 Arden G. Vanderwall
Arden G. Vanderwall Erin D. Milligan
Erin D. Milligan- 1Department of Neurosciences, University of New Mexico School of Medicine, Albuquerque, NM, United States
- 2Department of Anesthesiology and Critical Care, University of New Mexico School of Medicine, Albuquerque, NM, United States
Current pain therapeutics offer inadequate relief to patients with chronic pain. A growing literature supports that pro-inflammatory cytokine signaling between immune, glial, and neural cells is integral to the development of pathological pain. Modulation of these communications may hold the key to improved pain management. In this review we first offer an overview of the relationships between pro-inflammatory cytokine and chemokine signaling and pathological pain, with a focus on the actions of cytokines and chemokines in communication between glia (astrocytes and microglia), immune cells (macrophages and T cells), and neurons. These interactions will be discussed in relation to both peripheral and central nervous system locations. Several novel non-neuronal drug targets for controlling pain are emerging as highly promising, including non-viral IL-10 gene therapy, which offer the potential for substantial pain relief through localized modulation of targeted cytokine pathways. Preclinical investigation of the mechanisms underlying the success of IL-10 gene therapy revealed the unexpected discovery of the powerful anti-nociceptive anti-inflammatory properties of D-mannose, an adjuvant in the non-viral gene therapeutic formulation. This review will include gene therapeutic approaches showing the most promise in controlling pro-inflammatory signaling via increased expression of anti-inflammatory cytokines like interleukin-10 (IL-10) or IL-4, or by directly limiting the bioavailability of specific pro-inflammatory cytokines, as with tumor necrosis factor (TNF) by the TNF soluble receptor (TNFSR). Approaches that increase endogenous anti-inflammatory signaling may offer additional opportunities for pain therapeutic development in patients not candidates for gene therapy. Promising novel avenues discussed here include the disruption of lymphocyte function-associated antigen (LFA-1) activity, antagonism at the cannabinoid 2 receptor (CB2R), and toll-like receptor 4 (TLR4) antagonism. Given the partial efficacy of current drugs, new strategies to manipulate neuroimmune and cytokine interactions hold considerable promise.
Introduction
Pain is the evolutionarily protective perception of a nociceptive stimulus indicative of actual or potential bodily harm. However, when pain occurs outside the window of usefulness it is termed pathological. It is considered chronic when it lasts 3 months or greater (1). Chronic pain is a global problem affecting more than 1.5 billion people, and in 2015 was the leading cause of disability in most countries (2, 3). Unfortunately, clinically available medications offer insufficient long-term analgesia in many pain patients (4). Furthermore, as is the case with opioids like morphine, they can be associated with unwanted effects (e.g., sedation and constipation), rapid onset of tolerance, a high potential for abuse, and even risk of death (5).
Physiologic pain is mediated by neuron-to-neuron signaling with classical neurotransmitters and neuropeptides like glutamate, calcitonin-gene related protein (CGRP), and substance P (6). However, considerable evidence indicates that neurons are not alone in the development of pathological pain. Rather, numerous immunological pathways mediated by glia (e.g., astrocytes and microglia), immune cells, and pro-inflammatory cytokines and chemokines modify neuronal communication leading to pathological pain (7–9). Current pain therapeutics neglect the actions of these non-neuronal contributors. Therefore, development of interventions designed to target neuroimmune communication will likely improve patient outcomes.
The goal of this review is to provide an overview of the immune contributions to pathological pain with a particular focus on the role of cytokines and chemokines. The authors discuss advances in gene therapeutics and novel small molecule therapeutics that relieve chronic pain through modification of neuroimmune cytokine signaling pathways.
Overview of Immune Contributions to Pathological Pain
Peripheral Immune Actions in Pathological Pain
Following peripheral nerve injury or infection, receptors referred to as pattern recognition receptors expressed on immune cells are activated. Specifically, pattern recognition receptors are triggered by pathogen associated molecular patterns (PAMPs), exogenous signals indicative of pathogen invasion, or by danger associated molecular patterns (DAMPs), endogenous signals indicative of sterile tissue injury. Common pattern recognition receptors include toll-like receptors (TLRs) and Nod-like receptors (NLRs) (10). Ligand interaction with TLRs and NLRs activate pro-inflammatory intracellular pathways mediated by transcription factors like nuclear factor-κB (NF-κB) and activator protein-1 (AP-1), as well as activation of the inflammasome. This leads to increased synthesis of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-1β. Additionally, chemotactic cytokines, referred to as chemokines, like C-C motif chemokine ligand 2 (CCL2; aka MCP-1) and C-X3-C Motif Chemokine Ligand 1 (CX3CL1; aka Fractalkine), and other immune mediators like nitric oxide (NO) are also produced (9).
Within 3 days of injury, macrophages and neutrophils are recruited to the site and secrete cytokines which aid helper T cells and B cells as well as sensitize peripheral nociceptors (11). These inflammatory mediators include pro-inflammatory cytokines like TNF-α, IL-1β, IL-6, and IL-17, nerve growth factor (NGF), serotonin, histamine, and prostaglandins like prostaglandin E2 (PGE2) (12). Activated immune cells also release anti-inflammatory molecules, such as the cytokine IL-10 and pro-resolution mediators derived from omega-3 unsaturated fatty acids (e.g., resolvins, protectins, and maresins) as a method of autoregulation in the healing process and reestablishment of homeostasis (12–14).
Interestingly, sensory neurons express receptors for many of these mediators, such as cytokine receptors (IL-1βR, TNF-αR, IL-6R, and IL-17RA), NGF receptors (TrkA), and G protein coupled receptors for serotonin, histamine, and PGE2 (12, 15). Recent reports of single cell RNA-sequencing using peripheral nociceptors located in dorsal root ganglia (DRG) have identified numerous potential neuronally expressed cytokine and chemokine receptor targets, further supporting that direct immune modulation of neuronal action is a likely driver of pathological pain (16). Activation of these receptors expressed by nearby nociceptive neurons (e.g., C fibers and Aδ fibers) leads to enhanced membrane excitability and sensitization to subsequent stimulation (15, 17). This process is termed peripheral sensitization and results in primary nociceptors that become overly responsive to previously subthreshold levels of peripheral stimulus (6).
Peripheral nerve sensitization can in turn lead to enhanced nociceptor release of excitatory neurotransmitters at their terminal synapses within the spinal cord dorsal horn, a site of significant sensory signal modulation. This enhanced signaling within the dorsal horn further sensitizes second order sensory neurons through a mechanism known as central sensitization, a neurobiological plasticity process first characterized by Woolf in the early 1980s (18). Following development of central sensitization, nociceptive neurons now potently signal to higher order structures in the brain and brainstem, resulting in pathological pain perception including hyperalgesia (enhanced response to normally noxious stimuli) and allodynia (pain in response to normally non-noxious stimuli, such as to light touch) (19). Furthermore, expansion of receptive fields is often observed, resulting in pain perception from stimulation of uninjured tissue (secondary hyperalgesia) (6, 19). Pathological pain can arise if these nociceptive pathways remain excitable beyond the period of healing.
Central Immune Actions in Pathological Pain
Astrocytes arise from neuroepithelium and are found throughout the brain and spinal cord (20), outnumbering neurons and accounting for 40–50% of all central nervous system (CNS) glia (21). In contrast, microglia have mesodermal hematopoietic origin, constituting only 5–10% of all glia, and are considered the resident mononuclear phagocytes of the CNS (7, 20).
As with injury or infection of the CNS, damage to peripheral nerves incites glia in the spinal cord dorsal horn to express increased markers of activation (22). These spinal glia assume “activated” phenotypes, with changes in morphology (e.g., switch from highly ramified to amoeboid appearance) as well as upregulation of astrocytic activation markers like glial fibrillary acidic protein (GFAP), and microglial activation markers like complement receptor 3 (CR3), cluster of differentiation 11b (CD11b), or ionized calcium binding adaptor molecule 1 (Iba1) (8, 23, 24). Of note, the recently identified microglial-specific marker transmembrane protein 119 (TMEM119) (25) may offer newfound utility in discriminating between microglia vs. macrophages/monocytes (26–28), as prior markers are expressed by both.
