Catalytic Activity Origin and Design Principles of Graphitic Carbon Nitride Electrocatalysts for Hydrogen Evolution
 Yonghao Zhu
Yonghao Zhu Detao Zhang1
Detao Zhang1  Zhenhai Xia
Zhenhai Xia- 1College of Energy, Beijing University of Chemical Technology, Beijing, China
- 2Department of Materials Science and Engineering, University of North Texas, Denton, TX, United States
Graphitic carbon nitrogen (g-C3N4) has been shown to have huge potential in photo-/electrocatalytic hydrogen generation, owing to its unique electronic structure, high nitrogen content, and excellent thermal and chemical stabilities. We have, for the first time, developed a descriptor and design principle to accurately predict the HER electrocatalytic activities of heteroatom-doped g-C3N4. The new descriptor includes the dopant and its bonding environmental information, which provides a detailed guide to the rational design and screening of high-performance catalysts. A “volcano” relationship between the descriptor and the electrocatalytic activity was established, from which the most active dopant elements and related structures were identified. It was shown that doping near the edge of g-C3N4 could be a promising strategy in developing highly active metal-free g-C3N4-based catalysts. We also reveal the intrinsic origin of the catalytic activity of the doped g-C3N4 for different activity sites. Our work provides a basic design principle of heteroatom-doped g-C3N4 as an efficient HER catalyst and a fundamental understanding of the HER mechanism.
Introduction
Water-splitting is one of the most promising clean energy technologies to produce hydrogen fuel, using renewable energy such as wind and solar energy. This technology decomposes water into molecular hydrogen via electrolysis, in which a critical but sluggish chemical reaction, known as hydrogen evolution reaction (HER), occurs (Ni et al., 2007). To promote the HER, electrocatalysts, usually Pt and its derivatives, are used in water-splitting, owing to their superb electrocatalytic properties for HER (Subbaraman et al., 2011). However, the limited resources and high cost of platinum hamper the commercialization of the technology. Metal-free catalysts based on carbon materials for HER have attracted intensive interests because of their high activities and abundant resources (Zhang et al., 2010; Deng et al., 2014; Yeh et al., 2014; Ito et al., 2015; He et al., 2017a,b; Zhao et al., 2018b). Among carbon-based catalysts, graphitic carbon nitrogen (g-C3N4) is stable and efficient for use as an electrocatalyst, due to its unique electronic structure, high nitrogen content, and excellent thermal and chemical stabilities (Lyth et al., 2009; Wang et al., 2009, 2018; Jiao et al., 2017; Zheng et al., 2017; Zhao et al., 2018a). It has been demonstrated that with conductive carbon supports, g-C3N4 exhibited highly electrocatalytic activity for HER (He et al., 2014; Zheng et al., 2014; Duan et al., 2015; Shinde et al., 2015; Pei et al., 2016, 2017). In particular, doping of heteroatoms with p-block elements, such as P, S, Se, and B, can significantly improve its catalytic activity (Shinde et al., 2015; Pei et al., 2016, 2017).
In search for a better heteroatom doped g-C3N4, the trial-and-error approaches are used to date, which is costly and time consuming. Developing design principles or descriptors will be crucial to the rational design and rapid screening of highly efficient catalysts. Recently, a few descriptors were proposed for different types of catalysts for the particular chemical reactions For example, the d-band center theory for transition metals during the oxygen reduction reaction (ORR) (Hammer and Norskov, 1995) e.g., filling for transition-metal-oxide perovskites for ORR (Suntivich et al., 2011), crystal filed stabilization energy (CFSE) for covalent organic frameworks for ORR and oxygen evolution reaction (OER) (Lin et al., 2017), the highest peak position of density of state (DOS) of doped graphene for HER (Jiao et al., 2016), and the descriptor of doped carbon for ORR (Zhao et al., 2015). Although the doped g-C3N4 has been demonstrated to have improved the catalytic activity in HER experimentally, there is lack of a fundamental understanding surrounding the catalytic origin and a descriptor to accurately predict the catalytic activities. In this study we have, for the first time, identified an intrinsic descriptor to accurately predict the HER catalytic activities of heteroatom doped g-C3N4. A volcano relationship between the descriptor and the electrocatalytic activity was established, from which the most active dopant elements were found. Our results are consistent with the experimental results (Shinde et al., 2015; Pei et al., 2016, 2017). We also revealed the intrinsic origin of the catalytic activity of the doped g-C3N4 for different activity sites. Our work provides a basic design principle for the heteroatom doped g-C3N4 as an efficient HER catalyst.
Methods
The density functional theory (DFT) method was implemented with the Vienna ab-initio Simulation Package (VASP) to study the HER electrocatalytic activities on doped g-C3N4. This method was broadly employed to simulate the catalytic activities for different catalysts (Zheng et al., 2014; Zhao et al., 2015; Jiao et al., 2016; Lin et al., 2017). The Perdew-Burke-Ernzerhof (PBE) function was used as the exchange-correlation functional for structural optimization. The valence electron configurations were: C 2s2p2,N 2s22p3, O 2s22p4, F 2s22p5, Si 3s23sp2, P3s23sp3, S 3s23sp4, Cl 3s23sp5, Se 4s24p4, Br 4s24p5, I 5s25p5 (Zheng et al., 2014; Jiao et al., 2016; Lin et al., 2017; Xu et al., 2018). The pristine and doped g-C3N4 structures were built with the periodical boundaries in x and y directions, and a vacuum space of 16 Å in thickness was set in the z- direction to avoid interaction between slabs. For the calculation setting, the k-point sampling of the Brillion zone was obtained using 6x6x1gride, generating meshes with their origin centered at the gamma point. The cutoff energy for plane waves was 500 eV, and residual force and energy in each atom were converged to 1 meV and 1.0e−5 eV, respectively. The density of states were calculated by HSE06 methods, and were more accurate than PBE (Wang et al., 2015).
Heteroatom-doped g-C3N4 (D-g-C3N4) structures were built with substitutional, interstitial and chemisorption heteroatoms in pristine g-C3N4 (Figure 1), in which the heteroatoms D are the elements in p-block in the period table (D = B, C, Si, Se, N, P, O, S, F, Cl, Br, or I). These substitutional, interstitial and absorption doping structures are denoted with Sub-nx-D, Int-nx-ny-D, and Abs-nx-D, respectively, where nx (x = 1–8) is at the N or C position on pristine g-C3N4. For example, Sub-c3-P refers to substitutional doping on position c3 with the carbon atom replaced by dopant P, and Int-n2-n5-S refers to the interstitial doping with dopant S that bonds to two N atoms at positions n2 and n5.
