and their derivatives, in the quest for improved chemoenzymatic synthetic routes to enantiopure arene oxide and dioxide derivatives of quinoline 1.
Methods
Laboratory Studies
1H and 13C NMR spectra were recorded on Bruker Avance DPX-300 and DPX-500 instruments. Chemical shifts (δ) are reported in ppm relative to SiMe4 and coupling constants (J) are given in Hertz (Hz). Mass spectra were run at 70 eV, on an AE1-MS90 mass spectrometer updated by VG Autospec, using a heated inlet system. Accurate molecular weights were determined by the peak matching method, with perfluorokerosene as the standard. Elemental microanalyses were carried out on a Perkin-Elmer 2400 CHN microanalyser and IR spectra were recorded in KBr disc or in thin film, using a Perkin-Elmer Spectrum RX1 FT-IR spectrometer. ECD spectra were obtained using a Jasco J-720 instrument and MeCN as solvent. Optical rotations ([α]D) measurements (10−1 deg cm2 g−1) were carried out at ambient temperature on a Perkin-Elmer 214 polarimeter and specified solvent concentration (g/100 ml) at sodium D-line (589 nm). Melting points were recorded in degrees Celsius using a Stuart SMP10 melting point apparatus. Column chromatography and preparative layer chromatography (PLC) were performed on Merck Kieselgel type 60 (250–400 mesh) and PF254/366, respectively. Merck Kieselgel type 60F254 analytical plates were used for TLC.
The structures discussed in this section are found in Figures 8, 9, 11. Compounds 1, 4, 5, 6, 8, 13, 14, 16, 17, 19, 22, 23, 24 were purchased commercially and cis-dihydrodiols (+)-9 and (+)-10, arene oxides (–)-20 and (+)-25, were available from earlier studies (Agarwal et al., 1990; Boyd et al., 1991a, 1994, 2002).
(+)-(5R,6S)-2-Chloro-5,6-dihydroquinoline-5,6-diol 9 (Boyd et al., 1998, 2002)
mp 120–122°C (EtOAc/hexane); [α]D +140 (c 0.40, MeOH); 1H NMR δH (500 MHz, CDCl3) 2.05 (1H, br s, OH), 2.71 (1H, br s, OH), 4.38 (1H, dd, J 5.0, J 5.0, H-6), 4.77 (1H, d, J 5.0, H-5), 6.45 (1H, dd, J 5.0, J 9.9, H-7), 6.68 (1H, d, J 9.9, H-8), 7.21 (1H, d, J 8.0, H-3), 7.85 (1H, d, J 8.0, H-4).
(+)-(7S,8R)-2-Chloro-7,8-dihydroquinoline-7,8-diol 10 (Boyd et al., 1998, 2002)
Colorless crystalline solid, mp 112°C (from EtOAc/hexane); [α]D +148 (c 0.5, MeOH); 1H NMR δH (500 MHz, CDCl3) 2.61 (1H, br s, OH), 4.32 (1H, br s, OH), 4.46 (1H, dd, J 5.0, 5.0, H-7), 4.75 (1H, d, J 5.0, H-8), 6.30 (1H, dd, J 9.6, 5.0, H-6), 6.63 (1H, d, J 9.6, H-5), 7.25 (1H, d, J 8.0, H-3), 7.42 (1H, d, J 8.0, H-4).
(–)-(5R,6S)-5,6-Epoxy-5,6-dihydroquinoline 20 (Agarwal et al., 1990; Boyd et al., 1991a, 1994)
mp 68–71°C (pentane) [lit.rac. (Agarwal et al., 1990) 33–36°C]; [α]D −23 (c 0.9, CHCl3); 1H NMR δH (300 MHz, CDCl3) 4.16 (1H, m, H-6), 4.49 (1H, d, J 3.7, H-5), 6.73 (1H, dd, J 9.9, 3.8, H-7), 6.98 (1H, d, J 9.9, H-8), 7.24 (1H, m, H-3), 7.90 (1H, dd, J 7.6.0, 1.4 H-4), 8.62 1H, dd, J 4.9.6.0, 1.4 H-2).
(+)-(7S,8R)-7,8-Epoxy-7,8-dihydroquinoline 25 (Agarwal et al., 1990; Boyd et al., 1994)
mp 54–56°C (pentane) (lit.rac (Agarwal et al., 1990) 44–46°C); [α]D +55 (c 1.1, CHCl3); 1H NMR δH (300 MHz, CDCl3) 4.19 (1H, m, H-7), 4.66 (1H, d, J 3.7, H-8), 6.51 (1H, dd, J 9.6, 3.7, H-6), 6.72 (1H, dd, J 9.6,1.7 H-5), 7.31 (1H, dd, J 7.7, 4.8, H-3), 7.58 (1H, dd, J 7.7.0, 1.3, H-4), 8.53 1H, dd, J 4.8, 1.4, H-2).
Hydrogenation/Hydrogenolysis of cis-dihydrodiols (+)-9 and (+)-10 to Yield Tetrahydrodiols (–)-39 and (–)-40
(–)-(5R,6S)-5,6,7,8-Tetrahydroquinoline-5,6-diol 39 (Boyd et al., 1993)
cis-Dihydrodiol metabolite 9 (30 mg, 0.15 mmol) was dissolved in MeOH (8 cm3) and 10% Pd/C (0.5 mg) was added. The mixture was stirred under a H2 atmosphere at atmospheric pressure for 2 h, filtered and concentrated under reduced pressure. Aqueous Na2CO3 solution (5%) was added to the residue, the mixture saturated with NaCl and then extracted into EtOAc (3 × 10 cm3). The combined organic layers were dried (Na2SO4) and the solvent evaporated to furnish (5R,6S)-diol 39 (19 mg, 81%); mp 139–140°C (EtOAc) [lit. (Boyd et al., 1993) 138–140°C]; [α]D −7.0 (c 0.5, MeOH); 1H NMR (300 MHz, CDCl3) δH 1.96 (1H, m, H-7), 2.16 (1H, m, H-7′), 2.85 (1H, m, H-8), 3.09 (1H, m, H-8′), 4.1 (1H, m, H-6), 4.35 (2H, bs, 2 × OH) 4.67 (1H, d, J 3.4, H-5), 7.13 (1H, dd, J 4.8, 7.7, H-3), 7.79 (1H, dd, J 1.1, 7.2, H-4), 8.35 (1H, dd, J 1.5, 4.7, H-2).
(–)-(7S,8R)-5,6,7,8-Tetrahydroquinoline-7,8-diol 40 (Boyd et al., 1993)
Similar treatment of cis-dihydrodiol 10 (30 mg, 0.15 mmol) yielded (7S,8R)-diol 40 (21 mg, 81%), mp 137–139°C (EtOAc) [lit. (Boyd et al., 1993) 138–140°C]; [α]D −72.0 (c 0.47, MeOH); 1H NMR (300 MHz, CDCl3) δH 1.95 (1H, m, H-6), 2.22 (1H, m, H-6′), 2.75 (1H, m, H-5), 3.20 (1H, m, H-5′), 4.32 (1H, m, H-7), 4.92 (1H, d, J H-8), 7.17 (1H, dd, J 4.7, J 7.7, H-3), 7.46 (1H, d, J 7.7, H-4), 8.40 (1H, J 4.7, H-2).
Synthesis of (–)-(5R,6R,7R,8R)-quinoline-5,6:7,8-dioxide 29 (Boyd et al., 1991b)
Treatment of (–)-(5R,6S)-5,6-epoxy-5,6-dihydroquinoline 20 (120 mg, 0.8 mmol) in CH2Cl2 with aqueous sodium hypochlorite in the presence of phase transfer reagent (t-Bu4NHSO4) and workup, under identical conditions used earlier in the synthesis of racemic anti-quinoline dioxide 29, produced (–)-29 (51mg, 38%). White crystalline solid (ether-hexane); mp 85–86°C [lit.rac. (Boyd et al., 1991b), 44–46°C]; [α]D −100 (c 0.11, CHCl3); HRMS calcd for C9H7NO2, 161.0477, M+; found, 161.0472; 1H NMR (300 MHz, CDCl3) δH 3.73 (1 H, d, J 3.9, H-6b), 3.91 (1 H, d, J 4.0, H-2a), 4.05 (1 H, m, H-1a), 4.08 (1 H, m, H-1b), 7.28 (1 H, dd, J 7.7, 4.8, H-5), 7.73 (1 H, dd, J 7.5, 1.5, H-6), 8.53 (1 H, dd, J 4.8, 1.5, H-4); 13C NMR (125 MHz, CDCl3) δC 50.9, 53.2, 54.7, 55.5, 123.9, 127.7, 138.7, 149.8, 152.4; m/z 161 (M+, 15%), 132 (100). Electronic CD data: 269 nm (Δε −3.299), 226 nm (Δε −14.97), 197 nm (Δε 11.46).
Synthesis of (+)-(5S,6S,7R,8R)-quinoline-5,6:7,8-dioxide 28 (Boyd et al., 1991b)
(+)-(7S,8R)-7,8-Epoxy-7,8-dihydroquinoline 25 (120 mg, 0.8 mmol), it was reacted with N-bromoacetamide (130 mg, 1.0 mmol) in THF: water (2:1), under identical reaction conditions reported for a racemic arene oxide 25 (Vila et al., 2017). After extraction (CH2Cl2) drying and concentration, the crude product, the correct NMR spectrum for racemic 6-bromo-5-hydroxy-7,8-epoxy-5,6,7,8-tetrahydroquinoline 41, was then treated directly with sodium methoxide in THF to yield (+)-(5S,6S,7R,8R)-quinoline-5,6:7,8-dioxide 28. White solid mp 153–156°C [lit.rac. (Boyd et al., 1991b) 138–152°C]; [α]D +25 (c 0.2, CHCl3); HRMS calcd for C9H7NO2, 161.0477, M+; found, 161.0472; 1H NMR (500 MHz, CDCl3) δH 3.95–3.99 (2 H, m, H-1a and H-1b), 4.05 (1 H, d, J 3.5, H-6b), 4.18 (1 H, d, J 3.4, H-2a), 7.38 (1 H, m, H-5), 7.95 (1 H, dd, J 7.7, 1.6, H-6), 8.65 (1 H, dd, J 4.9, 1.6,H-4); m/z 161 (M+, 15%), 132 (100). Electronic CD data: 274 nm (Δε 15.93), 268 nm (Δε 14.31), 246 nm (Δε −3.425), 192 nm (Δε −16.08).
Cis-dihydroxylation of (+)-cis-diol 9 [and 10] and Acetylation to Yield Tetrahydroacetates, (+)-44, (–)-44 [(+)-42 and (+)-45]
To a stirred solution of (+)-cis-dihydrodiol 9 (200 mg, 1.02 mmol) and trimethylamine-N-oxide dihydrate (150 mg, 1.34 mmol) in CH2Cl2 (50 ml) was added a catalytic amount of OsO4 and the mixture stirred overnight at room temperature. After the addition of a 10% solution of sodium metabisulfite (2 ml), the reaction mixture was allowed to stir for another 0.5 h and then concentrated under reduced pressure to give a crude mixture of inseparable syn- and anti-tetraols. The mixture was acetylated by heating (75–80°C), overnight, with excess of acetic anhydride (ca. 2 ml) in pyridine (2 ml) solution. Concentration of the reaction mixture in vacuo, yielded a crude mixture (3:1) of anti-45 and syn-44 tetraacetates. After initial purification of the mixture, using flash chromatography (silica gel, 3% MeOH in CHCl3), followed by multiple elution PLC (20% EtOAc in hexane) separation gave major anti-tetraacetate (+)-45 and minor syn-tetraacetate (–)-44.