Glial Activation in Pathological Pain
Lumbar astrocytes undergo changes in morphology and increased GFAP expression, indicating a state of increased activation following chronic constriction injury (CCI) of the rat sciatic nerve (29). Since this initial study demonstrating anatomical evidence that spinal glia play a role in pain modulation, later studies documented that spinal glia are closely associated with neural synapses, express many of the same neurotransmitter receptors as neurons, and are known to assume “activated” phenotypes in response to abnormal or heightened neuronal signaling (8, 9, 15, 23, 30–32). Astrocytes and microglia express pain-relevant neurotransmitter receptors, like AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors, metabotropic glutamate receptors (mGluR), purinergic receptors (e.g., P2X4R or P2X7R), or neurokinin 1 receptors (NK1R), which are activated by neurotransmitters released from presynaptic nerve terminals. These neurotransmitters include glutamate, ATP and substance P (9). Following peripheral nerve damage, neurons also release chemokines like CCL2 (33) and CX3CL1(34), and other immune mediators like colony-stimulating factor 1 (CSF-1) (35) and ATP (36, 37). These released factors potently activate astrocytes and microglia (9), which in turn release pro-inflammatory cytokines (e.g., IL-1β, TNF-α, and IL-6) (38), chemokines (e.g., CCL2) (33), ATP, excitatory amino acids (EAAs), and NO (7). As in the periphery, dorsal horn neurons express receptors for many of these factors, including the pro-inflammatory cytokines, IL-1β, TNF-α, IL-6, and IL-17 (12, 15). Activation of neuronally-expressed cytokine receptors modifies neuronal function. For example, the pro-inflammatory cytokines TNF-α and IL-1β enhance excitatory synaptic transmission and suppress inhibitory synaptic transmission in neurons of spinal cord lamina II (31, 39). Whereas, TNF-α enhances spontaneous excitatory post-synaptic current (sEPSC) frequency, IL-6 reduces frequency of the spontaneous inhibitory post-synaptic current (sIPSC). Notably, IL-1β is able to enhance both sEPSC frequency and amplitude while reducing sIPSC frequency and amplitude. Additionally, TNF-α and IL-1β enhance excitatory AMPA- and NMDA-induced currents, and IL-1β and IL-6 suppress inhibitory GABA- and glycine-induced currents (39). Therefore, activation of these cytokine receptors further sensitizes local neurons leading to a feed-forward cycle of nociceptive signaling.
Trafficking Immune Cells and Pathological Pain
Macrophages in Pathological Pain
While it is clear that peripheral immune cells migrate to local sites of nerve damage, a growing body of evidence supports that they also traffic to more remote pain-relevant neural tissues and play a role in the development of pathological pain. For instance, macrophages are recruited to sites remote from the initial insult, such as to the spinal cord (40, 41) and DRG (41–43), following peripheral nerve trauma in response to chemotactic factors like CCL2 (42–44) and CX3CL1 (33, 45) derived from injured neurons with their cell bodies or central projections in these distant locations. Macrophages present in the DRG are most likely important for maintenance of chronic pain. This is supported by evidence that depletion of peripheral macrophages partially reverses established paclitaxel-induced or nerve ligation-induced mechanical hyperalgesia and reduces TNF-α expression in DRG (43, 46).
T Cells in Pathological Pain
In addition to macrophages, T cells may also be important in the mechanisms that underlie pathological pain. For instance, activated CD4+ T cells, specifically Th1 and Th17 cells, migrate to associated lumbar DRG and/or the lumbar spinal cord dorsal horn in animal models of sciatic nerve transection (47–49), spared nerve injury (50, 51), partial sciatic nerve ligation (52, 53), and sciatic nerve chronic constriction injury (54).
Increased T cell trafficking in these animal models of neuropathic pain has been associated with concurrent increases in pro-inflammatory T cell-related cytokine signaling. For example, Costigan et al. identified T cell infiltration of the spinal cord dorsal horn following a spared nerve injury (SNI) in rats with concurrent increases in spinal interferon-γ (IFN-γ) expression (50). IFN-γ is the signature pro-inflammatory cytokine released by activated Th1 cells (55). In a related study, neuropathic pain resulting from spared nerve injury is significantly diminished in both Rag1 knockout (Rag1−/−; T-cell deficient) mice and IFN-γ receptor null mutant mice as compared to wildtype controls (50).
T helper cell-mediated IL-17 expression also appears to be important for pain pathology. Compared to wild-type mice, IL-17 knockout mice display significantly less mechanical hypersensitivity as well as decreased T cell accumulation at both the site of partial sciatic nerve ligation and within the relevant lumbar DRG (L3-L5) (52). Similarly, increased CD4+ T cells were observed in the rat spinal cord in a model of spinal nerve ligation (e.g., peripheral nerve damage), with double-positive CD4+/IL-17+ T cells located in superficial laminae of the spinal cord dorsal horn by immunofluorescence staining (51). This was accompanied by significant upregulation of IL-17, IL-1β, and IL-6 (51).
Sexual Dimorphism in Pathological Pain
Much of what is known about the mechanisms of pain was identified in male animal models. However, recent reports indicate that mechanisms of pain may differ between the sexes (56, 57). While microglial activation is required for preclinical models of pathological pain in males, this does not seem to be the case for females who instead appear to be more vulnerable to the actions of T cells (54, 58–61).
Recent work from Noor et al. demonstrates that males and females exhibit divergent T cell-associated actions within discrete pain-relevant anatomic regions in a rodent model of sciatic neuropathy (54). During neuropathy, females display more profound Th17 specific responses with higher levels of IL-17A than males, both at the injured nerve and in the corresponding lumbar spinal cord. When neuropathic animals were treated with a β2-integrin antagonist, BIRT377, pain-relieved females expressed dramatically reduced IL-17A levels in sciatic and lumbar spinal cord tissues as compared to their male counterparts (for further discussion, see section “Lymphocyte Function-Associated antigen-1” below). This not only supports a role for IL-17A in mechanisms of neuropathic pain, it offers evidence of a T cell differentiation bias toward a pro-inflammatory status that is significantly greater in females than males (54).
There is growing evidence that other cytokines may also be differentially regulated in males vs. females following peripheral nerve injury. For instance, recent RNA-seq analysis of lumbar DRG following rat sciatic CCI identified that the cytokine CSF-1 is 1.7 times higher in females than males (62).
Gene Therapeutic Modulation of Cytokines for Control of Pathological Pain
Gene Therapeutic Approaches and Challenges
Gene therapy offers the advantage of anatomically targeted long-duration expression of a desired protein, thereby limiting possible unwanted systemic effects observed in other types of pharmacologic interventions. Currently available gene therapeutic approaches aim to treat disease through modification of gene expression by cells, often via viral transduction or non-viral transfection, though newer gene editing approaches, such as through CRISPR/Cas9, may become more common (63). To date, there are three FDA approved gene therapies, including Spinraza (nusinersen; anti-sense oligonucleotide for treatment of spinal muscular atrophy type 1) (64, 65), Kymriah (tisagenlecleucel; CAR T-cell therapy for treatment of B-cell precursor acute lymphoblastic leukemia) (66), and Luxturna (voretigene neparvovec; adeno-associated virus-mediated delivery of RPE65 for treatment of RPE65-mediated inherited retinal dystrophy) (67). Various gene therapies first began to enter clinical trials in the 1990s, but these early forays were plagued by unexpected toxicities, limited clinical benefit, and even cases of patient deaths (63). Further investigations revealed obstacles to using viral vectors, including insertional mutagenesis, immune-targeted destruction of genetically modified cells, and potentially fatal immunogenicity associated with the viral vectors themselves (68–70). While more modern viral vector approaches aim to limit these toxicities, non-viral approaches have become an appealing alternative. Specifically, viral approaches rely on viral vectors to mediate cellular transfection and subsequent gene expression. In contrast, non-viral gene therapies gain cellular access through a wide variety of mechanisms ranging from simple cellular uptake of naked plasmid DNA, to more formal packaging of genetic material within carriers like liposomes, polymers, or basic proteins to facilitate transfection (69–72). These packaging modifications are often aimed at limiting immune detection, protecting the payload (e.g., plasmid DNA) from degradation by endonucleases, targeting to the therapy to cells of interest, as well as promoting cellular entry (73).
Given that sustained functional suppression of pain-relevant pro-inflammatory cytokines may ameliorate chronic pain conditions, gene therapeutic expression of targets leading to improved anti-inflammatory function has been explored. A summary of reports in which gene therapeutics encoding anti-inflammatory mediators induce pain relief observed in pre-clinical pain models and in which changes in inflammatory mediators were also evaluated is provided here (Table 1).