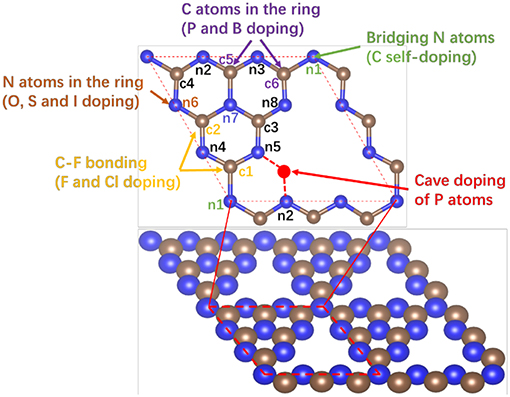
Figure 1. Schematic of a piece of pristine g-C3N4 with possible doping sites where there are three kinds of doping structures, substitution of carbon or nitrogen by heteroatoms at site c1 (Sub-c1-D), c3 (Sub-c3-D), n1(bridge nitrogen, Sub-n1-D), n5(edge nitrogen, Sub-n5-D), and n7(inner nitrogen, Sub-n7-D), interstitials between n2 and n5 in the cavity (Int-n2-n5-D), and chemisorption of heteroatoms on basal plane (Ads-c1-D and Ads-c3-D). c(c1-c5) and n(n1-n7) refer to the positions of carbon and nitrogen in pristine g-C3N4, respectively. The blue and gray balls represent N and C atoms.
The HER has two reaction mechanisms, Volmer-Heyrovsky reactions and Volmer-Tafel reactions.
In the Volmer-Heyrovsky reactions, the hydrogen (H2) is formed through two elementary steps,
In the Volmer-Tafel reactions, the hydrogen (H2) is formed through three elementary steps:
* refers to an active site on g-C3N4, g stands for gas phases, and H* and 2H* refer to adsorbed intermediates. Equation (2, 4, 5) are Volmer reaction (Hydrogen atom absorption) while Equations (3, 6) are Heyrovsky and Tafel reaction (Hydrogen atom desorption), respectively.
The density Functional theory (DFT) involving the SHE model was employed by Nørskov and co-workers to study the thermodynamic reaction process (Nørskov et al., 2004; Norskov et al., 2005). It was demonstrated that thermodynamically the electron prefers to single-electron transfer at one time instead of multiple-electron transfer. Therefore, when studying of HER thermodynamic process, only one electron transfer was considered in each sub-reaction, whether in the Volmer-Tafel or Volmer-Heyrovsky reaction.
The adsorption energy of nth hydrogen atom is calculated determined with following Equations:
E(nH*) and E[(n−1)H*] are the ground state energies of surface absorbed with nH* and (n−1)H*, respectively. E(H2) is the energy of H2 molecules in the gas phase (−6.764 eV). Equation (8) is computed for per hydrogen atom, an average value. Reaction free energy of the adsorption reaction step is determined with the following Equation:
Gads is the free energy of adsorption, T is the temperature, S is the entropy capacity, ZPE is the zero-point energy (ZPE), and is the binding energy calculated by Equation (7).
For the zero-point energy (ZPE), the vibrational frequency of adsorbed species (H, 2H, 3H) were calculated on g-C3N4. that remains fixed to obtain ZPE contribution in the free energy expression. The ZPE is determined with the following Equation:
, and are the ZPE of n-adsorbed hydrogen, (n−1) adsorbed hydrogen on the catalyst, and hydrogen in the gas phase (H2), respectively, and ZPEn = 1 = 0.15 eV.
The entropy of adsorption hydrogen atom is much less than the entropy in the gas phase (H2). Therefore, the entropy capacity is determined with the following Equation:
is the entropy of H2 gas at the standard condition and SH = −0.2 eV (Nørskov et al., 2004). In addition, the ZPE and entropy are not sensitive to the coverages (Gao et al., 2015). The overall corrections are:
ηHER is overpotential, and 0.35 is larger than 0.24 obtained from metal surface (Norskov et al., 2005) because of larger ZPE.
The d band center was used to characterize the catalytic properties of metal or metal oxide for ORR (Hammer, 2000; Nørskov et al., 2004). Similarly, the p band center was used as the descriptor of the catalytic activities of transition metal oxides for OER (Grimaud et al., 2013). In this work, the p band center for D-g-C3N4 was calculated with the following expression (Hammer, 2000; Jiao et al., 2016; Xu et al., 2018):
εp−orbital and E are the density of states projected onto heteroatoms p-orbital and the energy, respectively. Fermi level was set as zero. Jiao et al. employed the weighted density of states (DOS) center of graphene up to Fermi level (Jiao et al., 2016), as the descriptor of the catalytic activity of doped graphene for HER, which is analogous with the p band center in this work.
Results
Doping Structures and Reaction Pathways
Most of the doped g-C3N4 structures have been successfully synthesized experimentally, (Wang et al., 2010; Zhang and Antonietti, 2010; Dong et al., 2012; Hong et al., 2012; Li et al., 2012; Ma et al., 2012; Ge et al., 2013; Lin and Wang, 2013; Zhang et al., 2014; Cao et al., 2015; Fang et al., 2015; Martín-Ramos et al., 2015; Zhou et al., 2015; Guo et al., 2016; Lan et al., 2016; Pei et al., 2016, 2017; Su et al., 2016), but the detailed doping positions are still unclear. We have calculated the formation energy of all the possible doping structures using the DFT methods and determined the most likely structures according to the formation energy and experimental characterization. Figure 2 shows the most stable doping structures of D-g-C3N4 and Table 1 shows the lattice constant of these structures. The doping structures strongly depends on the number of valence electrons of dopants, nd. For nd < 4 (B, C, N, Si, P), the doping are usually substitutional on positions c1,c3, and n1, (Li et al., 2012; Lin and Wang, 2013; Fang et al., 2015; Su et al., 2016), but there is also the interstitial doping for P (Ma et al., 2012). For the chalcogens (nd = 4), the doping atoms (S, Se, and O) usually substitute the nitrogen at the edge positions (n5 and n8) (Ge et al., 2013; Cao et al., 2015). While Br and I also take the same edge positions as the chalcogens do (Zhang et al., 2014), other halogens (F and Cl) adsorb on carbon atoms in position c3 or c6 (Wang et al., 2010).