(+)-(5R,6R,7R,8R)-5,6,7,8-Tetraacetoxy-2-chloro-5,6,7,8-tetrahydroquinoline 45
White crystalline solid (121 mg, 30%); (Rf 0.3, 30% EtOAc in hexane); mp 87–88°C (from CHCl3); [α]D +14.6 (c 0.83, CHCl3); (Found: C, 50.6; H, 4.7; N, 3.3. C17H18ClNO8 requires C, 51.1; H, 4.5; N, 3.5%); 1H NMR (500 MHz, CDCl3) δH 2.07 (3H, s, OMe), 2.08 (1H, s, OMe), 2.11 (1H, s, OMe), 2.13 (1H, s, OMe), 5.63 (1 H, dd, J 10.1, 3.9, H-6), 5.73 (1 H, dd, J 10.1, J 4.1, H-7), 6.34 (1 H, d, J 3.9, H-5), 6.39 (1 H, d, J 4.1, H-8), 7.35 (1 H, d, J 8.3, H-3), 7.63 (1 H, d, J 8.3, H-4); 13C NMR (CDCl3, 125 MHz) δC 20.5, 20.6, 20.7, 20.8, 66.7, 67.1, 67.3, 67.6, 124.9, 127.1, 139.1, 151.5, 152.3, 169.7, 169.8, 169.9, 170.2, m/z 339 (M+, 15%, 35Cl), 43 (100); νmax/cm−1: 1,741 (C=O).
(–)-(5R,6R,7S,8S)-5,6,7,8-Tetraacetoxy-2-chloro-5,6,7,8-tetrahydroquinoline 44
White crystalline solid (53 mg, 13%); (Rf 0.25, 30% EtOAc-hexane); mp 177–178°C (CHCl3); [α]D −20 (c 0.69, CHCl3); (Found: C, 50.8; H, 4.3; N, 3.6. C17H18ClNO8 requires C, 51.1; H, 4.5; N, 3.5%); HRMS calcd for C17H18ClNO8, 399.0721, M+; found, 399.0727; 1H NMR (500 MHz, CDCl3) δH 2.07–2.17 (12 H, s, 4 × COCH3), 5.58 (1 H, dd, J 4.5, 2.2, H-7), 5.62 (1 H, dd, J 2.2, 4.7, H-5), 6.22 (1 H, d, J 4.4, H-8), 7.34 (1 H, d, J 8.2, H-3), 7.61 (1 H, d, J 8.2, H-4); 13C NMR (125 MHz, CDCl3) δC 19.6, 19.7, 19.8, 20.0, 65.6, 66.1, 66.3, 66.8, 124.1, 126.1, 139.3, 150.5, 151.3, 168.7, 168.8, 168.9, 169.1; m/z 399 (M+, 16%, 35Cl), 43 (100); νmax/cm−1: 1,741 (C=O).
(+)-(5S,6S,7R,8R)-5,6,7,8-Tetraacetoxy-2-chloro-5,6,7,8-tetrahydroquinoline 44
Employing a similar osmylation/acetylation procedure, used for metabolite (+)-9, to cis-dihydrodiol (+)-10 gave a mixture (1:3) of the opposite syn-tetraacetate enantiomer, (+)-44, [α]D +19.0 (c 0.74, CHCl3) and the anti-tetraacetate enantiomer (+)-45.
Hydrolysis of Chlorotetraacetates (–)-44, (+)-44, and (+)-45 to Yield Chlorotetrols (–)-42, (+)-42, and (+)-43
(–)-(5R,6R,7S,8S)-2-Chloro-5,6,7,8-tetrahydro-5,6,7,8-quinoline tetraol 42
A solution of tetraacetate (–)-44 (300 mg, 0.75 mmol) in MeOH (30 ml) was saturated with NH3 gas at 0°C and was left overnight in an ice bath. The solvent was evaporated under reduced pressure, and the ammonium acetate side product was removed by sublimation under high vacuum. Crystallization of the residue from a mixture of acetone-MeOH-CHCl3 gave pure tetrol (–)-42 (130 mg, 75%), white needles; mp 153–154°C; [α]D −8 (c 0.68, MeOH); (Found: C, 45.7; H, 4.3; N, 5.8. C9H10NO4Cl requires C, 46.7; H, 4.3; N, 6.1%); 1H NMR (500 MHz, D2O) δH 4.17 (1 H, m, J 2.1, 4.4, H-7), 4.19 (1 H, m, J 2.0, 4.13, H-6), 4.68 (1 H, d, J 4.4, H-8), 4.77 (1 H, d, J 4.13, H-5), 7.45 (1 H, d, J 8.3, H-3), 7.93 (1 H, d, J 8.3, H-4).
(+)-(5S,6S,7R,8R)-2-Chloro-5,6,7,8-tetrahydro-5,6,7,8-quinoline tetrol 42
Similar treatment of tetraacetate (+)-44 gave pure tetrol (+)-42 [α]D +7 (c 0.44, MeOH).
(+)-5R,6R,7R,8R)-2-Chloro-5,6,7,8-tetrahydro-5,6,7,8-quinoline tetrol 43
Tetraacetate (+)-45 (1.35 g, 3.41 mmol) was deprotected and crystallized in a similar manner to compound (-)-44 to yield pure tetraol (+)-43 (600mg, 76%), white crystals; mp 189-192 °C; [α]D +14.6 (c 0.57, MeOH); (Found: C, 46.6; H, 4.1; N, 6.0. C9H10NO4Cl requires C, 46.7; H, 4.3; N, 6.1%); Found: M+ 231.03090, C9H10ClNO4 requires 231.02984; νmax/cm−1: 3391 (OH); 1H NMR (500 MHz, D2O) δH 4.25 (1 H, dd, J 5.6, J 3.9, H-6), 4.30 (1 H, dd, J 5.6, J 4.0, H-7), 4.91 (1 H, d, J 4.0, H-8), 5.00 (1 H, d, J 3.9, H-5), 7.57 (1 H, d, J 8.3, H-3), 7.97 (1 H, d, J 8.3, H-4); 13C NMR (125 MHz, D2O) δC 67.9, 69.0, 69.1, 70.3, 125.6, 131.2, 142.5, 151.5, 154.6; m/z 231 (M+, 5%, 35Cl), 213 (34%), 171 (100).
Hydrogenolysis of Chlorotetrols (–)-42, (+)-42, and (+)-43 to Yield Tetrols (–)-46, (+)-46, and (–)-48
(+)-(5R,6R,7S,8S)-5,6,7,8-Tetrahydro-5,6,7,8-quinoline tetraol 46
A solution of chlorotetraol (–)-42 (200 mg, 0.86 mmol) in MeOH (20 ml) containing 10% Pd/C (50 mg) was stirred overnight at room temperature, under H2 atmosphere at 1 atm. pressure. The reaction mixture was basified (NH3 solution), the catalyst was filtered off and the filtrate concentrated under reduced pressure to yield tetraol (–)-46 (120 mg, 71%), white solid (MeOH); mp 164–165°C (MeOH); [α]D +25 (c 0.5, MeOH); (Found: C, 54.3; H, 5.3; N, 6.6. C9H11NO4 requires C, 54.8; H, 5.6; N, 7.1%); 1H NMR (500 MHz, D2O) δH 4.28 (1 H, dd, J 4.3, 1.4, H-7), 4.34 (1 H, dd, J 4.0, 1.4, H-6), 4.88 (1 H, d, J 4.3, H-8), 4.93 (1 H, d, J 4.0, H-5), 7.56 (1 H, dd, J 7.9, 4.5, H-3), 8.10 (1 H, d, J 7.9, H-4), 8.62 (1 H, d, J 4.5, H-2).
(–)-(5S,6S,7R,8R)-5,6,7,8-Tetrahydro-5,6,7,8-quinoline tetraol 46
A similar hydrogenolysis process on compound (+)-44 yielded tetraol (–)-45, [α]D −23 (c 0.4, MeOH).
(–)-(5R,6R,7R,8R)-5,6,7,8-Tetrahydro-5,6,7,8-quinoline tetrol 48
Hydrogenolysis of tetrol (+)-43 (0.2 g, 0.86 mmol) gave tetraol (–)-48, (0.12 g, 71%); mp 186-187 °C (MeOH); [α]D −104 (c 0.56, pyridine); (Found: C, 53.6; H, 5.8; N, 6.6. C9H11NO4 requires C, 54.8; H, 5.6; N, 7.1%); 1H NMR (500 MHz, D2O) δH 4.33 (1 H, dd, J 6.7, J 6.8, H-7), 4.38 (1 H, dd, J 6.8, J 6.5, H-6), 5.05 (1 H, d, J 6.7, H-8), 5.09 (1 H, d, J 6.5, H-5), 7.62 (1 H, dd, J 7.8, J 4.0, H-3), 8.06 (1 H, d, J 4.0, H-2), 8.69 (1 H, d, J 7.8, H-4); m/z 197 (M+, 7.5%), 137 (100).
Synthesis of Dibromohydroxyacetates (–)-47, (+)-47 and Dibromodiacetate 49 From tetrols (+)-46, (–)-46, and (–)-48
(–)-(5S,6S,7R,8R)-5,8-Dibromo-6-hydroxy-5,6,7,8-tetrahydro-7-quinolinyl acetate 47
To a stirred solution of tetrol (+)-46 (100 mg, 0.51 mmol) in dry CH3CN (5 ml) maintained at ice temperature, was added 1-bromocarbonyl-1-methylethyl acetate (0.15 ml, 1.04 mmol) and the stirring continued (2.5 h) at room temperature. The reaction mixture was concentrated in vacuo, the concentrate taken up into ether (20 ml) and washed repeatedly with 2.5% NaHCO3 solution until the aqueous layer was basic. The combined aqueous layer was back extracted with ether and the combined ether extract dried (Na2SO4) and concentrated under reduced pressure. Purification of the crude product by PLC yielded compound (–)-47 (80 mg, 65%), pale yellow oil; (Rf 0.25, 55% EtOAc-hexane); [α]D −31 (c 0.54, CHCl3); HRMS calcd for C11H10NO3Br, 282.9844, (M – HBr)+; found, 282.9839; 1H NMR (500 MHz, CDCl3) δH 2.08 (3 H, s, COCH3), 4.90 (1 H, dd, J 8.3, 2.3, H-6), 5.30 (1 H, d, J 4.0, H-8), 5.40 (1 H, d, J 8.3, H-5), 5.75 (1 H, dd, J 4.1, J 2.3, H-7), 7.32 (1 H, dd, J 8.9, H-4), 8.00 (1 H, d, J 8.9, H-4), 8.59 (1 H, d, J 5.6, H-2); 13C NMR (125 MHz, CDCl3) δC 20.9, 46.3, 51.5, 70.3, 74.3, 123.9, 130.2, 139.6, 150.3, 151.2, 169.9; m/z 285 (M-HBr)+, 79Br, 15%), 287 (M+-HBr, 81Br, 15%), 242 (100).
(+)-(5R,6R,7S,8S)-5,8-Dibromo-6-hydroxy-5,6,7,8-tetrahydro-7-quinolinyl acetate 47
A similar treatment procedure on tetrol (–)-46, yielded compound (+)-47 [α]D +30 (c 0.4, CHCl3).