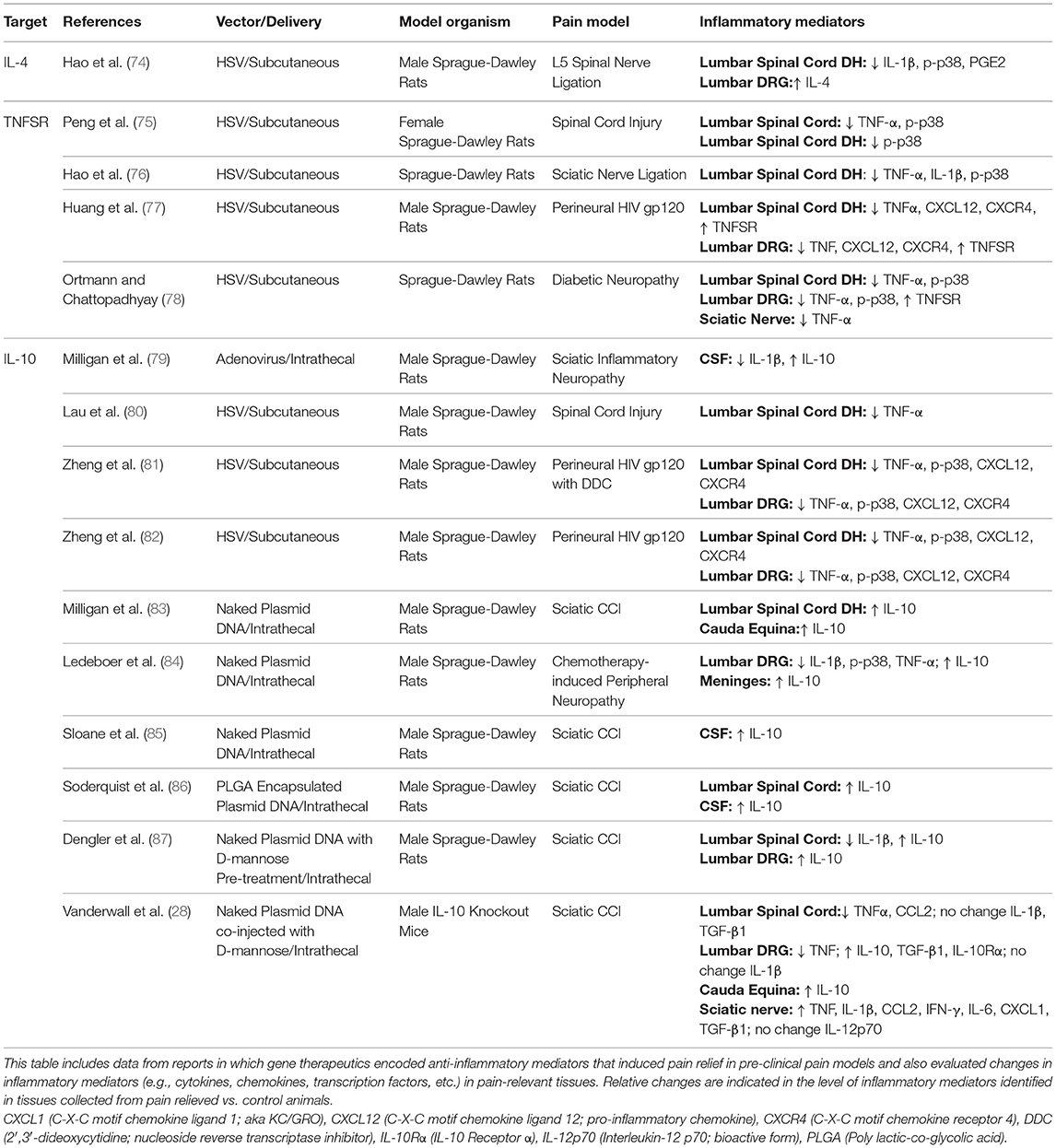
Table 1. Anti-inflammatory gene therapies for treatment of pathological pain.
Gene Therapy for Pathological Pain
Interleukin-4 Signaling
Interleukin-4 (IL-4) is an important regulator of immunity, with diverse roles in processes, such as T-cell proliferation, activated B-cell stimulation, activation of macrophages, chronic inflammation, and wound repair (88). IL-4 is primarily produced by macrophages, T-cells (especially Th2 cells), mast cells, eosinophils, and basophils (89). This cytokine exerts a variety of effects through actions at two receptor subtypes. The IL-4R-I receptor is expressed by T cells, and B cells, monocytes/macrophages, and fibroblasts (90), and is involved in the regulation of maturation and proliferation of Th2 cells, and in IgE synthesis by B cells (88).
IL-4 signaling has been shown to decrease the production and actions of several pain-relevant pro-inflammatory mediators. For instance, IL-4 suppresses NLR protein 3 (NLRP3) inflammasome-dependent caspase-1 activation and the subsequent IL-1β secretion by macrophages (91). NLRP3 serves as a sensor protein capable of initiating assembly of the intracellular inflammasome protein complex, resulting in caspase-1 activation and consequent conversion of pro-IL-1β to mature bioactive IL-1β (92, 93). IL-4 has also been shown to increase expression of endogenous IL-1 receptor antagonist (IL-1ra) mRNA and protein (94), thus diminishing the actions of pro-inflammatory IL-1β. Additionally, IL-4 directly inhibits the induction of nitric oxide synthase (NOS) and levels of cyclooxygenase 2 (COX-2), thereby decreasing the production of NO and PGE2, respectively (95).
In contrast to IL-10, which is predominantly characterized as an anti-inflammatory cytokine, IL-4 exerts some pro-inflammatory actions. In vascular endothelial cells, IL-4 induces expression of CCL2 and IL-6 (96). It also synergistically increases IL-1β, TNF-α, and vascular cell adhesion protein 1 (VCAM-1; a.k.a. CD106) in lipopolysaccharide-stimulated vascular endothelium (96). IL-4-induced IL-6 expression is mediated via a reactive oxygen species (ROS)-dependent mechanism in human aortic endothelial cells (96).
IL-4 Gene Therapy as a Pain Therapeutic
Despite some evidence that it can serves as a facilitator of pro-inflammatory cytokine expression (97–99), IL-4 is an attractive candidate for treatment of pathological pain due to its broad spectrum of anti-inflammatory actions. Additionally, IL-4 deficiency in naïve IL-4 knockout mice exhibit increased tactile allodynia (100). However, IL-4 protein replacement therapy is precluded from use for systemic administration due to IL-4's short protein half-life as well as its pleiotropic effects on immune and vascular function (74). Therefore, gene therapeutic approaches offer the best avenue for application.
IL-4 gene therapy has been examined for control of neuropathic pain in an animal model of spinal nerve ligation. Work by Hao et al. demonstrated that herpes simplex viral vector-mediated expression of IL-4 administered after the onset of mechanical allodynia results in 5 weeks of pain relief with concurrent increases in IL-4 protein expression in DRG (74). Similar effects were observed following a repeat injection. Interestingly, prophylactic treatment at 1 week prior to sciatic nerve ligation appeared to delay the onset of mechanical allodynia by about 5 weeks (74).
IL-4 gene therapy has also been examined in an animal model of painful inflammatory arthritis (101). Intra-articular injection of IL-4 gene therapy using a retroviral vector led to a reduction in paw swelling and decreased radiographic evidence of bone destruction, though differences only achieved significance about 2 weeks post-injection and paw edema measurements were not assessed throughout the entire timecourse of the inflammatory model (101), which can last several months (102), so duration of effect is speculative. Potential sex differences in IL-4 efficacy as a therapeutic to control chronic pathological pain has not been reported.
Tumor Necrosis Factor and Pathological Pain
Tumor necrosis factor-α (TNF-α) is a classic pro-inflammatory cytokine that exerts actions on two cell-surface receptors which are either constitutively expressed (TNFR1, p55-R) or inducible (TNFR2, p75-R), with most cells expressing constitutively low levels of TNFR1 (103). TNF-α signaling regulates apoptotic pathways, NF-κB-induced inflammation, and stress-induced activation of protein kinases (103, 104). Of note, TNF-α drives a positive regulatory feedback loop in which TNF-induced NF-κB signaling drives further expression of TNF-α (104).
Approaches that interfere with TNF-α signaling hold promise for the treatment of neuropathic pain, as demonstrated by pre-clinical work in animal models. Many studies have demonstrated that pain behaviors can be attenuated by local or spinal administration of agents that antagonize TNF-α (105–108). TNF soluble receptor (TNFSR), commercially known as etanercept, is previously approved by the FDA for treatment of painful inflammatory conditions including rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis (109). While preclinical trials with recombinant TNFSR have shown promise, randomized clinical trials have had limited success (109). Clinical translation may be limited by the potential loss of beneficial neuroprotective and neuroregenerative properties of TNF-α, and by its impermeability at the blood-brain-barrier (9). Interestingly, while TNFR1-mediated signaling is associated with neurotoxicity, TNFR2 signaling may by neuroprotective (110–112). Some controversy over the roles of TNFR1 vs. TNFR2 in chronic pain remains (113). The role of TNF-α in pathological pain has been extensively reviewed elsewhere (104, 113).
TNFSR Gene Therapy as a Pain Therapeutic
Gene therapeutic approaches may bypass some of the obstacles faced by application of recombinant TNFSR. When applied subcutaneously to the hindpaw 1 week following sciatic nerve ligation, herpes simplex virus (HSV)-mediated TNFSR is associated with transient improvements in mechanical allodynia and reversal of thermal latency (76). HSV-mediated TNFSR has also been shown to improve pain behaviors in models of diabetic neuropathy, spinal cord injury, HIV gp120-mediated inflammation of the sciatic nerve, and neuropathy secondary to antiretroviral therapy (75, 77, 78).