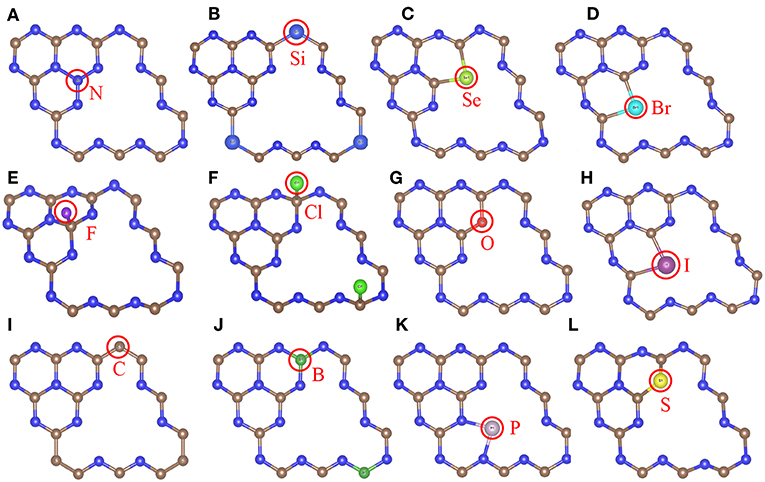
Figure 2. The most stable doping structures used in the calculations, gray, blue and other colors represent carbon, nitrogen and impurity atoms, respectively. (A) Sub-c3-N, (B) Sub-n1-Si, (C) Sub-n8-Se, (D) Sub-n5-Br, (E) Ads-c3-F, (F) Ads-c6-Cl, (G) Sub-n8-O, (H) Sub-n5-I, (I) Sub-n1-C, (J) Sub-c6-B, (K) Int-n2-n5-P, (L) Sub-n8-S models. The red circles indicate doping atoms.
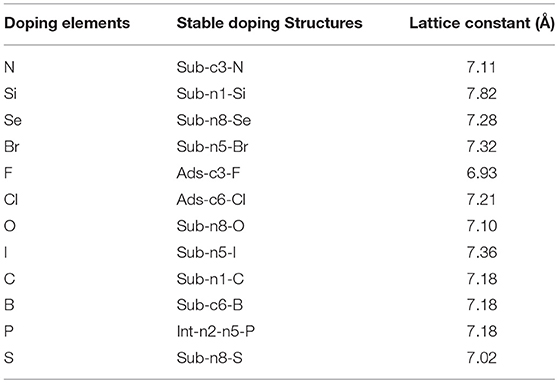
Table 1. The doping elements, stable doping structures and lattice constant.
HER on pristine and doped g-C3N4 structures were analyzed using the DFT methods following all the possible reaction pathways, as described in the Method section. HER can proceed in two different mechanisms, Volmer-Heyrovsky and Volmer-Tafel reactions (Zheng et al., 2014). In the first step of HER (Volmer reaction), protons adsorb on the active sites of the catalysts (H+ + e− → H*), where * stands for an adsorbed site. In the second step, the reaction either takes the Heyrovsky pathway ( or Tafel pathway (). Figures 3A,B show the Gibbs free-energy diagrams of Volmer-Heyrovsky and Volmer-Tafel reactions on pristine g-C3N4, respectively. For the Volmer-Heyrovsky reaction (shown in Figure 3A), the smallest average adsorption free energy of H is 0.2 eV, with 0.67 coverage rate due to the weaker adsorption of the second hydrogen atom (shown as Figure 3B), which is in line with pervious work (Gao et al., 2015). Figures 3C,D show the Gibbs free-energy diagrams of Volmer-Heyrovsky and Volmer-Tafel reactions on D-g-C3N4, respectively. As shown in Figure 3C, the Volmer step is a spontaneous reaction with large and negative Gibbs free energy because the edge nitrogen has lone pair electrons and large electronegativity. In Volmer-Heyrovsky mechanisms, the Heyrovsky reaction is the rate determining step because of strong adsorption for hydrogen atoms. In Tafel reactions (shown as Figure 3D), generally, the rate-limiting step is the Tafel step with the largest energy barriers. The Gibbs free-energy of the Volmer-Heyrovsky reaction (Figure 3C) was significantly lower compared with the Volmer-Tafel reaction (Figure 3D). Therefore, the HER on doped g-C3N4 favorably follows the Volmer-Heyrovsky reaction pathway. The average of relative Gibbs free-energy of two Volmer steps can be used to determine the catalytic activity, and the value equals the average of relative Gibbs free-energy of the Volmer-Tafel mechanism (Norskov et al., 2005; Zheng et al., 2014; Gao et al., 2015).
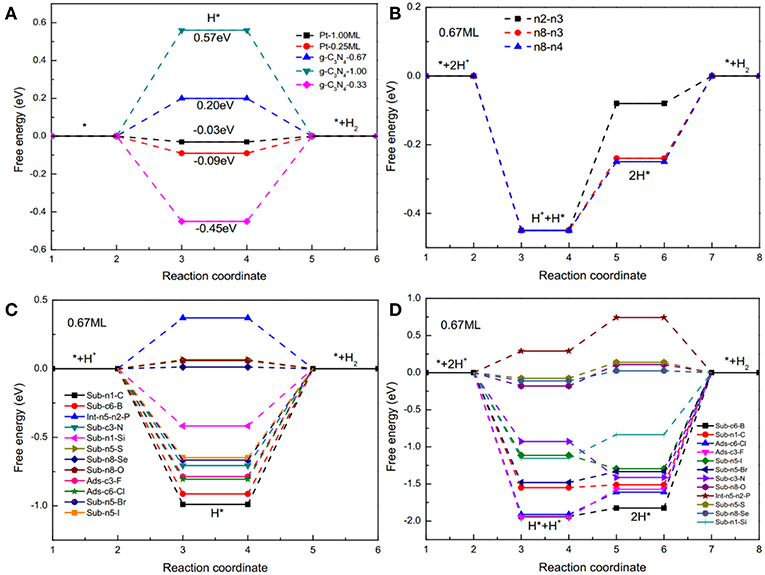
Figure 3. Free energy diagrams of the HER with (A) Volmer-Heyrovsky reactions on pristine g-C3N4 and Pt with different coverages, and (B) Volmer-Tafel reactions on pristine g-C3N4 with a coverage of 0.67 for two hydrogen atoms absorbing on n8 and n4 sites (edge nitrogen). Free energy diagrams of the HER with (C) Volmer-Heyrovsky mechanism with an intermediate (H*) on D-g-C3N4 under 0.67 coverage, and (D) Volmer-Tafel mechanism with two intermediates, H* and 2H* on D-g-C3N4 under 0.67 coverage.