(5S,6S,7S,8S)-6,7-(Diacetoxy)-5,6-dibromo-5,6,7,8-tetrahydroquinoline 49
Similar treatment of (–)-tetraol 48 (0.15 g, 0.78 mmol) with 1-bromocarbonyl-1-methylethyl acetate gave dibromodiacetate 49 (0.21 g, 65%), pale yellow syrup; (Rf 0.7, 55% EtOAc-hexane); (Found: M+-HBr, 324.996063. C13H12NO4Br requires M+, 324.994969); 1H NMR (500 MHz, CDCl3) δH 2.12 (3 H, s, COCH3), 2.15 (3 H, s, COCH3), 5.36 (1 H, d, J 6.1, H-8), 5.40 (1 H, d, J 7.5, H-5), 5.52 (1 H, m, H-6), 5.65 (1 H, m, H-7), 7.35 (1 H, dd, J 8.0, J 4.7, H-3), 7.99 (1 H, d, J 8.0, H-4), 8.65 (1 H, d, J 4.3, H-2); 13C NMR (125 MHz, CDCl3) δC 20.7, 23.9, 34.2, 46.4, 47.4, 58.8, 73.8, 124.2, 128.9, 134.1, 150.4, 150.6, 169.2, 169.3; m/z 325 (M+-HBr, 50%), 79 (100).
Synthesis of Quinoline-5,6: 7,8-dioxide (–)-28 and (+)-28 From 5,8-dibromo-6-hydroxy-5,6,7,8-tetrahydro-7-quinolinyl acetate (–)-47 and (+)-47
(–)-(5R,6R,7S,8S)-Quinoline-5,6:7,8-dioxide 28
A solution of bromohydrin (–)-47 (80 mg, 0.22 mmol) in THF (10 ml) was stirred at 0°C with an excess of anhydrous sodium methoxide (250 mg) for 1 h and at room temperature for another 1 h. After removal of the THF under reduced pressure ice-cold water (10 ml) was added to the residue and the aqueous mixture extracted with ether. The extract was dried (Na2SO4) concentrated and the crude product purified by PLC (Rf 0.2, 60% EtOAc in hexane) to give syn-quinoline dioxide (–)-28 (25 mg, 70%), white solid (CH2Cl2-ether-hexane); mp 153–156°C (lit (Boyd et al., 1991b) rac. mp 138–152°C); [α]D −26 (c 0.2, CHCl3); HRMS calcd for C9H7NO2, 161.0477, M+; found, 161.0472; 1H NMR (500 MHz, CDCl3) δH 3.95–3.99 (2 H, m, H-1a, and H-1b), 4.05 (1 H, d, J 3.5, H-6b), 4.18 (1 H, d, J 3.4, H-2a), 7.38 (1 H, m, H-5), 7.95 (1 H, dd, J 7.7, 1.6, H-6), 8.65 (1 H, dd, J 4.9, 1.6,H-4); m/z 161 (M+, 15%), 132 (100). Electronic CD data: 274 nm (Δε 15.93), 268 nm (Δε 14.31), 246 nm (Δε −3.425), 192 nm (Δε −16.08).
(+)-(5S,6S,7R,8R)-Quinoline-5,6:7,8-dioxide 28
Dibromoacetate (–)-46 yielded quinoline dioxide 28 [α]D +25 (c 0.2, CHCl3).
Synthesis of (5R,6R,7R,8R)-Quinoline-5,6:7,8-dioxide 29 and (5R,6S)-8-bromoquinoline-5,6-oxide 50 From diacetoxy-5,6-dibromo-5,6,7,8-tetrahydroquinoline 49
Similar treatment of dibromodiacetate 49 (200 mg, 0.48 mmol) with sodium methoxide gave a mixture of two products. PLC separation (50% EtOAc-hexane) yielded quinoline dioxide (–)-29 (46 mg, 60%) and bromoquinoline oxide (–)-50 (16 mg, 15%).
(–)-(5R,6R,7R,8R)-Quinoline-5,6:7,8-dioxide 29
Rf 0.4 (EtOAc- hexane); white solid; mp 85–86°C (ether-hexane) [lit. Boyd et al., 1991b rac.m.p. 44–46°C]; [α]D −100 (c 0.11, CHCl3); (Found: M+ 161.047158. C9H7NO2 requires 161.047679); 1H NMR (300 MHz, CDCl3) δH 3.73 (1 H, d, J 3.9, H-6b), 3.91 (1 H, d, J 4.0, H-2a), 4.05 (1 H, m, H-1a), 4.08 (1 H, m, H-1b), 7.28 (1 H, dd, J 7.7, J 4.8, H-5), 7.73 (1 H, dd, J 7.5, J 1.5, H-6), 8.53 (1 H, dd, J 4.8, J 1.5, H-4); 13C NMR (125 MHz, CDCl3) δC 50.9, 53.2, 54.7, 55.5, 123.9, 127.7, 138.7, 149.8, 152.4; m/z 161 (M+, 15%), 132 (100). Electronic CD data: 269 nm (Δε −3.30), 226 nm (Δε −14.97), 197 nm (Δε 11.46).
(–)-(5R,6S)-8-Bromo quinoline-5,6-oxide 50
Rf 0.6 (50% EtOAc-hexane); white solid; mp 107–108°C (ether- pet. ether), [α]D −104 (c 0.5, CHCl3), (Found: C, 48.9; H, 2.2; N, 6.0. C9H6NOBr requires C, 48.3; H, 2.7; N, 6.2); 1H NMR (500 MHz, CDCl3) δH 4.16 (1 H, dd, J 4.3, J 3.9, H-1a), 4.53 (1 H, d, J 3.8, H-7b), 7.25 (1 H, d, J 4.3, H-2), 7.35 (1 H, dd, J 7.7, J 4.9, H-6), 7.95 (1 H, d, J 7.6, H-7), 8.78 (1 H, d, J 4.9, H-5); 13C NMR (125 MHz, CDCl3) δC 53.7, 56.6, 123.3, 128.8, 129.1, 132.1, 137.8, 149.9, 150.0; m/z 223 (M+, 79Br, 60%), 225 (M+, 81Br, 60%), 89 (100); Electronic CD data: 302 nm (Δε _1.92), 276 nm (Δε 2.48), 239 nm (Δε _8.93), 233 nm (Δε −8.60), 225 nm (Δε −9.38).
Docking Process
All small molecule structures were created in.pdb format using UCSF Chimera (University of California). The crystal structures of TDO (PDB ID: 3en1) and NDO (1o7M) were obtained from the RCSB Protein Data Bank. The toluene contained in the TDO crystal structure was removed using UCSF Chimera. The dioxygen molecule was added to the iron prosthetic group of TDO (Fe, His222, His228, Asp376) from the iron prosthetic group (Fe, dioxygen, His-208, His-213, and Asp-362) of the NDO crystal structure using the “super” function of PyMol 2.4.0 to overlay the two partial structures at the position of 3en1 and copy the dioxygen atoms to the 3en1 model. All structures in.pdb format were then stripped of water molecules and converted to .pdbqt format using AutoDock Tools 1.5.6 (Scripps Research Institute).
The docking was performed using AutoDock Vina 1.1.2 (Scripps Research Institute) with the following configuration:
This configuration includes the amino acids within 6 Å of the toluene present in the 3en1 crystal structure: Gln215, Phe216, Met220, His222, Ala223, His228, Leu 272, Ile276, Ile232, Val309, His311, Leu321, Ile324, Phe366, Phe372, and Asp376. Only Met220 is not shown in the docking structures as it blocked the view on the docking orientation and was considered irrelevant for catalytic purposes.
Based on earlier publications and current understanding of the binding site and catalytic mechanism, His 222 is involved in edge-to-face binding of the substrate to direct a planar orientation, presenting a face to the active site. The probability for the methyl group of the toluene substrate to be oriented toward Phe216 is reduced by this residue, thus resulting in the observed enantiomeric excess. His311 and Asn215 are considered to be essential for the catalytic mechanism, as His311 is a H-acceptor and Asn215 is considered to be part of a water channel.
The docking model was assessed by docking toluene and comparing obtained orientations to the toluene position in the crystal structure. A total of nine orientations were obtained ranging from −5.3 to −4.2 kcal/mol−1. Three orientations emerged that present the 2,3-double bond to the dioxygen for dihydroxylation (see Supplementary Figures S-13A–C). These orientations showed minor differences in position compared to the orientation observed in the crystal structure (1–1.2 Å) and compared to each other. The difference in position between the docked and crystal structure was probably affected by the docking with dioxygen, as the crystal structure does not contain dioxygen, allowing the toluene to be closer to the iron prosthetic group. Two identical orientations emerged (see Supplementary Figure S-13D) which would result in the opposite enantiomer being formed. Given the enantiomeric excess of >98% of the dihydroxylated product of toluene, it appears unlikely that this orientation is readily dihydroxylated.
Results
Molecular Docking of TDO With Quinoline 1, 2-Chloroquinoline 8, 2-Chloropyridine 14 and Chlorobenzene 17
Molecular docking studies of TDO-catalyzed cis-dihydroxylations of mono- and di-substituted benzene substrates were conducted in other laboratories, using the GOLD program (Vila et al., 2016a,b, 2017). AutoDock and AutoDock Vina programs have similarly been used in our laboratories, to study TDO-catalyzed cis-dihydroxylation of other types of monocyclic arenes, e.g., substituted phenols and anilines and tricyclic heteroarenes, e.g., dibenzofuran and dibenzothiophene (Hoering et al., 2016; Boyd et al., 2017, 2019). Relative free binding energies (kcal/mol−1) and relative proximity of arene bonds to dioxygen, within the TDO active site (Å), were determined by applying the AutoDock Vina program to monocyclic arenes (toluene, chlorobenzene) and mono- and bicyclic- azarenes in the current study.
Using the AutoDock Vina docking model, to indicate binding energy (kcal/mol−1) and bond proximity (Å) values of TDO with quinoline substrate 1 (X = H), cis-dihydroxylation of the carbocyclic ring was predicted to occur preferentially at the 5,6- (Figure 4A, −6.4 kcal/mol−1, 3.6–3.7 Å) and 7,8- bonds (Figure 4B, −6.0 kcal/mol−1, 3.6 Å). This would be consistent with formation of similar quantities of the 5R,6S and 7S,8R cis-dihydrodiols 2 and 3, respectively. When the isolated yields of cis-dihydrodiol 3 and the derived 8-hydroxyquinoline 4 are combined, the relative proportions of originally formed cis-dihydrodiol regioisomers 2:3 were similar and 5R,6S and 7S,8R configurations were firmly established (Boyd et al., 1987, 1993).
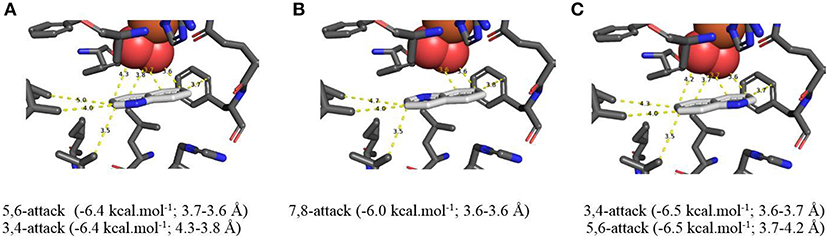
Figure 4. (A–C) TDO docking orientations of quinoline 1 (X = H) consistent with preferential cis-dihydroxylation at 5,6- (−6.4 kcal·mol−1, A), 7,8- (−6.0 kcal·mol−1, B), and 3,4-bonds (−6.5 kcal·mol−1, C). Hydrophobic substrate coordinating amino acids in the active site shown include Phe 326 and Val 309, The iron atom shown is coordinated by His 222, 228, and Asp 376. The positioning of the docked substrate relative to the positioned bound oxygen is consistent with experimentally observed product regio specificity.