Interleukin-10
Interleukin-10 Pleiotropically Inhibits Pro-inflammatory Signaling
Interleukin-10 (IL-10) is a powerful anti-inflammatory cytokine that binds to the IL-10 receptor (IL-10R), with IL-10R1 (a.k.a. IL-10Rα) being the high affinity subunit necessary for signal transduction (114). Importantly, the IL-10R1 subunit is expressed by astrocytes, microglia, oligodendrocytes, endothelial cells, and trafficking leukocytes (115). Despite IL-10 R1 having been identified by RNA-sequencing analysis of lumbar spinal and DRG neurons in several reports (16), these findings have not yet been convincingly validated by confirmed neuronal protein expression. Two separate reports provide evidence of IL-10R1 protein expression in embryonic (E16-18) rat neurons in vitro (116, 117). Post-natal rodent protein analyses have been somewhat mixed, with one report identifying IL-10R1 protein expression in DRG neurons (118), and another report failing to identify IL-10R1 in brains (115). Due to ongoing uncertainty of neuronal IL-10 receptor expression, it remains possible that the pain-relieving actions of IL-10 and other anti-inflammatory cytokines may provide therapeutic benefit through actions at neuronal receptors.
IL-10 pleiotropically inhibits the actions of many pro-inflammatory factors by numerous mechanisms briefly described here, but thoroughly discussed elsewhere [for review, see (119, 120)]. IL-10 signaling leads to decreased bioavailability of pro-inflammatory cytokine proteins through upregulation of both IL-1ra and TNF-α decoy receptor (119). This is also accomplished through reduced gene transcription, decreased mRNA stability, and reduced translation for pro-inflammatory cytokines and chemokines, many of which are downstream of toll-like receptor 4 (TLR4) (121, 122). Additionally, IL-10 destabilizes mRNA transcripts for the pro-inflammatory cytokines TNF-α and IL-1β (119, 123). IL-10 signaling also induces suppressor of cytokine signaling 1 (SOCS1) and SOCS3 production, thereby further suppressing pro-inflammatory cytokine production by targeting the p65 NF-κB subunit (a key transcriptional regulator in the inflammatory process) for degradation (124).
The anti-inflammatory effects of IL-10 come about by blocking downstream signaling of TLR4, the actions of which normally promote a pro-inflammatory environment critical for the development of neuropathic pain (125). For instance, IL-10 inhibits translation of MyD88, an adaptor protein used by TLR4 that leads to pro-inflammatory cytokine production and release (126). IL-10 also increases ubiquitination and subsequent proteasomal degradation of MyD88-dependent signaling molecules (127). Interestingly, IL-10 induces microRNA-146b, which negatively regulates TLR4-mediated pro-inflammatory cytokine and chemokine induction, with targets including IL-6, TNF-α, IL-8, CCL3, CCL2, CCL7, and CXCL10 (128). IL-10 signaling also increases transcription of numerous effector proteins that inhibit TLR-induced downstream signaling molecules. For instance, IL-10 increases expression of Abin-3 and DUSP-1, leading to inhibition of p38 MAP kinase (MAPK) phosphorylation as well as increased nuclear translocation and DNA binding of p50/p50 homodimers of NF-κB, a form of NF-κB insufficient for induction of pro-inflammatory cytokine transcription (129).
IL-10 Gene Therapy as a Pain Therapeutic
Several studies indicate that under some chronic pain states, patients suffer from suppressed IL-10 expression (114). Similarly, animal studies also support a link between IL-10 dysregulation and pathological pain. Following peripheral nerve injury, IL-10 is briefly upregulated in what is thought to be a compensatory effort for homeostatic conditions to return. However, as pathological pain develops, IL-10 expression falls below baseline levels in pain-relevant anatomic locations (130–134). Direct application of IL-10 protein to the site of pathology is capable of producing immediate symptom relief, effects are often transient and may require repeated injections (79). These findings support that returning IL-10 expression to near-basal levels may be key to treatment for pathological pain.
Like IL-4, IL-10 has a short half-life, thus making gene therapy a desirable route of administration, particularly in patients requiring local IL-10 expression, such as in individuals with low back radiculopathy. Initial studies utilized viral vectors for lumbar spinal (intrathecal) delivery of the IL-10 gene, including non-replicating adenovirus, adeno-associated virus, and herpes simplex viral vectors. While potent pain relief was observed in animal models of chronic sciatic pain, effects were short-lived (79–82, 135, 136). The transience of the pain relief in these studies may be a result of possible complications associated with the use of a viral vector itself, such as an immune response following exposure to viral antigens (68, 120). To circumvent these possible viral obstacles, non-viral IL-10 gene therapy was explored.
Paradoxically, though non-viral gene therapies are considered the least efficient methods of gene transfer, spinal non-viral IL-10 gene therapy has been repeatedly demonstrated to provide profound and long-lasting pain relief in a variety of animal models (28, 83–87, 114, 137–139). Spinal non-viral IL-10 gene delivery in neuropathic animals produces pain relief through elevated peri-spinal IL-10 production with corresponding reduction of pro-inflammatory mediators (84, 87, 138, 140). The intriguing possibility that the protein derived from the IL-10 transgene could be capable of dimerizing with available endogenous IL-10 protein followed by receptor binding was considered a potential mechanism for the observed enduring efficacy of this unique non-viral approach. Prior work identified a non-silent point mutation in the IL-10 gene being applied in the previously published reports, and there had been some concern that it might disrupt the ability of plasmid-derived IL-10 to engage endogenous IL-10 (28). However, a recent report demonstrates that non-viral IL-10 gene therapy applied to neuropathic IL-10 knockout mice following sciatic chronic constriction injury provides long-lasting relief of allodynia, indicating that endogenous IL-10 is not required for these effects (28). Expression of the IL-10 transgene expression in pain-relieved IL-10 knockout mice was identified in lumbar DRG with concurrent increases in anti-inflammatory transforming growth factor (TGF)-β1 and decreases in pro-inflammatory TNF-α mRNA. While no IL-10 transgene was identified in the lumbar spinal cord, anti-inflammatory changes in the DRG appear to drive concordant decreases in lumbar TNF-α, CCL2, and markers of both astrocytic (GFAP) and microglial (TMEM119) activation (28). This provides compelling evidence that modification of cytokines within the DRG may be sufficient for relief of spinal mechanisms of neuropathic pain following peripheral nerve damage. To date, sex differences in IL-10 efficacy for treating chronic pathological pain have not been reported.
Non-viral IL-10 gene therapy holds significant promise for clinical translation. At this time the non-viral IL-10 gene therapy XT-150 is currently in phase I/II clinical trials (ClinicalTrials.gov Identifiers: NCT03282149; NCT03769662; NCT03477487).
Adjuvants for Non-viral Gene Therapy
While non-viral IL-10 gene therapy has been shown to effectively relieve pathological pain in numerous animal models, initial studies required repeated large doses of DNA, thereby making clinical translation less feasible. To ameliorate the dose limitations, various carriers for non-viral gene therapy have been explored but are limited by adverse effects or added levels of complexity (71, 72, 141). Co-delivered adjuvants may offer a simpler alternative to carrier-type approaches.
D-mannose as an Adjuvant for Non-viral Gene Therapy
D-mannose is an inexpensive and commonly available dietary supplement (142) that has been assessed via phase III clinical trial for prevention of recurrent urinary tract infections (ClinicalTrials.gov Identifier: NCT01808755) (143). D-mannose is a known mannose receptor agonist. The mannose receptor (MR; CD206) is a c-type lectin receptor that functions in endocytosis and pathogen recognition (144). MR is expressed by macrophages, dendritic cells, and microvascular endothelial cells (145), as well as astrocytes, microglia, Schwann cells, and some neurons (146–148). MR is commonly used as a macrophage marker, though increased expression of MR is often associated with anti-inflammatory macrophages (149).
D-mannose was initially explored for use as an adjuvant due to the hypothesis that it may increase intracellular uptake of naked plasmid (87). Interestingly, D-mannose greatly increases gene therapeutic efficacy allowing for long-lasting pain relief following a single co-injection of D-mannose with plasmid DNA encoding the gene for IL-10 in neuropathic rats (87). Work by Dengler et al. also suggests that D-mannose exposure itself increases anti-inflammatory signaling, possibly through actions of endogenous IL-10. However, work by Vanderwall et al. identifies that D-mannose-mediated anti-inflammatory effects do not require IL-10, as intrathecal D-mannose alone induces transient pain relief in IL-10 knockout mice (28).
Historically MR was not considered to possess signaling ability due to the receptor's absence of a clear intracellular signaling motif. Further, if signaling was identified, it was reported as pro-inflammatory in nature. However, many of these studies were performed under inflammatory states which likely predispose MR toward pro-inflammatory signaling pathways (150–152). New evidence supports that MR activation is also capable of increasing anti-inflammatory cytokines under the right conditions (144, 153). For instance, treatment of dendritic cells with “activating” anti-MR monoclonal antibodies potently increases endogenous IL-10 and IL-1ra while decreasing IL-1β and TNF-α production (154). Furthermore, D-mannose treatment prior to LPS stimulation of RAW 264.7 mouse macrophages reduces release of pro-inflammatory TNF-α, IL-1β, and NO, but increases IL-10 secretion (87).