Catalytic Activity of the D-g-C3N4
To evaluate the catalytic activity of the D-g-C3N4 for HER, Pt was set as the benchmark. For Pt with a H coverage of 1.0 ML, the ΔG(H*) is −0.03 eV (Norskov et al., 2005). Thermodynamically, those catalysts with ΔG(H*) smaller than that of Pt would be more active than Pt for HER. Our results show that the electrocatalytic activities of D-g-C3N4 strongly depend on the types and doping positions of the heteroatoms in g-C3N4. The g-C3N4 doped with a chalcogen atom, by replacing the edged N, shows better activity compared with other doping types (Figure 4). Among the heteroatom-doped g-C3N4 considered in this study, the absolute value of ΔG(H*) for S-, Se-, and O-doped configurations are comparable to Pt, which is expected to have higher activity for HER. This result is consistent with the previous work on the same doped structures (Shinde et al., 2015; Pei et al., 2016, 2017). Thus, doping near the edge of g-C3N4 is a promising strategy in developing highly active metal-free g-C3N4-based catalysts.
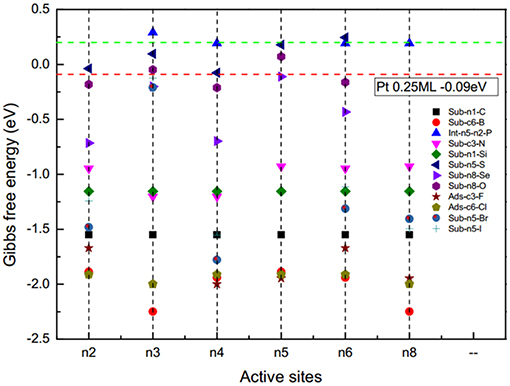
Figure 4. The adsorption energy of hydrogen on the doped g-C3N4, ΔG(H*) for different doping sites. Green and red lines represent the adsorption energy of pristine g-C3N4 and Pt (111), respectively.
The coverage of adsorbed H on the surface of the catalysts also affects the catalytic activity. For example, when hydrogen atoms are adsorbed on Pt (111), the is −0.09 eV is at a lower coverage of 0.25 ML, but it changes to −0.03 eV at a higher coverage of 1.0 ML (Norskov et al., 2005). We have calculated the of H on pristine g-C3N4 and D-g-C3N4 at different H coverage ratios, and found that the coverage ratios also have a significant effect on the catalytic activity (Figure 3). For pristine g-C3N4, the is −0.45 eV, indicating a strong adsorption on the edge nitrogen at a coverage of 0.33, and it changes to 0.20 eV (n8-n4) due to the existence of the first adsorbed H on site n8 (Figure 3A). As the coverage ratio further increases to 1, becomes 0.57 eV (Figure 3A). The adsorption strength of H gradually decreases with an increase in the coverage ratios. The relatively high coverage rate of H could be in favor of the HER.
The catalytic activity of the adsorption sites relates to the bond strength between the H and the adsorption site. It is well-known that the bond strength depends on the energy level position of bonding orbital and anti-bonding orbital. A low bonding orbital energy level or high anti-bonding orbital energy level results in a strong bond. The bonding orbital energy level could be represented by the energy range of the overlap between the partial density of states (PDOS), of adsorbed H and the adsorption site. To reveal the origin of the catalytic activity of the doped g-C3N4, the bonding orbital and anti-bonding orbital energy positions were studied by the PDOS of the adsorbed H and the adsorption site (Figures 5, 6). For g-C3N4, as a hydrogen atom adsorbed on an active site, the PDOS of the hydrogen and the adsorption site (Figure 5) has overlap in a specific range of energy, relating to the states of electron pairs formed between the hydrogen and the adsorption site. The overlap energy range of the hydrogen adsorbed on edge nitrogen (−7, −5 eV) (Figure 5A) is lower than that on carbon atom (−5, −2 eV) (Figure 5B), indicating that the edge nitrogen atom strongly absorbs hydrogen. These results show that these edge nitrogen atoms preferentially adsorb hydrogen, which is consistent with pervious work (Zheng et al., 2014).
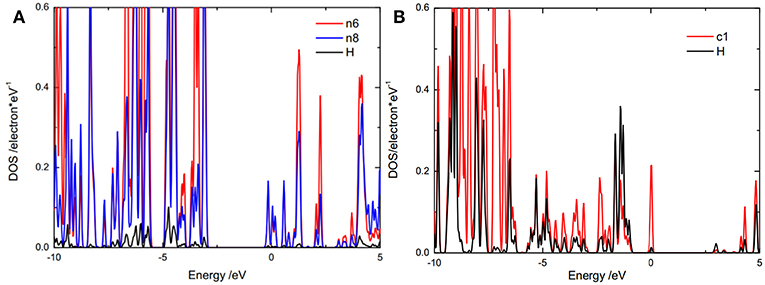
Figure 5. DOS of adsorbed hydrogen atom and the activity sites, (A) on edge nitrogen (n8) and (B) on c1 for pristine g-C3N4.
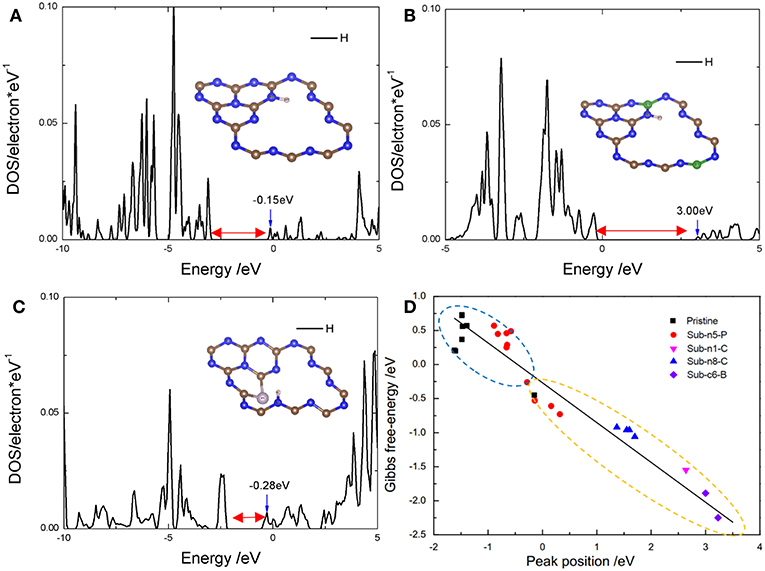
Figure 6. The peak position in DOS of adsorbed hydrogen atom on (A) pristine g-C3N4 with the adsorption site at edge nitrogen (n8), −0.45 eV and E(peak-position) = −0.15 eV, (B) Sub-c6-B with the adsorption site at edge nitrogen (n8), and E(peak-position) = 3.00 eV, and (C) Sub-n5-P with the adsorption site at edge nitrogen (n2), and E(peak-position) = −0.28 eV. Blue and gray balls represent N and C in the insets, respectively. (D) Linear relationship between the peak position and Gibbs free-energy. The blue circle and yellow circle represent 0.67 and 0.33 coverages, respectively.