Alternative in silico orientations of quinoline 1 within the TDO active site (Figure 4C, −6.5 kcal/mol−1, 3.7–4.2 Å), and (Supplementary Figure 4D, −5.8 kcal/mol−1, 3.8–4.2 Å), would lead to the incorrect 5S,6R and 7R,8S absolute configurations for isolated metabolites 2 and 3, respectively. Although these orientations had similar binding energies, compared with Figures 4A,B it was presumed that the larger distances between the relevant bonds and proximate dioxygen atom were the determining factors.
Support for the intermediacy of the undetected heterocyclic ring cis-dihydrodiol 7 was provided by the AutoDock Vina-derived prediction that TDO-catalyzed cis-dihydroxylation could occur at the 3,4-bond of the pyridine ring to yield transient intermediate (3S,4R)-7 (Figure 4C, −6.5 kcal/mol−1, 3.6–3.7 Å). The opposite (3R,4S) enantiomer 7 might also be expected from the orientation of quinoline 1 in Figure 4A. While a similar binding energy (−6.4 kcal/mol−1) is shown in Figure 4A, this orientation may be less favored, due to a larger distance from the nearest dioxygen atom (3.8–4.3 Å).
In silico data collected using the Autodock Vina TDO-docking program with 2-chloroquinoline 8 as substrate, similarly led to the prediction that cis-dihydroxylation should occur at the 5,6-position, to give (5R,6S)-dihydrodiol 9 (Figure 5A, −5.8 kcal/mol−1, 3.7–3.8 Å) and at the 7,8-bond to form (7S,8R)-dihydrodiol 10 (Figure 5B, −6.0 kcal/mol−1, 3.5–3.6 Å). Alternative docking orientations could also result in cis-dihydroxylation at the 5,6 bond (Figure 5C, −5.1 kcal/mol−1, 4.1–4.5 Å) and the 7,8 bond (Supplementary Figure 5D, −6.2 kcal/mol−1, 4.1–4.5 Å), but the resulting cis-diol metabolites 9 and 10 would have the opposite absolute configurations to the isolated metabolites (5R,6S and 7S,8R, respectively). The orientations in Figure 5C and Supplementary Figure 5D were again assumed to be less favored, due to the larger distances between substrate and nearby dioxygen atoms. Thus, the absolute configurations of the quinoline cis-dihydrodiols (2, 3, 9, 10) predicted by in silico results were identical to those found earlier for metabolites isolated from P. putida UV4 biotransformations (Boyd et al., 1987, 1993, 1998, 2002).
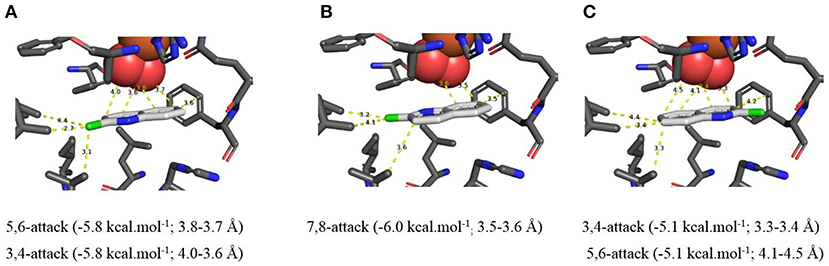
Figure 5. (A–C) TDO docking of 2-chloroquinoline 8 with cis-dihydroxylation at a 5,6- (A, −5.8 kcal·mol−1), 7,8- (B, −6.0 kcal·mol−1), and 3,4-bond (C, −5.1 kcal·mol−1). Hydrophobic substrate coordinating amino acids in the active site shown include Phe 326 and Val 309. The iron atom shown is coordinated by His 222, 228, and Asp 376. The positioning of the docked substrate relative to the positioned bound oxygen is consistent with experimentally observed product regio specificity.
More evidence for the TDO-catalyzed cis-dihydroxylation of a pyridyl ring was sought by docking studies of 2-chloropyridine 14 and comparison with chlorobenzene 17. Substrates 14 (Figure 6, −4.8 kcal/mol−1, 3.0–3.6 Å) and 17 (Figure 7, −4.9 kcal/mol−1, 3.0–3.6 Å) were found to adopt similar orientations, within the TDO active site, leading to the prediction that the derived (S, S) enantiomers of cis-dihydrodiols 15 and 18, would be formed preferentially. The (2S,3S) configuration for isolated metabolite 18, and its dehydration to yield phenol 16, was already established (Ziffer et al., 1977). The undetected intermediate 15, derived from 2-chloropyridine 14, was assumed to dehydrate more readily to yield the isolated hydroxypyridine 16 (Garrett et al., 2006). These docking results thus support the mechanism shown in Figure 3 (14→15→16).
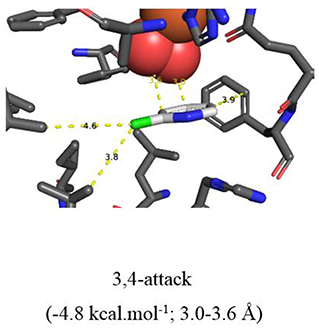
Figure 6. TDO docking of 2-chloropyridine 14 with cis-dihydroxylation at a 3,4-bond (−4.8 kcal·mol−1, this figure) and of chlorobenzene 17 at a 2,3 bond (−4.9 kcal·mol−1, Figure 7). Hydrophobic substrate coordinating amino acids in the active site shown include Phe 326 and Val 309. The iron atom shown is coordinated by His 222, 228, and Asp 376. The positioning of the docked substrate relative to the positioned bound oxygen is consistent with experimentally observed product regio specificity.
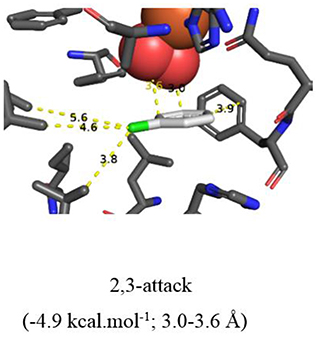
Figure 7. Same as Figure 6.
Based on the AutoDock Vina results, the orientations within the TDO active site, favored by heterocyclic arene substrates, e.g., 1 (X = H), 8, and 14, as well as substituted benzene substrates e.g., toluene and chlorobenzene 17, all revealed evidence of edge-to-face (T-bonding) interactions with Phe-216 and His-222. The simultaneous edge-to-face interactions, of amino acid residues Phe-216 and His-222, with both the phenyl and pyridyl rings of substrates 1, 8, and 14 in the present context, were noteworthy. Docking distances between the proximate T-bonding H atoms and arene faces of quinoline 1 (Supplementary Figures 4A1-A3) were estimated to be within the range 2.6–2.8 Å, assuming a C-H bond length of 1.08 Å. These inter-ring distances are in accord with the calculated interacting H-to-ring center perpendicular distances, of ca. 2.6–2.8 Å and observed distances ca. 2.70–2.86 Å, from X-ray crystallographic analysis, using a range of model systems (Hoering et al., 2016; Boyd et al., 2019). Keyhole pictures, looking through the rear face of the component aromatic ring, showed both edge to face-T (Supplementary Figures 4A3,B3,C3), and face tilted-T dockings (Supplementary Figures 4A2,B2,C2) of quinoline 1 with TDO. This was consistent with the attractive interactions between phenyl rings (Supplementary Figures 4A1,B1), phenyl/pyridyl rings (Supplementary Figure 4C1), phenyl/imidazoyl rings (Supplementary Figures 4A1,B1), and pyridyl/imidazoyl rings (Supplementary Figure 4C1).
It was concluded that the observed absolute configurations of the isolated carbocyclic ring cis-dihydrodiol metabolites 2, 3, 9, and 10 were in agreement with preferred in silico docking orientations of the corresponding substrates (Figures 4A,B, 5A,B). The preferred orientations of quinoline 1 (Figure 4C), 2-chloroquinoline 8 (Figure 5C), and 2-chloropyridine 14 (Figure 6), would also be supportive of TDO-catalyzed cis-dihydroxylation occurring at the pyridine rings to give the undetected cis-dihydrodiol intermediates 7, 12, and 15.
Chemoenzymatic Synthesis of Isolated and Potential Arene Oxide Metabolites of Quinoline
The docking studies described in Figures 4A,B, 5A,B enabled us to develop novel chemoenzymatic approaches for the preparation of different types of chiral azaarene derivatives. The availability of the cis-diol 10 as the major metabolite from TDO-catalyzed cis-dihydroxylation of 2-chloroquinoline 8, resulted in its first use in chemoenzymatic synthesis, when chiral 2,2′-bipyridines and 2,2′-bipyridine N-oxides were produced (Boyd et al., 2008, 2010). These compounds were found to be useful ligands for asymmetric oxidation (→ 97% ee) (Boyd et al., 2008), cyclopropanation (→ 95% ee) (Boyd et al., 2008), aminolysis (→ 84% ee) (Boyd et al., 2010), and allylation reactions (→ 86% ee) (Boyd et al., 2010). Further possible applications of both cis-diol metabolites 9 and 10, to the chemoenzymatic synthesis of detected and possible mammalian metabolites of quinolines, were also developed.
The identification of a 1,2-epoxide, a mammalian metabolite of naphthalene (Jerina et al., 1968), resulting from cytochrome P-450 (CYP-450)-catalyzed epoxidation, led to a quest for other examples of arene oxide metabolites. A similar type of monooxygenase-catalyzed dearomatization was later found to yield epoxide metabolites of both monocyclic arenes (e.g., benzene 1,2-oxide) and polycyclic azaarenes (e.g., quinoline 5,6-oxide) (Agarwal et al., 1986). Arene dioxides, trans-dihydrodiols, trans-tetrahydrodiol epoxides, and tetrahydroarene tetrols were later identified as other mammalian metabolites of naphthalene (Stillwell et al., 1978, 1982; Horning et al., 1980). Further studies established that liver metabolism of larger (tetracyclic and pentacyclic) bay-region arene and heteroarene substrates often involved epoxide intermediates associated with their mutagenicity and tumorigenicity. The metabolic sequence: arenes → arene oxides → trans-dihydrodiols → trans-tetrahydrodiol epoxides → DNA-adducts, became known as the Bay-region Mechanism (Buening et al., 1978; Karle et al., 2004; Chang et al., 2013).