While D-mannose was initially explored as an adjuvant for gene therapy, its modulation of pain-relevant cytokine signaling pathways make it a novel pain therapeutic in its own right. D-mannose and MR-mediated anti-inflammatory signaling may be clinically relevant for the treatment of a variety of chronic pain conditions due to its engagement of both IL-10-independent and IL-10-mediated anti-inflammation (28, 87). These characteristics may make D-mannose useful for pain relief in patients with known IL-10 gene polymorphisms that would prevent normal IL-10 production in response to other therapeutic approaches. It may also be beneficial for treatment of pain pathologies associated with decreased IL-10 expression, like fibromyalgia (114, 155).
Harnessing Endogenous Anti-Inflammatory Capacity for Treatment of Chronic Pain
While gene therapy offers many advantages in for treatment of chronic pain, it may not be appropriate for all patients. Therapeutics must balance between homeostasis vs. immunocompromise. For example, excessive or mistimed IL-10 secretion can inhibit a sufficient pro-inflammatory response to pathogens like Leishmania spp. (leishmaniasis), Plasmodium spp. (malaria), T. cruzi (Chagas disease), Mycobacterium spp. (tuberculosis), and lymphocytic choriomeningitis virus (lymphocytic choriomeningitis), effectively enabling the pathogens to escape immune control (156). This can result in fatal or chronic infections.
Whereas gene therapeutics are designed to be expressed independently of host transcriptional regulation, therapeutics that engage expression of endogenous anti-inflammatory functions may instead allow for dampened pro-inflammatory signaling without detriment to the body's immune-activating homeostatic mechanisms. A number of therapeutics exist that support the intriguing approach of harnessing the cell's anti-inflammatory processes. We have chosen only a subset based on recent preclinical evidence of altering glial and immune cell proinflammatory activation and corresponding regulation of anti-inflammatory cytokine production. Several promising targets for treatment of chronic neuropathic pain through engagement of endogenous anti-inflammatory capacity are reviewed below.
Cannabinoid Receptors
The two best characterized cannabinoid receptors are CB1R and CB2R, both of which belong to the G-protein coupled receptor (GPCR) superfamily and couple to the inhibitory Gi/o and Gi proteins, respectively (157). CB1R was first discovered as the receptor for Δ9-tetrahydrocannabinol (THC), the major psychoactive ingredient in Cannabis sativa (158). Peripherally, CB1R is primarily expressed by neurons within the heart, small intestine, urinary bladder and vas deferens, while centrally CB1R is expressed at the highest concentrations by neurons in the cerebellum, hippocampus, basal ganglia and cerebral cortex (157, 159, 160). Clinical application of CB1R agonists are limited by development of tolerance, psychotropic effects (e.g., cognitive impairment, catalepsy, and negative impacts on learning and memory), and risk of hypothermia (157).
CB2R is abundantly expressed by peripheral immune cells (e.g., lymphocytes, neutrophils, and macrophages), and at lower levels within the CNS primarily on microglia, but has also been identified on some neurons (e.g., in the hippocampus, cortex, and substantia nigra) (159–163). CB2R signaling, while less well-understood, appears to primarily promote anti-inflammatory effects (164), and lack the unwanted psychotropic effects observed with CB1R activation (132). CB2R agonism has been associated with increased expression of IL-10 and decreased production of pro-inflammatory mediators like TNF-α, CCL2, and nitric oxide (164). These characteristics make CB2R a potential target for pain therapeutic development.
CB2R agonists reduce pathological pain with corresponding decreases in pro-inflammatory signaling in several animal pain models, including chronic constriction injury (132), paw incision (165), L5 spinal nerve transection (166), diabetic peripheral neuropathy (167), and chemotherapy induced neuropathic pain (168–171). The pre-clinical and clinical use of cannabinoid receptor agonists in pain has been well-reviewed elsewhere (157, 172–174).
A key CB2R ligand used in these studies, AM1710, does not cross the blood brain barrier and is 57-fold more selective for the CB2R than CB1R (175). Intrathecal AM1710 was shown to both reverse bilateral mechanical hypersensitivity in a model of peripheral nerve injury, and to prevent bilateral mechanical hypersensitivity in a model if spinal gp120 inflammatory pain. While IL-10 immunoreactivity was decreased in neuropathic animals, relief of mechanical sensitivity was associated with a return to basal levels of IL-10 expression (132). This supports that targeting the body's endogenous anti-inflammatory capabilities may relieve pathological pain through re-establishment of homeostatic conditions.
More recently, several orphan receptors, including the GPCRs GPR18, GPR55, and GPR119, have been identified as putative members of the cannabinoid receptor family, with growing evidence that GPR18 and GPR55 may be novel targets for therapeutic modulation of chronic pain (160). GPR18 is expressed in spleen, lymphocytes, neutrophils, macrophages, and lymph nodes (176–179), while GPR55 is expressed in tonsil, spleen, and by CNS neurons (160, 180, 181). The immune-relevant roles of these receptors is not well-understood, though there is some evidence to suggest that GPR55 may alleviate inflammatory pain through altered expression of IL-4, IL-10, and IFN-γ (180).
Lymphocyte Function-Associated Antigen-1
Lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18) is a β2-integrin expressed on myeloid cells, T cells, and microglia (182–184). LFA-1 is most widely understood to be important for the leukocyte–endothelial cell interaction, which involves leukocytic rolling, firm adhesion, transmigration, and sub-endothelial migration (185). In addition to the widely characterized roles of LFA-1 in cell adhesion and migration, evidence reveals that LFA-1 expression levels following immune challenge are inversely correlated with serum IL-10 expression in experimental mice (186). Reports demonstrate LFA-1 engagement with T cells results in T cell activation, T cell differentiation, and stabilization of T cell cytokine mRNA transcripts including TNF-α (186–189). For example, LFA-1 deficient mice are resistant to LPS-induced acute liver injury, and the mechanism appears to rely on increased IL-10 expression (186). In vitro pretreatment of RAW 264.7 macrophages with BIRT-377, an LFA-1 antagonist, prior to LPS stimulation results in a dose-dependent increase in IL-10 protein with a simultaneous decrease in IL-1β and TNF-α (190). Activation of LFA-1 beyond T cell interaction stimulates a variety of downstream signaling cascades in a context-dependent manner modified by the local cytokine milieu (191). In point of fact, these recent reports demonstrate in vivo pro-inflammatory cytokine contribution driven by activated LFA-1 on innate immune cells (e.g., macrophages) and T cells during chronic disease.
The dual actions of LFA-1 in cell migration and pro-inflammatory cytokine signaling make it a reasonable target for development of pain therapeutics. The role of LFA-1 in pathological pain was first explored in work by Noor et al. that revealed prenatal alcohol exposure (PAE) predisposes adult rats to develop neuropathic pain in response to a minor injury. They found that LFA-1 expression is increased on microglia and trafficking immune cells in the lumbar spinal cords and on peripheral immune cells of neuropathic PAE rats (192). It was later identified that intrathecal or intravenous administration of BIRT-377, a small molecule non-competitive LFA-1 antagonist, relieves allodynia in neuropathic rats, which was associated with decreased immunoreactivity for spinal markers of glial activation and IL-1β and concurrent increases in IL-10 immunoreactivity (193, 194). Additionally, BIRT-377 intravenous administration in female neuropathic rats reduces Th17 cytokines and elevates FOXP3, a T cell-specific transcription factor necessary for inducing the generation of T regulatory cells characterized by their secretion of IL-10 [(193); Noor et al., under review].
Given the potential for sexual dimorphism in pathological pain, it is noteworthy that intravenous administration of BIRT-377 relieves sciatic neuropathic pain in both male and female mice to a similar degree (54). Pain relief in neuropathic mice of both sexes was concurrent with reductions in pro-inflammatory IL-1β and TNF-α and increases in anti-inflammatory IL-10 and TGF-β1. Interestingly, female-derived T cell cytokines were found to be transcriptionally regulated by LFA-1 antagonism, as demonstrated by reduced pro-inflammatory IL-17A production and increases in IL-10, TGF-β, as well as FOXP3 (54).
The Potential Role of TLR4 Underlying Opioid-Induced Hyperalgesia
A common and unfortunate clinical side effect of chronic opioid use is the development of a paradoxical increase in pain sensitivity, termed opioid-induced hyperalgesia (195–201). Studies demonstrate in both humans and animal models that a number of key factors affect the development of opioid-induced hyperalgesia including anatomical projections from supra-spinal regions onto spinal pain projection neurons (an axonal route where pain facilitation can occur), variants in the mu-opioid receptor, sex differences, prior experience with pain, and even local activation of microglia and astrocytes (201). Given this work's emphasis on glial and immune factors, a brief review of the literature that supports opioid-induced hyperalgesia resulting from the production of pro-inflammatory immune mediators (e.g., cytokines, chemokines, and NO) that produce hyperalgesia (202) is provided below.