The highest peak position of PDOS corresponding to the anti-bonding energy level of the adsorbed H 1s-p is associated with the bond strength between the adsorbed H and the active site (Kitchin et al., 2004; Jiao et al., 2016), and thus, can represent the catalytic activity of the active center. We found that there was a linear relationship between the Peak Position (Figures 6A–C) and for different D-g-C3N4 configurations (Figure 6D). The definition of Peak Position (PP) is the highest peak position of adsorbate, hydrogen atoms, at the bottom of conduction band. For example, as shown in Figures 6A–C, the PPs of three models, pristine, B- and P-g-C3N4, are −0.15, 3.00, and −0.28 eV, respectively. Obviously, B-g-C3N4 will strongly adsorb hydrogen atoms, because the high PP with weaker antibonding orbital will generate the strong bonding between the hydrogen and the adsorption site.
Since the PP is important in determining the bonding strength, we plotted the adsorption free energy, , as a function of the PP (Figure 6D). The -PP relationship curve can be separated into two parts. One part shows negative , the active center strongly absorbs the hydrogen atoms on the doped g-C3N4 with a coverage ratio of 0.33. For the other part, is positive, suggesting that the strength of absorption is weak, with the coverage value of 0.67. Therefore, the Heyrovsky reaction would process more easily after H adsorbed on D-g-C3N4 structures. The Peak Position is −0.5eV, corresponding to 0. Thus, the active site with PP at −0.5 eV is favorable to HER.
Intrinsic Descriptor for Doped g-C3N4
The Gibbs free-energy of HER varies significantly after the g-C3N4 is doped. Usually, as the Gibbs free-energy changes for intermediate H* chemisorption (Volmer reaction), , is used as a rational descriptor for characterizing the catalytic activity of the catalyst for HER (Norskov et al., 2005). The overpotential of the HER is defined by , because the standard reaction free energy of H+ + 2e− → H2 is 0. The volcano relationships between the overpotential and the free energy of H adsorption can then be established (Figures 7A,B), from which the best doping structure can be identified near the volcano top.
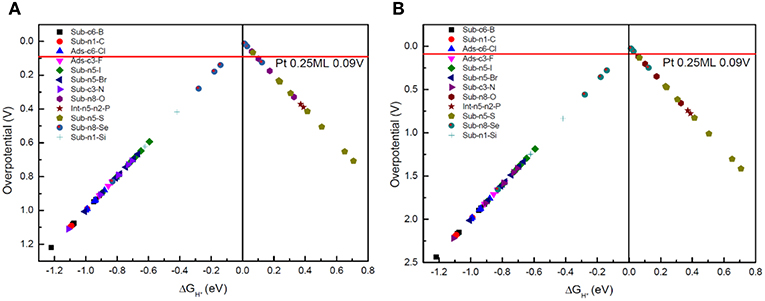
Figure 7. (A) The volcano relationships between the overpotential and the free energy of H adsorption for Volmer-Heyrovsky reaction mechanism, and (B) for Volmer-Tafel reaction mechanism.
Although the can be used as a descriptor to characterize the catalytic activity of D-g-C3N4, it is desirable to find an intrinsic descriptor that is directly related to the doping structures and intrinsic material properties so that it can guide the rational design and screening of high-performance catalysts. To identify a new descriptor for D-g-C3N4 structures, we explored the relationship between the catalytic activity of D-g-C3N4 and the intrinsic properties of the dopants and their bonding environments. We expect that the bond structure-related properties, such as the number of p-electrons and electronegativity of dopant, would have a strong effect on hydrogen adsorption and consequently determine the catalytic activity. It was found that the catalytic activity of D-g-C3N4 for HER could be well-described by a descriptor:
n, E, and I are the number of electrons occupying the outermost p orbital (valence electrons of p-orbital), the electronegativity and the electronic affinity, respectively, and subscripts d, N, and b represent the dopant, nitrogen and replaced/adsorption-site atom, respectively.
The for all the D-g-C3N4 vs. the descriptor ϕ was plotted in Figure 8, where a volcano relationship is established, which distinguishes the doping structures and the catalytic activity of D-g-C3N4. On the left side of the “volcano” is the doping with nd < 4 (B, C, N, Si, and P), in which P is identified to be the best dopant ( = −0.37 eV) with the activity in the order of P>Si>N>B>C. On the right side is the halogen doping, in which I stands out ( = 0.64 eV), with the catalytic activity in the order of I>Br>F>Cl. The chalcogens-doping sits on the top of the volcano, which has the highest catalytic activities of HER.
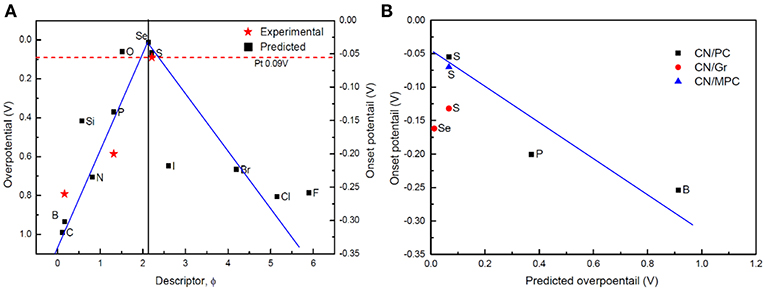
Figure 8. (A) “volcano”-shaped relationship between overpotential (Gibbs free-energy of H*) and descriptor ϕ for the most stable D-g-C3N4. The measured onset potential [CN/PC (Pei et al., 2017)] is also plotted as a function of ϕ. (B) relationship between the predicted overpotential and the experimental onset potential of D-g-C3N4 [CN/MPC (Shinde et al., 2015; Pei et al., 2016, 2017), CN/PC (Pei et al., 2017), and CN/Gr (Shinde et al., 2015)].
Our new descriptor well-describes the catalytic activities of different doping structures of D-g-C3N4. It is well-known that the onset potential measured in the experiments is an indicator of the catalytic activity of the catalysts. The smaller the onset potential, the more active the catalysts. We have compared our predictions (the overpotential) with the onset potential measured in the experiments (Duan et al., 2015; Shinde et al., 2015; Pei et al., 2016, 2017). The predictions are consistent with the experimental data (Figure 8).