Identification of mammalian metabolites of quinoline 1 and their role in its mutagenicity and tumorigenicity, has been of interest for more than 40 years (Hollstein et al., 1978; Tada et al., 1980, 1982; LaVoie et al., 1983; Agarwal et al., 1986, 1990; Willems et al., 1992; Saeki et al., 1993; Reigh et al., 1996; Suzuki et al., 2000; Dowers et al., 2004; Hakura et al., 2007; Diaz Duran et al., 2015; Matsumoto et al., 2018). Several quinoline arene oxides, e.g., 20 and 27, were initially postulated as possible liver microsomal metabolites of quinoline 1 (Hollstein et al., 1978; Tada et al., 1980, 1982; LaVoie et al., 1983), before 5,6-arene oxide 20 was finally confirmed as a major metabolite (Figure 8) (Agarwal et al., 1986). Epoxide hydrolyase-catalyzed hydrolysis of the identified 5,6-arene oxide 20 would also account for the isolated trans-dihydrodiol metabolite 21, and aromatization of the undetected 3,4-arene oxide 27 could yield the isolated 3-hydroxyquinoline 5. Transient arene oxides, including compound 27, resulting from CYP-450-catalyzed epoxidation of quinoline 1, were initially postulated to be responsible for its mutagenicity and carcinogenicity (Hollstein et al., 1978; Tada et al., 1980, 1982; LaVoie et al., 1983). Liver microsomal metabolism of quinoline 1 also yielded DNA adducts, which on hydrolysis, under acidic or basic conditions, gave 3-hydroxyquinoline 5. Efforts to identify mammalian metabolic pathways responsible for the mutagenicity (and carcinogenicity) of quinoline 1 (Willems et al., 1992; Suzuki et al., 2000), required the chemical synthesis of identified, and possible, metabolites of similar structures to those derived earlier from naphthalene (e.g., phenols, arene oxides, arene dioxides, trans-dihydrodiols, and trans-tetrahydrodiol epoxides) (Jerina et al., 1968; Stillwell et al., 1978, 1982; Horning et al., 1980).
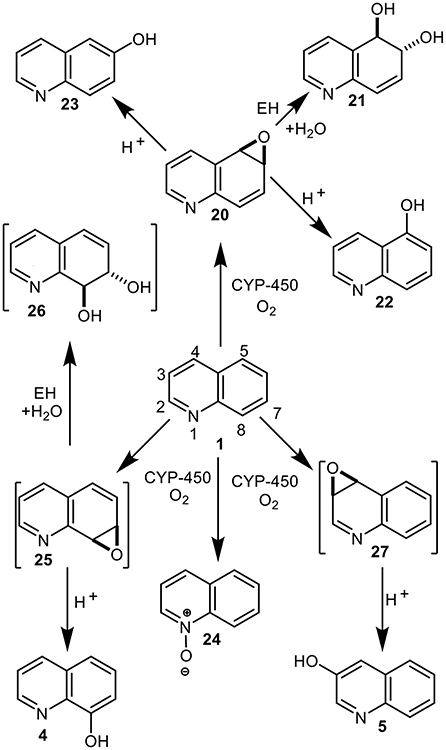
Figure 8. Mammalian metabolites of quinoline 1, both isolated (4, 5, 20–24) and postulated (25–27).
The syntheses of racemic quinoline arene oxides (20, 25), trans-dihydrodiols (21, 26), syn- and anti- arene dioxides (28 and 29), and trans-diol epoxides (30 and 31), were previously reported (Agarwal et al., 1986; Boyd et al., 1989, 1994; Bushman et al., 1989; Willems et al., 1992) (Figures 8, 10). These chiral compounds, along with achiral metabolites including N-oxide 24 and phenols 4, 5, 22, and 23, were tested for mutagenicity, using the Ames/Salmonella microsomes method (Willems et al., 1992; Suzuki et al., 2000). As none of these metabolites or possible metabolites, were found to be more mutagenic than quinoline 1, it was assumed that a metabolic sequence similar to that in the Bay-region Mechanism, involving racemic arene oxides, e.g., 20→trans-dihydrodiols 21→trans-tetrahydrodiol epoxides 31, was unlikely to be involved in its mutagenicity. It was later established that the particular absolute configurations of polycyclic arene metabolites, associated with the Bay-region Mechanism, had a marked effect on their carcinogenicity (Buening et al., 1978; Karle et al., 2004; Chang et al., 2013). The mutagenicity of the enantiopure derivatives of quinoline 20, 21, 25, 26, 28, 29, 30, and 31 was not investigated.
An alternative metabolic pathway for quinoline 1 in mammals was proposed involving dearomatization of the pyridyl ring and formation of the undetected quinoline hydrate 36 (Saeki et al., 1993, 1996, 1997a,b; Suzuki et al., 2000) (Figure 9). While relatively stable arene hydrate derivatives of quinoline were isolated earlier as metabolites of 5,6- and 7,8-dihydroquinolines (Agarwal et al., 1994), compound 36, being both an enamine and an arene hydrate, would be expected to be less stable. The CYP-450-catalyzed epoxidation of enamine intermediate 36 was postulated to give the undetected epoxide 37 and to react with DNA to yield unstable adduct 38, which rearomatized under both acidic and basic conditions to yield 3-hydroxyquinoline 5 (Saeki et al., 1993, 1996, 1997a,b; Suzuki et al., 2000). The metabolic pathway 1→36→37→38 (Enamine Epoxide Theory, Figure 9) was thus proposed to account for the mutagenicity and carcinogenicity of quinoline 1. It is noteworthy that while some substituted quinolines were found to be carcinogenic, 2-chloroquinoline 8 was not (Hirao et al., 1976; Hakura et al., 2007).
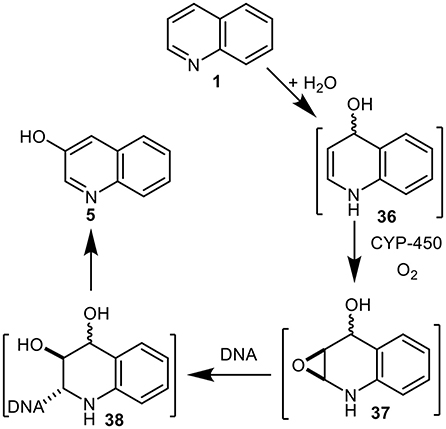
Figure 9. Proposed metabolic sequence for quinoline 1 involving intermediates 36–38.
In the context of mutagenesis studies of known and possible mammalian metabolites of quinoline 1 (Willems et al., 1992), our multistep chemical synthesis routes to racemic quinoline oxides (20 and 25) and quinoline dioxides (28 and 29), were achieved via tetrahydrobromohydrins (32, 34) and tetrahydroepoxides (33, 35) intermediates (Figure 10) (Agarwal et al., 1986, 1990; Boyd et al., 1989, 1991b).
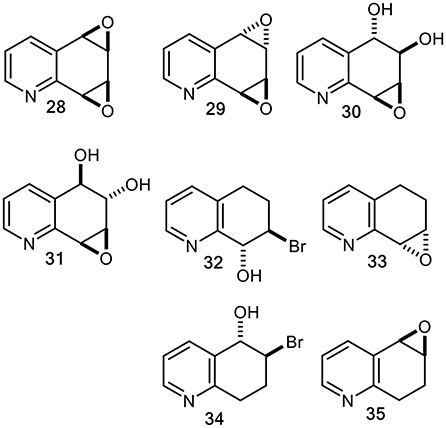
Figure 10. Chemically synthesized racemic quinoline syn- and anti-dioxides (28 and 29), diol epoxides (30 and 31), tetrahydrobromohydrins (32 and 34), and derived tetrahydroepoxides (33 and 35).
While the individual enantiomers of arene oxides 20 and 25 were separable by analytical chiral stationary phase (CSP) HPLC, this analytical scale approach was unsuitable for further chemoenzymatic synthesis studies and therefore alternative preparative methods for obtaining these enantiopure compounds were developed. The chemical resolution of bromohydrin derivatives of 1,2,3,4-polycyclic tetrahydroarenes as MTPA esters provided a generally applicable route to a range of enantiopure arene oxide derivatives, e.g., naphthalene 1,2-oxide and anthracene 1,2-oxide (Akhtar et al., 1979). Further application of this MTPA ester resolution method to racemic bromohydrin 32, yielded enantiopure tetrahydroepoxides (7S,8R)-33 ([α]D +157) and (7R,8S,)-33 ([α]D −157) intermediates and finally quinoline 7,8-oxide, (7S,8R)-25 ([α]D +55) and (7R,8S)-25 ([α]D −55, unpublished data). The racemic tetrahydroquinoline bromohydrin 34 was also resolved, but as dibenzotartrate salts to give enantiomers of quinoline 5,6-oxide (5R,6S)-20 ([α]D −23) and (5S,6R)-20 ([α]D +23) via tetrahydroepoxides (5R,6S)-35 and (5S,6R)-35 (Akhtar et al., 1979). The enantiopurities (>98% ee) of arene oxides 20 and 25 were confirmed by CSPHPLC analysis; more than ten synthetic steps from quinoline 1 were required using these chemical resolution methods.
An alternative chemoenzymatic approach to the synthesis of arene oxide enantiomers 20 and 25, using the quinoline cis-dihydrodiol metabolites (5R,6S)-2 and (7S,8R)-3 (X = H) as precursors was then developed (Boyd et al., 1991a). This again produced the quinoline oxide enantiomers (–)-(5R,6S)-20 and (+)-(7S,8R)-25, in seven steps from quinoline 1, via tetrahydroepoxides (5R,6S)-35 and (7S,8R)-33. Although this chemoenzymatic approach required fewer steps than the earlier methods involving the chemical resolution of bromohydrins 32 and 34 (Boyd et al., 1994), the very low (<5%) isolated yields of the less stable cis-dihydrodiol precursors 2 and 3 obtained by P. putida UV biotransformation (Boyd et al., 1993), was a limiting factor. As the isolated yields of the more stable cis-dihydrodiol metabolites 9 (9%) and 10 (21%), obtained from of 2-chloroquinoline 8 were higher, under similar conditions, they were used as alternative starting compounds to yield cis-tetrahydrodiol intermediates (5R,6S)-39 and (7S,8R)-40 in the current study (Figure 11).
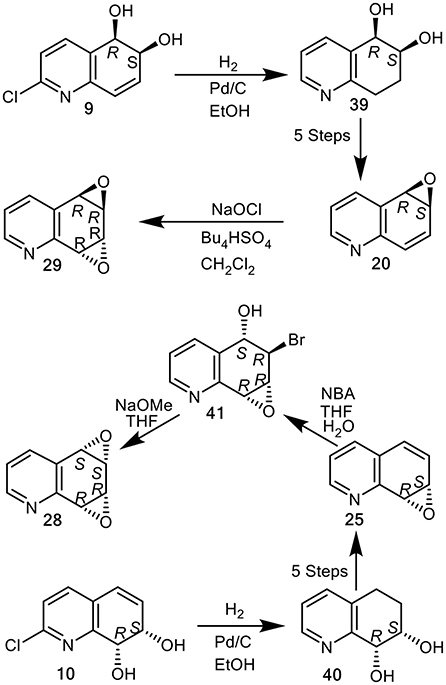
Figure 11. Synthesis of quinoline arene oxides (–)-20, (+)-25, and arene dioxides (+)-syn-28 and (–)-anti-29 from cis-dihydrodiols (+)-10 and (+)-9.
(5R,6S)-cis-Tetrahydrodiol 39 ([αD] −7, CHCl3) and (7S,8R)-cis-tetrahydrodiol 40 ([αD] −72, CHCl3), known precursors of arene oxides 20 and 25 (Boyd et al., 1994) were efficiently synthesized (ca. 80% yield), by catalytic hydrogenation/hydrogenolysis (H2, Pd/C, EtOH) of the (5R,6S)-cis-dihydrodiol 9 ([αD] +140, MeOH) and (7S,8R)-cis-dihydrodiol 10 ([αD] +148, MeOH), respectively. This new approach avoided use of the less stable cis-dihydrodiol metabolites 2 and 3 and provided an improved chemoenzymatic route the corresponding arene oxides (–)-20 and (+)-25 (Figure 11) (Boyd et al., 1994).