TLR4 actions may play a role in opioid-induced hyperalgesia. For instance, TLR4 signaling blockade attenuates opioid-induced hypersensitivity due to morphine or morphine metabolites (e.g., M3G) (203–208). Further, inhibition of glial activation (e.g., with pharmacologic glial inhibitors like minocycline and ibudilast, or by utilization of a microglial targeted Designer Receptor Exclusively Activated by a Designer Drug [DREADD] system) or direct inhibition of specific pro-inflammatory mediators (e.g., TNF-α, IL-1β, IL-6, CX3CL1, NOS, and p38 MAPK) after repeated morphine attenuates opioid-induced hyperalgesia (207–212).
Not only do opioids induce hyperalgesia, recent work by Grace et al., implicates that repeated opioid exposure prolongs neuropathic pain symptoms. They showed that morphine administered to neuropathic rats leads to persistent nociceptive sensitization through induction of spinal microglial NLRP3 inflammasome via activation of TLR4. This sensitization can be prevented or reversed. Naloxone is famous for its application as a rescue agent in cases of opioid overdose. While (–)-naloxone has antagonistic actions at both the μ-opioid receptor and TLR4, the (+)-naloxone isomer retains only the ability to block TLR4 signaling due to (–)-naloxone stereoselectivity at the μ-opioid receptor (213, 214). TLR4-mediated signaling has been repeatedly implicated in opioid tolerance and opioid-induced hyperalgesia (203–208, 215). These findings indicate that use of (+)-naloxone as an adjuvant to opioid therapy may enable opioid-analgesia while limiting the negative effects caused by TLR4-mediated pro-inflammatory signaling that would ordinarily result in opioid tolerance and opioid-induced hyperalgesia.
Finally, given the growing literature that supports pro-inflammatory cytokine actions in the development of opioid-induced hyperalgesia, gene therapeutics that aim to decrease such pro-inflammatory factors or increase anti-inflammatory cytokines may offer a novel tool for both the study and treatment of this unwanted side effect.
Concluding Remarks
The worldwide opioid crisis has brought public awareness to the concept that clinically available therapeutics for pain management are not adequate and come with significant health risks. To address this, new approaches must be developed, but this cannot occur without a better understanding of the underlying mechanisms of various chronic pain pathologies. There is now a greater appreciation for actions by non-neuronal support and immune cells, including astrocytes, microglia, and leukocytes that mediate the development and maintenance of chronic pain. Here we have described what is known about the methods of immunological communication between neurons, glia, and immune cells leading to pathological pain, with a focus on the role of pro- and anti-inflammatory cytokines. We also offer potential targets for development of novel non-opioid pain therapeutics that aim to relieve pathological pain by augmenting the body's own anti-inflammatory potential. Future studies will continue to benefit from exploring the roles of neuro-immune interactions and allow for improved patient outcomes.
Author Contributions
AV and EM contributed to all aspects of the manuscript.
Funding
This work was supported by National Institute of Alcohol Abuse and Alcoholism R21-AA023051: Exploration of neural immune mechanisms, pain, and prenatal alcohol exposure. National Institute of Alcohol Abuse and Alcoholism R01-AA025967: Prenatal alcohol exposure potentiates pain via lifelong spinal immune changes, National Institute of Alcohol Abuse and Alcoholism T32-AA014127: Graduate student training grant in prenatal alcohol exposure, National Institute of Alcohol Abuse and Alcoholism P50-AA022534: Fetal ethanol-induced behavioral deficits: mechanisms diagnosis and intervention, and National Institute of Drug Abuse R01-DA018156: Spinal neuroimmune mechanisms underlying interleukin-10 gene therapy for pain control.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, et al. Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the international classification of diseases (ICD-11). Pain. (2019) 160:19–27. doi: 10.1097/j.pain.0000000000001384
2. Yaqub F. Pain in the USA: states of suffering. Lancet. (2015) 386:839. doi: 10.1016/S0140-6736(15)00041-0
3. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1545–602. doi: 10.1016/S0140-6736(16)31678-6
4. Med I. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academy of Sciences (2011). p. 1–364.
5. Lembke A, Humphreys K, Newmark J. Weighing the risks and benefits of chronic opioid therapy. Am Fam Physician. (2016) 93:982–90. doi: 10.1001/jamapsychiatry.2016.1390
6. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. (2009) 139:267–84. doi: 10.1016/j.cell.2009.09.028
7. Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. (2007) 21:131–46. doi: 10.1016/j.bbi.2006.10.011
8. Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. (2009) 10:23–36. doi: 10.1038/nrn2533
9. Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. (2014) 14:217–31. doi: 10.1038/nri3621
10. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
11. Totsch SK, Sorge RE. Immune system involvement in specific pain conditions. Mol Pain. (2017) 13:1744806917724559. doi: 10.1177/1744806917724559
12. Chavan SS, Pavlov VA, Tracey KJ. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity. (2017) 46:927–42. doi: 10.1016/j.immuni.2017.06.008
13. Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med. (2010) 16:592–7, 591p following 597. doi: 10.1038/nm.2123
14. Ji R-R, Xu Z-Z, Gao Y-J. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. (2014) 13:533. doi: 10.1038/nrd4334
15. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. (2007) 10:1361–8. doi: 10.1038/nn1992
16. Crosson T, Roversi K, Balood M, Othman R, Ahmadi M, Wang JC, et al. Profiling of how nociceptor neurons detect danger–new and old foes. J Intern Med. (2019) 286:268–89. doi: 10.1111/joim.12957
17. Littlejohn G. Neurogenic neuroinflammation in fibromyalgia and complex regional pain syndrome. Nat Rev Rheumatol. (2015) 11:639. doi: 10.1038/nrrheum.2015.100
18. Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. (1983) 306:686–8. doi: 10.1038/306686a0
19. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. (2009) 10:895–926. doi: 10.1016/j.jpain.2009.06.012
20. Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. (2010) 468:214–22. doi: 10.1038/nature09611
21. Lee JC, Mayer-Proschel M, Rao MS. Gliogenesis in the central nervous system. Glia. (2000) 30:105–21. doi: 10.1002/(SICI)1098-1136(200004)30:2<105::AID-GLIA1>3.0.CO;2-H
22. Watkins LR, Maier SF. Immune regulation of central nervous system functions: from sickness responses to pathological pain. J Intern Med. (2005) 257:139–55. doi: 10.1111/j.1365-2796.2004.01443.x
23. Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new players in pain. Pain. (2001) 93:201–5. doi: 10.1016/S0304-3959(01)00359-1
24. Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. (2008) 135:37–47. doi: 10.1016/j.pain.2007.05.002
25. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
26. Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology. (2016) 36:39–49. doi: 10.1111/neup.12235
27. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of ‘homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
28. Vanderwall AG, Noor S, Sun MS, Sanchez JE, Yang XO, Jantzie LL, et al. Effects of spinal non-viral interleukin-10 gene therapy formulated with d-mannose in neuropathic interleukin-10 deficient mice: behavioral characterization, mRNA and protein analysis in pain relevant tissues. Brain Behav Immun. (2018) 69:91–112. doi: 10.1016/j.bbi.2017.11.004
29. Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. (1991) 565:1–7. doi: 10.1016/0006-8993(91)91729-K
30. De Leo JA, Tawfik VL, Lacroix-Fralish ML. The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain. (2006) 122:17–21. doi: 10.1016/j.pain.2006.02.034
31. Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. (2016) 354:572–7. doi: 10.1126/science.aaf8924
32. Li T, Chen X, Zhang C, Zhang Y, Yao W. An update on reactive astrocytes in chronic pain. J Neuroinflammation. (2019) 16:140. doi: 10.1186/s12974-019-1524-2
33. Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci. (2017) 74:3275–91. doi: 10.1007/s00018-017-2513-1
34. Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. (2007) 21:642–51. doi: 10.1016/j.bbi.2006.11.003
35. Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. (2016) 19:94–101. doi: 10.1038/nn.4189
36. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. (2003) 424:778. doi: 10.1038/nature01786
37. Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nat Neurosci. (2012) 15:1068–73. doi: 10.1038/nn.3155
38. Chen G, Zhang Y-Q, Qadri YJ, Serhan CN, Ji R-R. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. (2018) 100:1292–311. doi: 10.1016/j.neuron.2018.11.009
39. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. (2008) 28:5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008
40. Hu P, Bembrick AL, Keay KA, Mclachlan EM. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav Immun. (2007) 21:599–616. doi: 10.1016/j.bbi.2006.10.013
41. Raoof R, Willemen HLDM, Eijkelkamp N. Divergent roles of immune cells and their mediators in pain. Rheumatology. (2017) 57:429–40. doi: 10.1093/rheumatology/kex308
42. Kwon MJ, Shin HY, Cui Y, Kim H, Thi AHL, Choi JY, et al. CCL2 mediates neuron–macrophage interactions to drive proregenerative macrophage activation following preconditioning injury. J Neurosci. (2015) 35:15934–47. doi: 10.1523/JNEUROSCI.1924-15.2015
43. Zhang H, Li Y, De Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, et al. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain. (2016) 17:775–86. doi: 10.1016/j.jpain.2016.02.011
44. Schreiber RC, Krivacic K, Kirby B, Vaccariello SA, Wei T, Ransohoff RM, et al. Monocyte chemoattractant protein (MCP)-1 is rapidly expressed by sympathetic ganglion neurons following axonal injury. Neuroreport. (2001) 12:601–6. doi: 10.1097/00001756-200103050-00034
45. Huang ZZ, Li D, Liu CC, Cui Y, Zhu HQ, Zhang WW, et al. CX3CL1-mediated macrophage activation contributed to paclitaxel-induced DRG neuronal apoptosis and painful peripheral neuropathy. Brain Behav Immun. (2014) 40:155–65. doi: 10.1016/j.bbi.2014.03.014
46. Barclay J, Clark AK, Ganju P, Gentry C, Patel S, Wotherspoon G, et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. (2007) 130:225–34. doi: 10.1016/j.pain.2006.11.017
47. Cao L, Deleo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. (2008) 38:448–58. doi: 10.1002/eji.200737485
48. Cao L, Beaulac H, Eurich A. Differential lumbar spinal cord responses among wild type, CD4 knockout, and CD40 knockout mice in spinal nerve L5 transection-induced neuropathic pain. Mol Pain. (2012) 8:88. doi: 10.1186/1744-8069-8-88
49. Draleau K, Maddula S, Slaiby A, Nutile-Mcmenemy N, De Leo J, Cao L. Phenotypic identification of spinal cord-infiltrating CD4(+) T lymphocytes in a murine model of neuropathic pain. J Pain Relief Suppl. (2014) 3:003. doi: 10.4172/2167-0846.S3-003
50. Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. (2009) 29:14415–22. doi: 10.1523/JNEUROSCI.4569-09.2009
51. Sun C, Zhang J, Chen L, Liu T, Xu G, Li C, et al. IL-17 contributed to the neuropathic pain following peripheral nerve injury by promoting astrocyte proliferation and secretion of proinflammatory cytokines. Mol Med Rep. (2017) 15:89–96. doi: 10.3892/mmr.2016.6018
52. Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain. (2011) 12:370–83. doi: 10.1016/j.jpain.2010.08.003
53. Lopes DM, Malek N, Edye M, Jager SB, Mcmurray S, Mcmahon SB, et al. Sex differences in peripheral not central immune responses to pain-inducing injury. Sci Rep. (2017) 7:16460. doi: 10.1038/s41598-017-16664-z
54. Noor S, Sun MS, Vanderwall AG, Havard MA, Sanchez JE, Harris NW, et al. LFA-1 antagonist (BIRT377) similarly reverses peripheral neuropathic pain in male and female mice with underlying sex divergent peripheral immune proinflammatory phenotypes. Neuroimmunol Neuroinflamm. (2019) 6:10. doi: 10.20517/2347-8659.2019.18
55. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. (2018) 9:847. doi: 10.3389/fimmu.2018.00847
56. Mapplebeck JC, Beggs S, Salter MW. Molecules in pain and sex: a developing story. Mol Brain. (2017) 10:9. doi: 10.1186/s13041-017-0289-8
57. Rosen S, Ham B, Mogil JS. Sex differences in neuroimmunity and pain. J Neurosci Res. (2017) 95:500–8. doi: 10.1002/jnr.23831
58. Sorge RE, Lacroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin JS, Ritchie J, et al. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. (2011) 31:15450–4. doi: 10.1523/JNEUROSCI.3859-11.2011
59. Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. (2015) 18:1081–3. doi: 10.1038/nn.4053
60. Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, et al. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: sex-dependent microglial signaling in the spinal cord. Brain Behav Immun. (2016) 55:70–81. doi: 10.1016/j.bbi.2015.10.006
61. Mapplebeck JCS, Dalgarno R, Tu Y, Moriarty O, Beggs S, Kwok CHT, et al. Microglial P2X4R-evoked pain hypersensitivity is sexually dimorphic in rats. Pain. (2018) 159:1752–63. doi: 10.1097/j.pain.0000000000001265
62. Stephens KE, Zhou W, Ji Z, Chen Z, He S, Ji H, et al. Sex differences in gene regulation in the dorsal root ganglion after nerve injury. BMC Genomics. (2019) 20:147. doi: 10.1186/s12864-019-5512-9
63. Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. (2018) 359:eaan4672. doi: 10.1126/science.aan4672
64. Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) 377:1723–32. doi: 10.1056/NEJMoa1702752
65. Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) 378:625–35. doi: 10.1056/NEJMoa1710504
66. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
67. Russell S, Bennett J, Wellman JA, Chung DC, Yu Z-F, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. (2017) 390:849–60. doi: 10.1016/S0140-6736(17)31868-8
68. Wood MJA, Charlton HM, Wood KJ, Kajiwara K, Byrnes AP. Immune responses to adenovirus vectors in the nervous system. Trends Neurosci. (1996) 19:497–501. doi: 10.1016/S0166-2236(96)10060-6
69. Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet. (2005) 6:299–310. doi: 10.1038/nrg1577
70. Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. (2011) 11:321–30. doi: 10.2174/156652311796150354
71. Jayant RD, Sosa D, Kaushik A, Atluri V, Vashist A, Tomitaka A, et al. Current status of non-viral gene therapy for CNS disorders. Expert Opin Drug Deliv. (2016) 13:1433–45. doi: 10.1080/17425247.2016.1188802
72. Slivac I, Guay D, Mangion M, Champeil J, Gaillet B. Non-viral nucleic acid delivery methods. Expert Opin Biol Ther. (2017) 17:105–18. doi: 10.1080/14712598.2017.1248941
73. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. (2014) 15:541. doi: 10.1038/nrg3763
74. Hao S, Mata M, Glorioso JC, Fink DJ. HSV-mediated expression of interleukin-4 in dorsal root ganglion neurons reduces neuropathic pain. Mol Pain. (2006) 2:6. doi: 10.1186/1744-8069-2-6
75. Peng XM, Zhou ZG, Glorioso JC, Fink DJ, Mata M. Tumor necrosis factor-alpha contributes to below-level neuropathic pain after spinal cord injury. Ann Neurol. (2006) 59:843–51. doi: 10.1002/ana.20855
76. Hao S, Mata M, Glorioso JC, Fink DJ. Gene transfer to interfere with TNFα signaling in neuropathic pain. Gene Ther. (2007) 14:1010. doi: 10.1038/sj.gt.3302950
77. Huang W, Zheng W, Ouyang H, Yi H, Liu S, Zeng W, et al. Mechanical allodynia induced by nucleoside reverse transcriptase inhibitor is suppressed by p55TNFSR mediated by herpes simplex virus vector through the SDF1alpha/CXCR4 system in rats. Anesth Analg. (2014) 118:671–80. doi: 10.1213/ANE.0000000000000079
78. Ortmann KLM, Chattopadhyay M. Decrease in neuroimmune activation by HSV-mediated gene transfer of TNFα soluble receptor alleviates pain in rats with diabetic neuropathy. Brain Behav Immun. (2014) 41:144–51. doi: 10.1016/j.bbi.2014.05.009
79. Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O'connor K, et al. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci. (2005) 21:2136–48. doi: 10.1111/j.1460-9568.2005.04057.x
80. Lau D, Harte SE, Morrow TJ, Wang S, Mata M, Fink DJ. Herpes simplex virus vector-mediated expression of interleukin-10 reduces below-level central neuropathic pain after spinal cord injury. Neurorehabil Neural Repair. (2012) 26:889–97. doi: 10.1177/1545968312445637
81. Zheng W, Huang W, Liu S, Levitt RC, Candiotti KA, Lubarsky DA, et al. IL-10 mediated by herpes simplex virus vector reduces neuropathic pain induced by HIV gp120 combined with ddC in rats. Mol Pain. (2014) 10:49. doi: 10.1186/1744-8069-10-49