Discussion
The descriptor proposed above captures the major factors that influence the catalytic activity of D-g-C3N4. Generally, the electronegativity describes the tendency of an atom to attract a shared pair of electrons (or electron density) toward itself, while the electronic affinity is the amount of energy released or spent when an electron is added to a neutral atom or molecule in the gaseous state to form a negative ion. Unlike most intrinsic descriptors that only consider dopant properties (Hammer and Norskov, 1995; Suntivich et al., 2011; Zhao et al., 2015; Jiao et al., 2016; Lin et al., 2017), here the bonding environment (e.g. the substituted/adsorption-site atoms) are also included in the new descriptor. The descriptor also includes the number of outermost p electrons that greatly affects the chemical properties of the elements, such as the number of bonds with other elements and the ability to bind to the outermost electrons. These basic material parameters provide the direct information on the doping structures and active centers for rational design of D-g-C3N4 catalysts.
From the viewpoint of electronic structures, the new descriptor is closely related to the orbital electrons, especially p-orbital electrons shared with carbon or nitrogen atoms of g-C3N4. As can be seen in Figures 9C–E, doping leads to electric charges redistributing around the doping site in g-C3N4, which can alter the adsorption mode and binding energy of H. For example, the Bader charge of edge nitrogen atoms is −1.15e−, indicating that additional 1.15 electrons locate on the atoms. Since the whole p orbital electron integral is associated with the ability to accept excess electrons during the process of HER, we have calculated the whole orbital integral and p orbital integral of dopant under the Fermi level in the DOS. There is almost a linear relationship between the orbital integral and ϕ (Figure 9A). With increasing magnitude of the descriptor, the orbital electrons increase, consequently enhancing the interactions or adsorption energy of H. Since the electronegativity, electronic affinity and the number of outermost p electrons are all associated with the p orbital electrons in some way, the new descriptor describes the catalytic activity of doped g-C3N4 well.
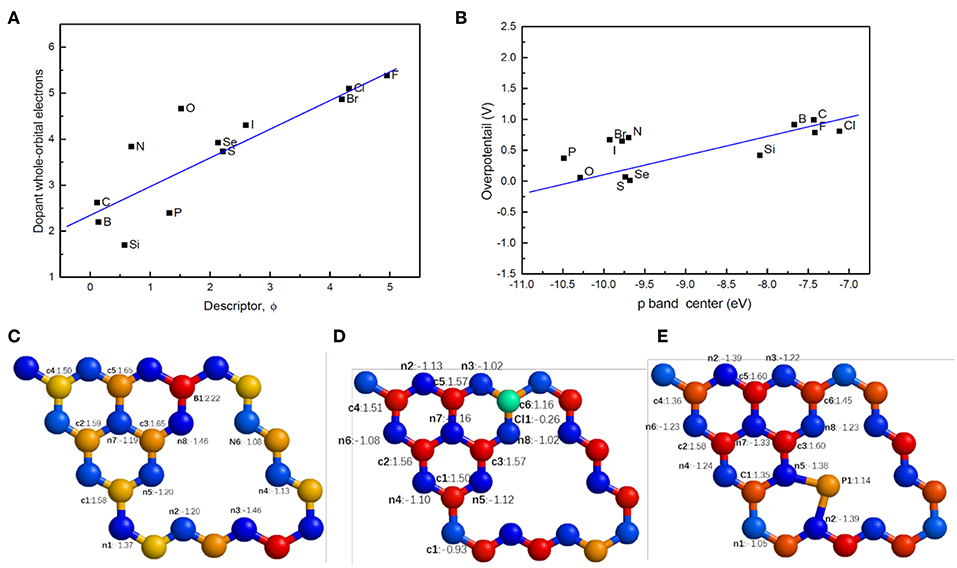
Figure 9. (A) The relationship between the descriptor and dopant whole-orbital electrons (integral of DOS under the Fermi level). (B) The relationship between p band center and the overpotential of doped g-C3N4. Typical electron charge distributions, bader charge, for (C) Sub-c6-B, (D) Ads-c6-Cl, (E) Int-n5-n2-P, where c (c1-c3) and n (n1-n7) refer to the positions of carbon and nitrogen in pristine g-C3N4, respectively.
The catalytic activities of the doped g-C3N4 can also be understood via the p-band center, defined analogous to the well-known d-band center theory for metals (Hammer and Norskov, 1995). Similarly to metals, on the surface of the doped g-C3N4, the hybridized energy levels of the two atoms split into two groups: the bonding orbital and anti-bonding states. The anti-bonding states are essential for the strength of absorption with occupation of electrons and the peak position of anti-bonding H 1s-p. Like the d-band center, the p-band center determines the adsorption strength of the hydrogen atoms. As shown in Figure 9B, the overpotential almost linearly increases with an increase in the p-band center. Therefore, the p-band center represents the origin of the catalytic activity of the doped g-C3N4.
Conclusion
An intrinsic descriptor has been discovered to accurately predict the electrocatalytic activities of heteroatom doped g-C3N4 catalysts for HER. This descriptor combines the information on doping structures, the number of p electrons, electronegativity and the electronic affinity energy of the dopants and substituted/adsorption-site atom, which can provide a detailed guide for the screening of high performance C3N4 catalysts. A volcano relationship between the overpotential and the descriptor φ was established, from which the most active dopants are predicted. The S, Se and O sit at the summit of the volcano and are expected to be the most active doping elements. The predictions are consistent with the experimental results (Shinde et al., 2015; Pei et al., 2016, 2017). Doping near the edge of g-C3N4 is a promising strategy in developing highly active metal-free g-C3N4-based catalysts. Besides the descriptor φ, we also found the peak position and p-band center related to anti-bonding of H1s-p on the DOS of adsorbed H. These activity-center parameters show a liner relationship with and an overpotential of the doped g-C3N4, and could serve as an additional descriptor at the level of the electronic structures. These relationships reveal the influence of p-block element doping from two different aspects. The approach in this study could also be applied for other two-dimensional materials.
Author Contributions
YZ wrote the paper. YZ, DZ, and LG did the calculations. ZX designed the calculations. ZX and LZ reviewed and revised the paper. All the authors joined discussion of this paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the National Key Research and Development Program of China (2017YFA0206500), National Natural Science Foundation of China (51732002), Distinguished Scientist Program at BUCT (buctylkxj02), and US National Science Foundation (1561886, 1363123, and 1662288) for the support of this research. Computational resources were provided by UNT high performance computing initiative.