Attempted epoxidation of available arene oxides 20 and 25 to yield the arene dioxide isomers 28 and 29, using several oxidants, including m-chloroperoxybenzoic acid and dimethyl dioxirane, resulted in the formation of N-oxides. The epoxidation of arene oxides 20 and 25 in CH2Cl2 solution was achieved using sodium hypochlorite solution (pH 8.6) in the presence of tetrabutylammonium hydrogen sulfate and gave anti-quinoline dioxide 29 exclusively. With arene oxide (–)-20 available from earlier studies (Boyd et al., 1991a, 1994), NaOCl oxidation gave the (5R,6R,7R,8R)—arene dioxide 29 ([α]D −100, 38% yield, Figure 11). Reaction of the (+)-(7S,8R) enantiomer of arene oxide 25 with N-bromoacetamide under reported conditions, yielded the crude trans-bromohydrin intermediate 41, which upon treatment with sodium methoxide furnished the syn-quinoline dioxide (5S,6S,7R,8R)-28 ([α]D +25, Figure 11) (Boyd et al., 1991b).
A further approach to the synthesis of the syn-quinoline dioxide enantiomers (–) and (+)-28 involved the cis-dihydroxylation of the 5,6-(+)-9 and 7,8 cis-dihydrodiol (+)-10. Catalytic cis-hydroxylation (OsO4, Me3NO) of 2-chloroquinoline cis-dihydrodiol (+)-9, produced an isomeric mixture of polar tetrols 42 and 43 that could not be separated by chromatography and were converted directly to the corresponding tetraacetate derivatives 44 and45 (1:3, Figure 12). Purification of the crude tetraacetates was achieved by flash chromatography followed by multiple elution PLC.
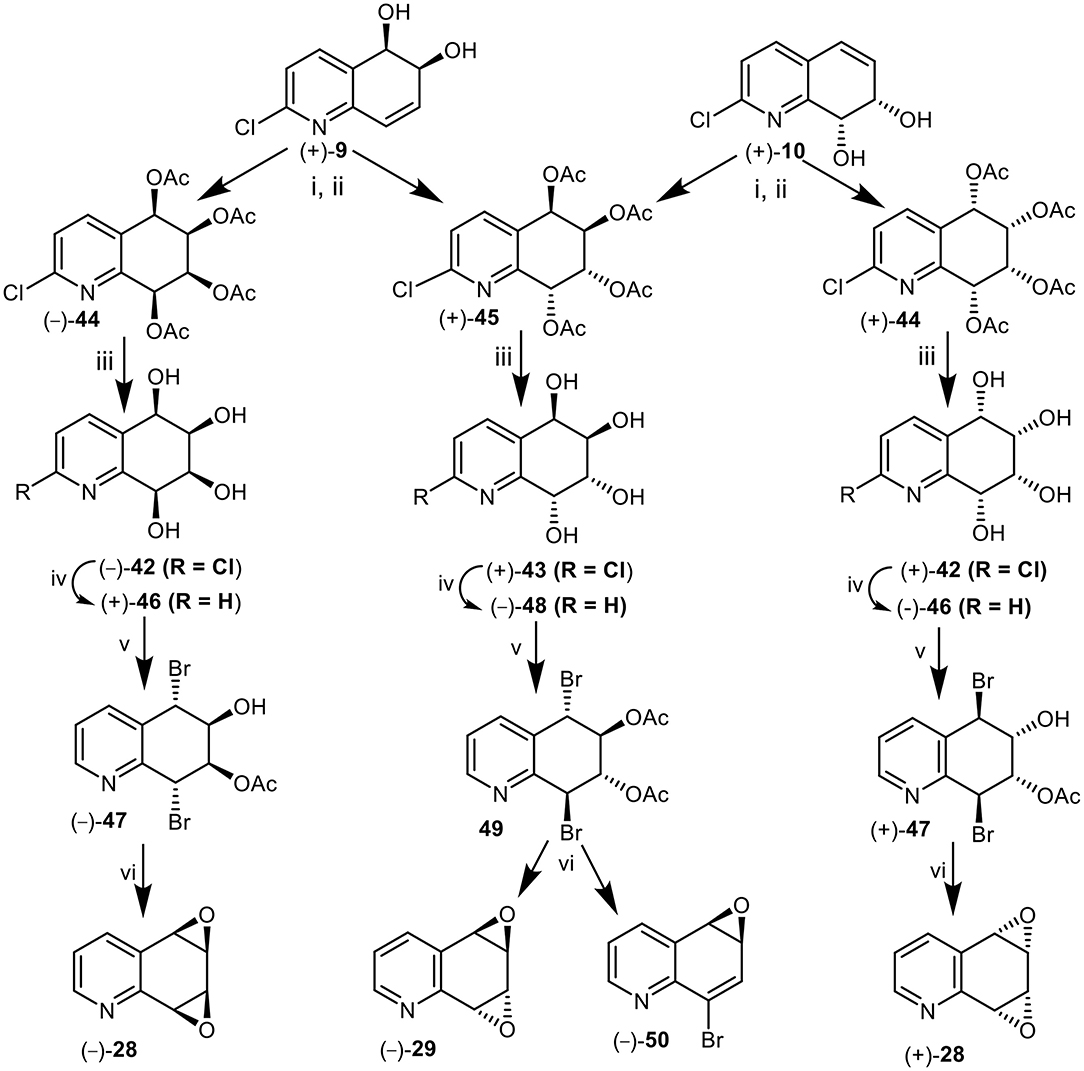
Figure 12. Synthesis of anti (–)-29 and syn-quinoline dioxides (–)-28 and (+)-28 from cis-dihydrodiols (+)-9 and (+)-10. Reagents: (i) OsO4, Me3NO-CH2Cl2; (ii) Ac2O-pyridine; (iii) NH3, MeOH; (iv) H2, Pd-C, MeOH; (v) AcOMe2COBr, MeCN; (vi) NaOMe, Et2O.
Hydrolysis of the minor syn-tetraacetate enantiomer (–)-44, under basic conditions (NH3, MeOH) yielded the 2-chloroquinoline syn-tetrol (–)-42 which was converted to the corresponding quinoline tetrol (+)-46, by hydrogenolysis (Pd-C). Treatment of tetrol (+)-46 with acetoxyisobutrylbromide in MeCN solution yielded mainly a dibromoacetate, tentatively assigned as (–)-47; it was separated from trace amounts of the expected dibromodiacetate by PLC. Dibromoacetate 47 was formed by partial hydrolysis of the dibromodiacetate during the isolation process. Treatment of dibromomonoacetate (–)-47 with NaOMe in THF yielded the syn-arene dioxide enantiomer (–)-28 from 5,6-cis-dihydrodiol (+)-9 in the sequence (+)-9 → (–)-44 → (–)-42 → (+)-46 → (–)-47 → (–)-28 (Figure 12).
In a similar manner metabolite (+)-10 was converted into the mixture of tetraacetates 44 and 45 (1:3). The minor syn-tetraacetate (+)-44, was separated by PLC; it was assigned the opposite (5S,6S,7R,8R) absolute configuration. The syn-tetraacetate (+)-44 was used in an enantiocomplementary synthetic sequence (+)-10 → (+)-44 → (+)-42 → (–)-46 → (+)-47 → (+)-28 (Figure 12).
anti-Tetraacetate (5R,6R,7R,8R) enantiomer (+)-45 obtained from 5,6-(+)-9 and 7,8-(+)-10 cis-dihydrodiol metabolites, was treated in a similar manner to the syn-tetraacetate enantiomers (+)-44 and (–)-44 to yield tetrols (+)-43, (–)-48, and dibromodiacetate -49 (Figure 12). Treatment of bromoacetate 49 with sodium methoxide yields a mixture of the expected anti-quinoline dioxide (–)-29 as the major product accompanied by a minor compound which was identified as 8-bromoquinoline-5,6-oxide 50. A competing E1cB mechanism could account for the unexpected loss of acetic acid to form the minor product, arene oxide 50. These products were thus formed via the synthetic sequence (+)-9 or (+)-10 → (+)-45 → (+)-43 → (–)-48→49 → (–)-29 + (–)-50 (Figure 12).
Discussion
Both Autodock and Gold programs proved to be very successful in matching the preferred in silico orientations of arene substrates with the experimentally confirmed regiochemistry and absolute stereochemistry of the isolated cis-diol metabolites. Previous docking studies of arene substrates with TDO indicated that the preferred orientations were controlled by: (i) attractive edge-to-face T shaped interactions with the orthogonal phenyl group of Phe-216 and imidazole ring of His 222 and (ii) Van der Waals interactions with the proximate hydrophobic amino acids Ile-276, Leu-272, Ile-324, Val-309, Leu-272, Phe-352 (Hoering et al., 2016; Vila et al., 2016a,b, 2017; Boyd et al., 2017, 2019). Theoretical, crystallographic and experimental support for phenyl-phenyl and phenyl-pyridyl edge-to-face bonding (T-bonding) interactions, between arene and heteroarene rings, has been reported (Jennings et al., 2001; Escudero et al., 2009; Gonzalez-Rosende et al., 2017; Aliev and Motherwell, 2019). Little evidence was available, from crystalline protein structures, for similar edge-to-face interactions between the imidazole ring of histidine with other aromatic residues, e.g., Phe, Tyr, Trp, His (Bhattacharyya et al., 2003). The possibility of further types of edge-to-face interactions between heteroarene substrates 1 (Figures 4A–C), 8 (Figures 5A–C), and 14 (Figure 6) with Phe-216 and His-222 in the active site of TDO (Figures 4–6), is also apparent.
Based on the docking orientations of 2-chloroquinoline 8 (Figures 5A,C), cis-dihydroxylation could also occur at the 3,4-bond of the pyridine ring. The shorter distance between this bond and dioxygen in Figure 5C (3.3–3.4 Å), would appear to favor formation of the undetected (3R,4R)-dihydrodiol 12, over the (3S,4S) configuration resulting from the orientation of substrate 8 shown in Figure 5A (3.6–4.0 Å). Conversely, the higher binding energy, −5.8 kcal/mol−1, associated with Figure 5A relative to Figure 5C (−5.1 kcal/mol−1), would suggest a preference for the (3S,4S) absolute configuration, thus making a prediction of the preferred stereochemistry of cis-dihydrodiol 11 less reliable.
To account for the unexpected isolation of (3S,4S)-diol 11 as a product from P. putida UV4 biotransformations of 2-chloroquinoline 8, the undetected cis-dihydrododiol 12 of unknown absolute configuration, was originally proposed as a possible intermediate in the metabolic sequence 8→12→11 (Figure 2) (Boyd et al., 1998). Later GC-MS identification of 2-quinolone 13 as a very minor metabolite of substrate 8, was consistent with an alternative or additional metabolic pathway (8→13→11, Figure 2) (Boyd et al., 2002). Stronger evidence for the latter biosynthetic route was provided when 2-quinolone 13 was used as substrate and (3S,4S)-diol 11 was found to be the only identifiable metabolite. Since 2-hydroxyquinoline is a minor tautomer of 2-quinolone 13, the formation of cis-diol metabolite 11 could also be considered as a TDO-catalyzed cis-dihydroxylation within a substituted pyridyl ring. It is therefore possible that cis-diol metabolite 11 was formed from 2-chloroquinoline 8, via both 2-quinolone 13 and cis-dihydrodiol 12 intermediates.