References
Cao, L., Wang, R., and Wang, D. (2015). Synthesis and characterization of sulfur self-doped g-C3N4 with efficient visible-light photocatalytic activity. Mater. Lett. 149, 50–53. doi: 10.1016/j.matlet.2015.02.119
Deng, J., Ren, P., Deng, D., Yu, L., Yang, F., and Bao, X. (2014). Highly active and durable non-precious-metal catalysts encapsulated in carbon nanotubes for hydrogen evolution reaction. Energy Environ. Sci. 7, 1919–1923. doi: 10.1039/c4ee00370e
Dong, G., Zhao, K., and Zhang, L. (2012). Carbon self-doping induced high electronic conductivity and photoreactivity of g-C3N4. Chem. Commun. 48, 6178–6180. doi: 10.1039/c2cc32181e
Duan, J., Chen, S., Jaroniec, M., and Qiao, S.Z. (2015). Porous C3N4 nanolayers@N-graphene films as catalyst electrodes for highly efficient hydrogen evolution. ACS Nano 9, 931–940. doi: 10.1021/nn506701x
Fang, J., Fan, H., Li, M., and Long, C. (2015). Nitrogen self-doped graphitic carbon nitride as efficient visible light photocatalyst for hydrogen evolution. J. Mater. Chem. A 3, 13819–13826. doi: 10.1039/c5ta02257f
Gao, G., Jiao, Y., Ma, F., Jiao, Y., Waclawik, E., and Du, A. (2015). Metal-free graphitic carbon nitride as mechano-catalyst for hydrogen evolution reaction. J. Catal. 332, 149–155. doi: 10.1016/j.jcat.2015.10.006
Ge, L., Han, C., Xiao, X., Guo, L., and Li, Y. (2013). Enhanced visible light photocatalytic hydrogen evolution of sulfur-doped polymeric g-C3N4 photocatalysts. Mater. Res. Bull. 48, 3919–3925. doi: 10.1016/j.materresbull.2013.06.002
Grimaud, A., May, K.J., Carlton, C.E., Lee, Y.L., Risch, M., Hong, W.T., et al. (2013). Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat Commun 4:2439. doi: 10.1038/ncomms3439
Guo, S., Deng, Z., Li, M., Jiang, B., Tian, C., Pan, Q., et al. (2016). Phosphorus-doped carbon nitride tubes with a layered micro-nanostructure for enhanced visible-light photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 55, 1830–1834. doi: 10.1002/anie.201508505
Hammer, B. (2000). Theoretical surface science and catalysis—calculations and concepts. Adv. Catal. 45, 71–129. doi: 10.1016/S0360-0564(02)45013-4
Hammer, B., and Norskov, J.K. (1995). Why gold is thte noblest of all the metals. Nature 376, 238–240.
He, D., Kou, Z., Xiong, Y., Cheng, K., Chen, X., Pan, M., et al. (2014). Simultaneous sulfonation and reduction of graphene oxide as highly efficient supports for metal nanocatalysts. Carbon 66, 312–319. doi: 10.1016/j.carbon.2013.09.005
He, D., Tang, H., Kou, Z., Pan, M., Sun, X., Zhang, J., et al. (2017a). Engineered graphene materials: synthesis and applications for polymer electrolyte membrane fuel cells. Adv Mater. 29:1601741. doi: 10.1002/adma.201601741
He, T., Gao, G., Kou, L., Will, G., and Du, A. (2017b). Endohedral metallofullerenes (M@C60) as efficient catalysts for highly active hydrogen evolution reaction. J. Catal. 354, 231–235. doi: 10.1016/j.jcat.2017.08.025
Hong, J., Xia, X., Wang, Y., and Xu, R. (2012). Mesoporous carbon nitride with in situ sulfur doping for enhanced photocatalytic hydrogen evolution from water under visible light. J. Mater. Chem 22:15006. doi: 10.1039/c2jm32053c
Ito, Y., Cong, W., Fujita, T., Tang, Z., and Chen, M. (2015). High catalytic activity of nitrogen and sulfur co-doped nanoporous graphene in the hydrogen evolution reaction. Angew. Chem. Int. Ed. 54, 2131–2136. doi: 10.1002/anie.201410050
Jiao, Y., Zheng, Y., Chen, P., Jaroniec, M., and Qiao, S.Z. (2017). Molecular scaffolding strategy with synergistic active centers to facilitate electrocatalytic CO2 reduction to hydrocarbon/alcohol. J. Am. Chem. Soc. 139, 18093–18100. doi: 10.1021/jacs.7b10817
Jiao, Y., Zheng, Y., Davey, K., and Qiao, S.-Z. (2016). Activity origin and catalyst design principles for electrocatalytic hydrogen evolution on heteroatom-doped graphene. Nat. Energy. 1:16130. doi: 10.1038/nenergy.2016.130
Kitchin, J.R., Nørskov, J.K., Barteau, M.A., and Chen, J.G. (2004). Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces. Phys. Rev. Lett. 93:156801. doi: 10.1103/PhysRevLett.93.156801
Lan, Z.-A., Zhang, G., and Wang, X. (2016). A facile synthesis of Br-modified g-C3N4 semiconductors for photoredox water splitting. Appl. Catal. B Environ. 192, 116–125. doi: 10.1016/j.apcatb.2016.03.062
Li, J., Shen, B., Hong, Z., Lin, B., Gao, B., and Chen, Y. (2012). A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity. Chem. Commun. 48, 12017–12019. doi: 10.1039/c2cc35862j
Lin, C.Y., Zhang, L., Zhao, Z., and Xia, Z. (2017). Design principles for covalent organic frameworks as efficient electrocatalysts in clean energy conversion and green oxidizer production. Adv. Mater. 29:1606635. doi: 10.1002/adma.201606635
Lin, Z., and Wang, X. (2013). Nanostructure engineering and doping of conjugated carbon nitride semiconductors for hydrogen photosynthesis. Angew. Chem. Int. Ed. 52, 1735–1738. doi: 10.1002/anie
Lyth, S.M., Nabae, Y., Moriya, S., Kuroki, S., Kakimoto, M.-A., Ozaki, J.-I., et al. (2009). Carbon nitride as a nonprecious catalyst for electrochemical oxygen reduction. J. Phys. Chem. C 113, 20148–20151. doi: 10.1021/jp907928j
Ma, X., Lv, Y., Xu, J., Liu, Y., Zhang, R., and Zhu, Y. (2012). A strategy of enhancing the photoactivity of g-C3N4 via doping of nonmetal elements: a first-principles study. J. Phy. Chem. C 116, 23485–23493. doi: 10.1021/jp308334x
Martín-Ramos, P., Martín-Gil, J., Dante, R.C., Vaquero, F., Navarro, R.M., and Fierro, J.L.G. (2015). A simple approach to synthesize g-C3N4 with high visible light photoactivity for hydrogen production. Int. J. Hydrogen Energy 40, 7273–7281. doi: 10.1016/j.ijhydene.2015.04.063