The previously reported regioselective and enantioselective cis-dihydroxylation, of the carbocyclic ring of quinoline 1, 2-chloroquinoline 8 and 2-chloropyridine 14, was rationalized by molecular docking (Autodock Vina) studies of these substrates. The in silico results revealed novel edge-to-face attractive interactions at the active site of TDO. These studies also provided support for the TDO-catalyzed cis-dihydroxylation of: (i) the pyridyl ring in substrates, 1, 8, and 14, to yield the corresponding unstable cis-dihydrodiol metabolites, 7, 12, and 15, and (ii) the carbocyclic ring to give stable cis-dihydrodiol metabolites 2, 3, 9, and 10 having the correct absolute configurations. Evidence for the monooxygenase-catalyzed epoxidation and dioxygenase-catalyzed cis-dihydroxylation of a pyridyl ring was discussed, in the context of both bacterial and mammalian metabolism of quinoline 1.
Our findings are consistent with the current thinking on the catalytic mechanism for these enzymes, which is not fully understood. Barry and Challis (2013) discuss an important point, i.e., whether the Fe(III)-OOH complex in the TDO catalytic cycle reacts directly with the substrate or first undergoes rearrangement to an Fe(V)-O(OH) complex. They highlight the question of which arene carbon atom first forms a bond with an oxygen atom.
In conclusion, new synthetic applications of the relatively stable cis-diol metabolites 9 and 10, derived from 2-chloroquinoline 8 were successfully investigated. These cis-dihydrodiols were utilized in the chemoenzymatic synthesis of the corresponding cis-tetrahydrodiols 39 and 40, required in the synthesis of enantiopure quinoline arene oxides, (–)-20 and (+)-25. This process was greatly facilitated by molecular docking preliminary studies. The unexpected 8-bromoquinoline oxide (–)-50 was obtained during the synthesis of anti-(–)-29 and syn-quinoline dioxide enantiomers (–)-28 and (+)-28 from cis-dihydrodiols 9 and 10.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
CA, DB, NS, and PS supervised the research, designed the research, obtained the funding, wrote the manuscript, and designed the experiments. PH conducted the modeling and docking. JC, PL, and NS conducted the laboratory synthesis/biotransformations. All authors contributed to the article and approved the submitted version.
Funding
Financial support for post-graduate studentships, RA funding, and academic staff funding was gratefully received from BBSRC (grant number 81/ABC09451 to DB and NS for RA and academic funding, grant number BB/E013848/1 to CA for academic funding) and the Leverhulme Trust (to CA/PH for Ph.D. and academic funding), the European Social Fund (JC for Ph.D. funding) and Queen's University of Belfast, Oxford Glycosciences, and the Overseas Research Studentship (to PL for Ph.D. funding).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor Brian Jennings for helpful discussion and Dr. Lynne Hamilton for preliminary results.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.619175/full#supplementary-material
References
Agarwal, R., Boyd, D. R., Sharma, N. D, McMordie, R. A. S., Porter, H. P., van Ommen, B., et al. (1994). Chemical and enzyme-catalysed synthesis of quinoline arene hydrates. Bioorg. Med. Chem. 2, 439–446. doi: 10.1016/0968-0896(94)80013-8
Agarwal, S. K., Boyd, D. R., Davies, R. J. H., Hamilton, L., Jerina, D. M., McCullough, J. J., et al. (1990). Synthesis of arene oxide and trans-dihydrodiol metabolites of quinoline. J. Chem. Soc. Perkin Trans. 1, 1969–1974. doi: 10.1039/p19900001969
Agarwal, S. K., Boyd, D. R., Porter, H. P., Jennings, W. B., Grossman, S. J., and Jerina, D. M. (1986). Arene oxides and trans-dihydrodiols of quinoline. Tetrahedron Lett. 27, 4253–4256. doi: 10.1016/S0040-4039(00)94245-X
Akhtar, M. N., Boyd, D. R., Hamilton, J. G., and Hamilton, J. C. (1979). Synthesis of (+)- and (–)-naphthalene and anthracene 1,2-oxides. J. Chem. Soc. Perkin Trans. 1, 2437–2440. doi: 10.1039/P19790002437
Aliev, A. E., and Motherwell, W. B. (2019). Some recent advances in the design and use of molecular balances for the experimental quantification of intramolecular noncovalent interactions of pi systems. Chem. Eur. J. 25, 10516–10530. doi: 10.1002/chem.201900854
Barr, S. C., Bowers, N., Boyd, D. R., Sharma, N. D., Hamilton, L., McMordie, R. A. S., et al. (1998). The potential role of cis-dihydrodiol intermediates in bacterial aromatic hydroxylation and the NIH Shift. J. Chem. Soc. Perkin Trans. 1, 3443–3451. doi: 10.1039/a805077e
Barry, S. M., and Challis, G. L. (2013). Mechanism and catalytic diversity of Rieske non-heme iron-dependant oxygenases. ACS Catal. 3, 2363–2370. doi: 10.1021/cs400087p
Bhattacharyya, R., Saha, R. P., Samanta, U., and Chakrabarti, P. (2003). Geometry of interaction of the histidine ring with other planar and basic residues. J. Proteome Res. 2, 255–263. doi: 10.1021/pr025584d
Bott, G., Schmidt, M., Rommel, T. O., and Lingens, F. (1990). Microbial metabolism of quinoline and related compounds. V. Degradation of 1H-4-oxoquinoline by Pseudomonas putida 33/1. Biol. Chem. 371, 999–1003. doi: 10.1515/bchm3.1990.371.2.999
Boyd, D. R., Bushman, D. R., Davies, R. J. H., Dorrity, M. R. J., Hamilton, L., Jerina, D. M., et al. (1991a). Absolute configurations of the arene oxide, trans-dihydrodiol and cis-dihydrodiol products resulting from metabolism of quinoline at the 5,6-bond. Tetrahedron Lett. 32, 2963–2966. doi: 10.1016/0040-4039(91)80663-Q
Boyd, D. R., Davies, R. J. H., Dunlop, R., Porter, H. P., Hamilton, L., and McCullough, J. J. (1989). Non-enzymatic hydration, amine addition, and oxidation reactions of aza-arene oxides. J. Chem. Soc. Chem. Commun. 1989, 163–165. doi: 10.1039/c39890000163
Boyd, D. R., Davies, R. J. H., Hamilton, L., McCullough, J. J., and Porter, H. P. (1991b). Arene oxides of quinoline: epoxidation, N-oxidation and N-methylation reactions. J. Chem. Soc. Perkin Trans. 1, 2189–2192. doi: 10.1039/p19910002189
Boyd, D. R., McMordie, R. A. S., Porter, H. P., Dalton, H., Jenkins, R. O., and Howarth, O. W. (1987). Metabolism of bicyclic aza-arenes by Pseudomonas putida to yield vicinal cis-dihydrodiols and phenols. J. Chem. Soc. Chem. Commun. 1987, 1722–1724. doi: 10.1039/c39870001722
Boyd, D. R., Sharma, N. D., Agarwal, R., Kerley, N. A., McMordie, R. A. S., Smith, A., et al. (1994). A new synthetic route to non-K and bay region arene oxide metabolites from cis-diols. J. Chem. Soc. Chem. Commun. 1994, 1693–1694. doi: 10.1039/c39940001693
Boyd, D. R., Sharma, N. D., Brannigan, I. N., Evans, T. A., Haughey, S. A., McMurray, B. T., et al. (2012). Toluene dioxygenase-catalyzed cis-dihydroxylation of benzo[b]thiophenes and benzo[b]furans: synthesis of benzo[b]thiophene 2,3-oxide. Org. Biomol. Chem. 10, 7292–7304. doi: 10.1039/c2ob26120k
Boyd, D. R., Sharma, N. D., Brannigan, I. N., McGivern, C. J., Nockemann, P., Stevenson, P., et al. (2019). Cis-dihydroxylation of tricyclic arenes and heteroarenes catalyzed by toluene dioxygenase: a molecular docking study and experimental validation. Adv. Synth. Catal. 361, 2526–2537. doi: 10.1002/adsc.201900147
Boyd, D. R., Sharma, N. D., Carroll, J. G., Malone, J. F., Mackerracher, D. G., and Allen, C. C. R. (1998). Dioxygenase-catalysed cis-dihydrodiol formation in the carbo- and hetero-cyclic rings of quinolines. Chem. Commun. 1998, 683–684. doi: 10.1039/a709138i
Boyd, D. R., Sharma, N. D., Dorrity, M. R. J., Hand, M. V., McMordie, R. A. S., Malone, J. F., et al. (1993). Structure and stereochemistry of cis-dihydrodiol and phenol metabolites of bicyclic azaarenes from Pseudomonas putida UV4. J. Chem. Soc. Perkin Trans. 1, 1065–1071. doi: 10.1039/p19930001065
Boyd, D. R., Sharma, N. D., McIntyre, P. B. A., Stevenson, P. J., McRoberts, W. C., Gohil, A., et al. (2017). Enzyme-catalysed synthesis of cyclohex-2-en-1-one cis-diols from substituted phenols, anilines and derived 4-hydroxycyclohex-2-en-1-ones. Adv. Synth. Catal. 359, 4002–4014. doi: 10.1002/adsc.201700711
Boyd, D. R., Sharma, N. D., Modyanova, L. V., Carroll, J. G., Malone, J. F., Allen, C. C. R., et al. (2002). Dioxygenase-catalyzed cis-dihydroxylation of pyridine-ring systems. Can. J. Chem. 80, 589–600. doi: 10.1139/v02-062
Boyd, D. R., Sharma, N. D., Sbircea, L., Murphy, D., Belhocine, T., Malone, J. F., et al. (2008). Azaarene cis-dihydrodiol-derived 2,2′-bipyridine ligands for asymmetric allylic oxidation and cyclopropanation. Chem. Commun. 2008, 5535–5537. doi: 10.1039/b814678k
Boyd, D. R., Sharma, N. D., Sbircea, L., Murphy, D., Malone, J. F., James, S. L., et al. (2010). Chemoenzymatic synthesis of chiral 2,2′-bipyridine ligands and their N-oxide derivatives: applications in the asymmetric aminolysis of epoxides and asymmetric allylation of aldehydes. Org. Biomol. Chem. 8, 1081–1090. doi: 10.1039/b919894f
Buening, M. K., Wislocki, P. G., Levin, W., Yagi, H., Thakker, D. R., Akagi, H., et al. (1978). Tumorigenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides in newborn mice: exceptional activity of (+)-7b,8a-dihydroxy-9a,10a epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Proc. Natl. Acad. Sci. U.S.A. 75, 5358–5361. doi: 10.1073/pnas.75.11.5358
Bushman, D. R., Sayer, J. M., Boyd, D. R., and Jerina, D. M. (1989). Solvolysis of the quinoline 5,6- and 7,8-oxides: effect of the ring nitrogen. J. Am. Chem. Soc. 111, 2688–2691. doi: 10.1021/ja00189a051
Chang, R. L., Wood, A. W., Huang, M. T., Xie, J. G., Cui, X. X., Reuhl, K. R., et al. (2013). Mutagenicity and tumorigenicity of the four enantiopure bay-region 3,4-diol-1,2-epoxide isomers of dibenz[a,h]anthracene. Carcinogenesis 34, 2184–2191. doi: 10.1093/carcin/bgt164
Diaz Duran, L. T., Rincon, N. O., Galvis, C. E. P., Kouznetsov, V. V., and Lorenzo, J. L. F. (2015). Genotoxicity risk assessment of diversely substituted quinolines using the SOS chromotest. Environ. Toxicol. 30, 278–292. doi: 10.1002/tox.21905
Dowers, T. S., Rock, D. A., Perkins, B. N. S., and Jones, J. P. (2004). An analysis of the regioselectivity of aromatic hydroxylation and N-oxygenation by cytochrome P450 enzymes. Drug. Metab. Dispos. 32, 328–332. doi: 10.1124/dmd.32.3.328
Escudero, D., Estarellas, C., Frontera, A., Quinonero, D., and Deya, P. M. (2009). Theoretical and crystallographic study of edge-to-face aromatic interactions between pyridine moieties and benzene. Chem. Phys. Lett. 468, 280–285. doi: 10.1016/j.cplett.2008.12.007
Fetzner, S., Volger, B., and Lingens, F. (1993). Transformation of 2-chloroquinoline to 2-chloro-cis-7,8-dihydro-7,8-dihydroxyquinoline by quinoline-grown resting cells of Pseudomonas putida 86. FEMS Microbiol. Lett. 112:151. doi: 10.1111/j.1574-6968.1993.tb06441.x
Garrett, M. D., Scott, R., Sheldrake, G. N., Dalton, H., and Goode, P. (2006). Biotransformation of substituted pyridines with dioxygenase-containing microorganisms. Org. Biomol. Chem. 4, 2710–2715. doi: 10.1039/b606113c
Gonzalez-Rosende, M. E., Castillo, E., Jennings, W. B., and Malone, J. F. (2017). Stereodynamics and edge-to-face CH–π aromatic interactions in imino compounds containing heterocyclic rings. Org. Biomol. Chem. 15, 1484–1494. doi: 10.1039/C6OB02618D
Hakura, A., Kadoi, M., Suzuki, T., and Saeki, K. I. (2007). Clastogenicity of quinoline derivatives in the liver micronucleus assay using rats and mice. J. Health Sci. 53, 470–474. doi: 10.1248/jhs.53.470
Hirao, K., Shinohara, Y., Tsuda, H., Fukushima, S., Takahashi, M., and Ito, N. (1976). Carcinogenic activity of quinoline on rat liver. Cancer Res. 36, 329–335.
Hoering, P., Rothschild-Mancinelli, K., Sharma, N. D., Boyd, D. R., and Allen, C. C. R. (2016). Oxidative biotransformations of phenol substrates catalysed by toluene dioxygenase: a molecular docking study. J. Mol. Catal. B. Enzym. 134, 396–406. doi: 10.1016/j.molcatb.2016.10.013
Hollstein, M., Talcott, R., and Wei, E. (1978). Quinoline: conversion to a mutagen by human and rodent liver. J. Natl. Cancer Inst. 60, 405–410. doi: 10.1093/jnci/60.2.405
Horning, M. G., Stillwell, W. G., Griffin, G. W., and Tsang, W. S. (1980). Epoxide intermediates in the metabolism of naphthalene by the rat. Drug Metab. Disposit. 8, 404–414.
Jennings, W. B., Farrell, B. M., and Malone, J. F. (2001). Attractive intramolecular edge-to-face aromatic interactions in flexible organic molecules. Acc. Chem. Res. 34, 885–894. doi: 10.1021/ar0100475
Jerina, D. M., Daly, J. W., Witkop, B., Zaltz-Nirenberg, P., and Udenfriend, S. (1968). The role of arene oxide-oxepin systems in the metabolism of aromatic substrates. III. Formation of 1,2-naphthalene oxide from naphthalene by liver microsomes. J. Am. Chem. Soc. 90, 6525–6527. doi: 10.1021/ja01025a058
Kaiser, J. P., Feng, Y., and Bollag, J-M. (1996). Microbial metabolism of pyridine, quinoline, acridine, and their derivatives under aerobic and anaerobic conditions. Microbiol. Rev. 60:493. doi: 10.1128/MR.60.3.483-498.1996
Karle, I. L., Yagi, H., Sayer, J. M., and Jerina, D. M. (2004). Crystal and molecular structure of a benzo[a]pyrene 7,8-diol 9,10-epoxide N2-deoxyguanosine adduct: absolute configuration and conformation. Proc. Natl. Acad. Sci. U.S.A. 101, 1433–1438. doi: 10.1073/pnas.0307305101
LaVoie, E. J., Adams, E. A., Shigematsu, A., and Hoffmann, D. (1983). On the metabolism of quinoline and isoquinoline: possible molecular basis for differences in biological activities. Carcinogenesis 4, 1169–1173. doi: 10.1093/carcin/4.9.1169
Lewis, S. E. (2016). “Asymmetric dearomatization under enzymatic conditions”, in Asymmetric Dearomatization Reactions, ed S. L. You (Weinheim: Wiley-VHF), 279–346. doi: 10.1002/9783527698479.ch12
Matsumoto, M., Kano, H., Suzuki, M., Noguchi, T., Umeda, Y., and Fukushima, S. (2018). Carcinogenicity of quinoline by drinking-water administration in rats and mice. J. Toxicol. Sci. 43, 113–127. doi: 10.2131/jts.43.113
Reigh, G., McMahon, H., Ishizaki, M., Ohara, T., Shimane, K., Esumi, Y., et al. (1996). Cytochrome P450 species involved in the metabolism of quinoline. Carcinogenesis 17, 1989–1996.
Saeki, K., Kawai, H., Kawazoe, Y., and Hakura, H. (1997b). Dual stimulatory and inhibitory effects of fluorine-substitution on mutagenicity: an extension of the enamine epoxide theory for axtivation of the quinoline nucleus. Biol. Pharm. Bull. 20, 646–650. doi: 10.1248/bpb.20.646
Saeki, K. I., Kadoi, M., Kawazoe, Y., Futakuchi, M., Tiwawech, D., and Shirai, T. (1997a). Modification of the carcinogenic potency of quinoline, a hepatocarcinogen, by fluorine atom substitution: evaluation of carcinogenicity by a medium-term assay biol. Pharm. Bull. 20, 40–43. doi: 10.1248/bpb.20.40
Saeki, K. I., Takahashi, K., and Kawazoe, Y. (1993). Metabolism of mutagenicity-deprived 3-fluoroquinoline: comparison with mutagenic quinoline. Biol. Pharm. Bull. 16, 232–234. doi: 10.1248/bpb.16.232
Saeki, K. I., Takahashi, K., and Kawazoe, Y. (1996). Potent mutagenic potential of 4-methylquinoline : metabolic and mechanistic considerations. Biol. Pharm. Bull. 19, 541–546. doi: 10.1248/bpb.19.541
Stillwell, W. G., Bouwsma, O. J., Thenot, J. P., Horning, M. G., Griffin, G. W., Ishikawa, K., et al. (1978). Methylthio metabolites of naphthalen excreted by the rat. Res. Commun. Chem. Pathol. Pharmacol. 20, 509–530. doi: 10.1016/B978-0-08-023768-8.52191-5
Stillwell, W. G., Horning, M. G., Griffin, G. W., and Tsang, W. S. (1982). Identification and synthesis of the isomeric tetrahydroxytetrahydronaphthalene metabolites excreted in rat urine. Drug Metab. Disposit. 10, 11–14.
Suzuki, T., Wang, X., Miyata, Y., Saeki, K. I., Kohara, A., Kawazoe, Y., et al. (2000). Hepatocarcinogen quinolineinduces G:C to C:G transversions in the cII gene in the liver of lambda/lacZ transgenic mice (MutaMouse). Mutat. Res. 456, 73–81. doi: 10.1016/S0027-5107(00)00128-7
Tada, M., Takahashi, K., and Kawazoe, Y. (1982). Binding of quinoline to nucleic acid in a subcellular microsomal system. Chem. Pharm. Bull. 30, 3834–3837. doi: 10.1248/cpb.30.3834
Tada, M., Takahashi, K., Kawazoe, Y., and Ito, N. (1980). Binding of quinoline to nucleic acid in a subcellular microsomal system. Chem. Biol. Interact. 29, 257–266. doi: 10.1016/0009-2797(80)90145-3
Vila, M. A., Pazos, M., Iglesias, C., Veiga, N., Seoane, G., and Carrera, I. (2016a). Toluene dioxygenase-catalysed oxidation of benzyl azide to benzonitrile: mechanistic insights for an unprecedented enzymatic transformation. Chem. Bio. Chem. 17, 291–295. doi: 10.1002/cbic.201500653
Vila, M. A., Umpierrez, D., Seoane, G., Rodriguez, S., Carrera, I., and Veiga, N. (2016b). Computational insights into the oxidation of mono- and 1,4 disubstituted arenes by the toluene dioxygenase enzymatic complex. J. Mol. Catal. B. Enzym. 133, S410–S419. doi: 10.1016/j.molcatb.2017.03.003
Vila, M. A., Umpierrez, D., Veiga, N., Seoane, G., Carrera, I., and Giordano, S. R. (2017). Site-directed mutagenesis studies on the toluene dioxygenase enzymatic system: role of phenylalanine 366, threonine 365 and isoleucine 324 in the chemo-, regio-, and stereoselectivity. Adv. Synth. Catal. 359, 2149–2157. doi: 10.1002/adsc.201700444
Willems, M. I., Dubois, G., Boyd, D. R., Davies, R. J. H., Hamilton, L., McCullough, J. J., et al. (1992). Comparison of the mutagenicity of quinoline and all monohydroxyquinolines with a series of arene oxide, trans-dihydrodiol, diol epoxide, N-oxide and arene hydrate derivatives of quinoline in the Ames/Salmonella microsome test. Mutat. Res. 278, 227–236. doi: 10.1016/S0165-1218(10)80002-3
Zia, M. F., Vasko, A. G., Riedl, Z., Hametner, C., Hajos, G., Mereiter, K., et al. (2016). Biodihydroxylation of substituted quinolines and isoquinolines by recombinant whole-cell mediated biotransformations. Tetrahedron 72, 7348–7355. doi: 10.1016/j.tet.2016.06.077
Keywords: arene oxides, cis-dihydrodiols, dioxygenase, docking, biocatalysis
Citation: Boyd DR, Sharma ND, Loke PL, Carroll JG, Stevenson PJ, Hoering P and Allen CCR (2021) Toluene Dioxygenase-Catalyzed cis-Dihydroxylation of Quinolines: A Molecular Docking Study and Chemoenzymatic Synthesis of Quinoline Arene Oxides. Front. Bioeng. Biotechnol. 8:619175. doi: 10.3389/fbioe.2020.619175
Received: 19 October 2020; Accepted: 22 December 2020;
Published: 12 February 2021.
Edited by:
Ligia R. Rodrigues, University of Minho, PortugalReviewed by:
Jin-Song Gong, Jiangnan University, ChinaSergio F. Sousa, University of Porto, Portugal
Copyright © 2021 Boyd, Sharma, Loke, Carroll, Stevenson, Hoering and Allen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derek R. Boyd, dr.boyd@qub.ac.uk; Christopher C. R. Allen, c.allen@qub.ac.uk