Ni, M., Leung, M.K.H., Leung, D.Y.C., and Sumathy, K. (2007). A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sust. Energy. Rev. 11, 401–425. doi: 10.1016/j.rser.2005.01.009
Norskov, J.K., Bligaard, T., Logadottir, A., Kitchin, J.R., Chen, J.G., Pandelov, S., et al. (2005). Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 215, J23–J26. doi: 10.1149/1.1856988
Nørskov, J.K., Rossmeisl, J., Logadottir, A., and Lindqvist, L. (2004). Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108:17886. doi: 10.1021/jp047349j
Pei, Z., Gu, J., Wang, Y., Tang, Z., Liu, Z., Huang, Y., et al. (2017). Component matters: paving the roadmap toward enhanced electrocatalytic performance of graphitic C3N4-based catalysts via atomic tuning. ACS Nano 11, 6004–6014. doi: 10.1021/acsnano.7b01908
Pei, Z., Zhao, J., Huang, Y., Huang, Y., Zhu, M., Wang, Z., et al. (2016). Toward enhanced activity of a graphitic carbon nitride-based electrocatalyst in oxygen reduction and hydrogen evolution reactions via atomic sulfur doping. J. Mater. Chem. A 4, 12205–12211. doi: 10.1039/c6ta03588d
Shinde, S.S., Sami, A., and Lee, J.-H. (2015). Electrocatalytic hydrogen evolution using graphitic carbon nitride coupled with nanoporous graphene co-doped by S and Se. J. Mater. Chem. A 3, 12810–12819. doi: 10.1039/c5ta02656c
Su, F.-Y., Xu, C.-Q., Yu, Y.-X., and Zhang, W.-D. (2016). Carbon Self-doping induced activation of n-π* electronic transitions of g-C3N4 nanosheets for efficient photocatalytic H2 evolution. Chem. Cat. Chem. 8, 3527–3535. doi: 10.1002/cctc.201600928
Subbaraman, R., Tripkovic, D., Strmcnik, D., Chang, K.C., Uchimura, M., Paulikas, A.P., et al. (2011). Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 334, 1256–1260. doi: 10.1126/science.1211934
Suntivich, J., Gasteiger, H.A., Yabuuchi, N., Nakanishi, H., Goodenough, J.B., and Shao-Horn, Y. (2011). Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 3, 546–550. doi: 10.1038/nchem.1069
Wang, D., Wang, H., and Hu, P. (2015). Identifying the distinct features of geometric structures for hole trapping to generate radicals on rutile TiO2 (110) in photooxidation using density functional theory calculations with hybrid functional. Phys. Chem. Chem. Phys. 17, 1549–1555. doi: 10.1039/c4cp04159c
Wang, X., Maeda, K., Thomas, A., Takanabe, K., Xin, G., Carlsson, J.M., et al. (2009). A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76–80. doi: 10.1038/nmat2317
Wang, X., Wang, W., Qiao, M., Wu, G., Chen, W., Yuan, T., et al. (2018). Atomically dispersed Au1 catalyst towards efficient electrochemical synthesis of ammonia. Sci. Bull. 63, 1246–1253. doi: 10.1016/j.scib.2018.07.005
Wang, Y., Di, Y., Antonietti, M., Li, H., Chen, X., and Wang, X. (2010). Excellent visible-light photocatalysis of fluorinated polymeric carbon nitride solids. Chem. Mate. 22, 5119–5121. doi: 10.1021/cm1019102
Xu, H., Cheng, D., Cao, D., and Zeng, X.C. (2018). A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 1, 339–348. doi: 10.1038/s41929-018-0063-z
Yeh, T.F., Teng, C.Y., Chen, S.J., and Teng, H. (2014). Nitrogen-doped graphene oxide quantum dots as photocatalysts for overall water-splitting under visible light illumination. Adv. Mater. 26, 3297–3303. doi: 10.1002/adma.201305299
Zhang, G., Zhang, M., Ye, X., Qiu, X., Lin, S., and Wang, X. (2014). Iodine modified carbon nitride semiconductors as visible light photocatalysts for hydrogen evolution. Adv. Mater. 26, 805–809. doi: 10.1002/adma.201303611
Zhang, J., Chen, X., Takanabe, K., Maeda, K., Domen, K., Epping, J.D., et al. (2010). Synthesis of a carbon nitride structure for visible-light catalysis by copolymerization. Angew. Chem. Int. Ed. 49, 441–444. doi: 10.1002/anie.200903886
Zhang, Y., and Antonietti, M. (2010). Photocurrent generation by polymeric carbon nitride solids: an initial step towards a novel photovoltaic system. Asian J. Chem. 5, 1307–1311. doi: 10.1002/asia.200900685
Zhao, G., Cheng, Y., Wu, Y., Xu, X., and Hao, X. (2018a). New 2D carbon nitride organic materials synthesis with huge-application prospects in CN photocatalyst. Small 14:e1704138. doi: 10.1002/smll.201704138
Zhao, G., Yang, H., Liu, M., and Xu, X. (2018b). Metal-free graphitic carbon nitride photocatalyst goes into two-dimensional time. Front. Chem. 6:551. doi: 10.3389/fchem.2018.00551
Zhao, Z., Li, M., Zhang, L., Dai, L., and Xia, Z. (2015). Design principles for heteroatom-doped carbon nanomaterials as highly efficient catalysts for fuel cells and metal-air batteries. Adv. Mater. 27, 6834–6840. doi: 10.1002/adma.201503211
Zheng, Y., Jiao, Y., Zhu, Y., Cai, Q., Vasileff, A., Li, L.H., et al. (2017). Molecule-level g-C3N4 coordinated transition metals as a new class of electrocatalysts for oxygen electrode reactions. J. Am. Chem. Soc. 139, 3336–3339. doi: 10.1021/jacs.6b13100
Zheng, Y., Jiao, Y., Zhu, Y., Li, L.H., Han, Y., Chen, Y., et al. (2014). Hydrogen evolution by a metal-free electrocatalyst. Nat. Commun. 5:3783. doi: 10.1038/ncomms4783
Keywords: carbon nitride, DFT calculation, hydrogen evolution reaction, descriptor, electrocatalysts
Citation: Zhu Y, Zhang D, Gong L, Zhang L and Xia Z (2019) Catalytic Activity Origin and Design Principles of Graphitic Carbon Nitride Electrocatalysts for Hydrogen Evolution. Front. Mater. 6:16. doi: 10.3389/fmats.2019.00016
Received: 28 November 2018; Accepted: 28 January 2019;
Published: 19 February 2019.
Edited by:
Dong Wook Chang, Pukyong National University, South KoreaReviewed by:
Gang Zhao, University of Jinan, ChinaShichun Mu, Wuhan University of Technology, China
Copyright © 2019 Zhu, Zhang, Gong, Zhang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lipeng Zhang, zhanglipeng2011@gmail.com
Zhenhai Xia, Zhenhai.xia@unt.edu