82. Zheng W, Huang W, Liu S, Levitt RC, Candiotti KA, Lubarsky DA, et al. Interleukin 10 mediated by herpes simplex virus vectors suppresses neuropathic pain induced by human immunodeficiency virus gp120 in rats. Anesth Analg. (2014) 119:693–701. doi: 10.1213/ANE.0000000000000311
83. Milligan ED, Sloane EM, Langer SJ, Hughes TS, Jekich BM, Frank MG, et al. Repeated intrathecal injections of plasmid DNA encoding interleukin-10 produce prolonged reversal of neuropathic pain. Pain. (2006) 126:294–308. doi: 10.1016/j.pain.2006.07.009
84. Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. (2007) 21:686–98. doi: 10.1016/j.bbi.2006.10.012
85. Sloane E, Langer S, Jekich B, Mahoney J, Hughes T, Frank M, et al. Immunological priming potentiates non-viral anti-inflammatory gene therapy treatment of neuropathic pain. Gene Ther. (2009) 16:1210–22. doi: 10.1038/gt.2009.79
86. Soderquist RG, Sloane EM, Loram LC, Harrison JA, Dengler EC, Johnson SM, et al. Release of plasmid DNA-encoding IL-10 from PLGA microparticles facilitates long-term reversal of neuropathic pain following a single intrathecal administration. Pharm Res. (2010) 27:841–54. doi: 10.1007/s11095-010-0077-y
87. Dengler EC, Alberti LA, Bowman BN, Kerwin AA, Wilkerson JL, Moezzi DR, et al. Improvement of spinal non-viral IL-10 gene delivery by D-mannose as a transgene adjuvant to control chronic neuropathic pain. J Neuroinflammation. (2014) 11:92. doi: 10.1186/1742-2094-11-92
88. Busch-Dienstfertig M, González-Rodríguez S. IL-4, JAK-STAT signaling, and pain. JAK-STAT. (2013) 2:e27638. doi: 10.4161/jkst.27638
89. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. (2012) 189:4213–9. doi: 10.4049/jimmunol.1202246
90. Andrews A-L, Holloway JW, Holgate ST, Davies DE. IL-4 receptor α is an important modulator of IL-4 and IL-13 receptor binding: implications for the development of therapeutic targets. J Immunol. (2006) 176:7456–61. doi: 10.4049/jimmunol.176.12.7456
91. Hwang I, Yang J, Hong S, Ju Lee E, Lee S-H, Fernandes-Alnemri T, et al. Non-transcriptional regulation of NLRP3 inflammasome signaling by IL-4. Immunol Cell Biol. (2015) 93:591–9. doi: 10.1038/icb.2014.125
92. Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J. (2015) 282:435–44. doi: 10.1111/febs.13133
93. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. (2019) 40:1035–52. doi: 10.1016/j.it.2019.09.005
94. Vannier E, Miller LC, Dinarello CA. Coordinated antiinflammatory effects of interleukin 4: interleukin 4 suppresses interleukin 1 production but up-regulates gene expression and synthesis of interleukin 1 receptor antagonist. Proc Natl Acad Sci USA. (1992) 89:4076–80. doi: 10.1073/pnas.89.9.4076
95. Hart PH, Vitti GF, Burgess DR, Whitty GA, Piccoli DS, Hamilton JA. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2. Proc Natl Acad Sci USA. (1989) 86:3803–7. doi: 10.1073/pnas.86.10.3803
96. Lee YW, Lee WH, Kim PH. Oxidative mechanisms of IL-4-induced IL-6 expression in vascular endothelium. Cytokine. (2010) 49:73–9. doi: 10.1016/j.cyto.2009.08.009
97. Pousset F, Cremona S, Dantzer R, Kelley K, Parnet P. Interleukin-4 and interleukin-10 regulate IL1-beta induced mouse primary astrocyte activation: a comparative study. Glia. (1999) 26:12–21. doi: 10.1002/(SICI)1098-1136(199903)26:1<12::AID-GLIA2>3.0.CO;2-S
98. Van Kampen C, Gauldie J, Collins SM. Proinflammatory properties of IL-4 in the intestinal microenvironment. Am J Physiol Gastrointest Liver Physiol. (2005) 288:G111–7. doi: 10.1152/ajpgi.00014.2004
99. Park KW, Baik HH, Jin BK. Interleukin-4-induced oxidative stress via microglial NADPH oxidase contributes to the death of hippocampal neurons in vivo. Curr Aging Sci. (2008) 1:192–201. doi: 10.2174/1874609810801030192
100. Uceyler N, Topuzoglu T, Schiesser P, Hahnenkamp S, Sommer C. IL-4 deficiency is associated with mechanical hypersensitivity in mice. PLoS ONE. (2011) 6:e28205. doi: 10.1371/journal.pone.0028205
101. Boyle DL, Nguyen KHY, Zhuang S, Shi Y, Mccormack JE, Chada S, et al. Intra-articular IL-4 gene therapy in arthritis: anti-inflammatory effect and enhanced Th2activity. Gene Ther. (1999) 6:1911–8. doi: 10.1038/sj.gt.3301049
102. Pearson CM, Wood FD. Studies of polyarthritis and other lesions induced in rats by injection of mycobacterial adjuvant. I. General clinical and pathologic characteristics and some modifying factors. Arthritis Rheum. (1959) 2:440–59. doi: 10.1002/1529-0131(195910)2:5<440::AID-ART1780020510>3.0.CO;2-N
103. Sedger LM, Mcdermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants–past, present and future. Cytokine Growth Factor Rev. (2014) 25:453–72. doi: 10.1016/j.cytogfr.2014.07.016
104. Zhang J-M, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. (2007) 45:27–37. doi: 10.1097/AIA.0b013e318034194e
105. Lindenlaub T, Teuteberg P, Hartung T, Sommer C. Effects of neutralizing antibodies to TNF-alpha on pain-related behavior and nerve regeneration in mice with chronic constriction injury. Brain Res. (2000) 866:15–22. doi: 10.1016/S0006-8993(00)02190-9
106. Sommer C, Lindenlaub T, Teuteberg P, Schafers M, Hartung T, Toyka KV. Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res. (2001) 913:86–9. doi: 10.1016/S0006-8993(01)02743-3
107. Sommer C, Schafers M, Marziniak M, Toyka KV. Etanercept reduces hyperalgesia in experimental painful neuropathy. J Peripher Nerv Syst. (2001) 6:67–72. doi: 10.1046/j.1529-8027.2001.01010.x
108. Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. (2005) 379:209–13. doi: 10.1016/j.neulet.2004.12.064
109. Hung AL, Lim M, Doshi TL. Targeting cytokines for treatment of neuropathic pain. Scand J Pain. (2017) 17:287–93. doi: 10.1016/j.sjpain.2017.08.002
110. Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. (2002) 22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002
111. Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. (2002) 22:3025–32. doi: 10.1523/JNEUROSCI.22-08-03025.2002
112. Figiel I. Pro-inflammatory cytokine TNF-alpha as a neuroprotective agent in the brain. Acta Neurobiol Exp (Wars). (2008) 68:526–34.
113. Leung L, Cahill CM. TNF-alpha and neuropathic pain–a review. J Neuroinflammation. (2010) 7:27. doi: 10.1186/1742-2094-7-27
114. Milligan ED, Penzkover KR, Soderquist RG, Mahoney MJ. Spinal interleukin-10 therapy to treat peripheral neuropathic pain. Neuromodulation. (2012) 15:520–6; discussion 526. doi: 10.1111/j.1525-1403.2012.00462.x
115. Gonzalez P, Burgaya F, Acarin L, Peluffo H, Castellano B, Gonzalez B. Interleukin-10 and interleukin-10 receptor-I are upregulated in glial cells after an excitotoxic injury to the postnatal rat brain. J Neuropathol Exp Neurol. (2009) 68:391–403. doi: 10.1097/NEN.0b013e31819dca30
116. Zhou Z, Peng X, Insolera R, Fink DJ, Mata M. Interleukin-10 provides direct trophic support to neurons. J Neurochem. (2009) 110:1617–27. doi: 10.1111/j.1471-4159.2009.06263.x
117. Chen H, Lin W, Zhang Y, Lin L, Chen J, Zeng Y, et al. IL-10 promotes neurite outgrowth and synapse formation in cultured cortical neurons after the oxygen-glucose deprivation via JAK1/STAT3 pathway. Sci Rep. (2016) 6:30459. doi: 10.1038/srep30459
118. Shen K-F, Zhu H-Q, Wei X-H, Wang J, Li Y-Y, Pang R-P, et al. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol. (2013) 247:466–75. doi: 10.1016/j.expneurol.2013.01.018
119. Moore KW, De Waal Malefyt R, Coffman RL, O'garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
120. Kwilasz AJ, Grace PM, Serbedzija P, Maier SF, Watkins LR. The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacology. (2015) 96:55–69. doi: 10.1016/j.neuropharm.2014.10.020
121. Kishore R, Tebo JM, Kolosov M, Hamilton TA. Cutting edge: clustered AU-rich elements are the target of IL-10-mediated mRNA destabilization in mouse macrophages. J Immunol. (1999) 162:2457–61.
122. Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. (2005) 102:8686–91. doi: 10.1073/pnas.0500419102
123. Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M. Balancing the immune response in the brain: IL-10 and its regulation. J Neuroinflammation. (2016) 13:297. doi: 10.1186/s12974-016-0763-8
124. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. (2007) 7:454–65. doi: 10.1038/nri2093
125. Tanga FY, Nutile-Mcmenemy N, Deleo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. (2005) 102:5856–61. doi: 10.1073/pnas.0501634102
126. Dagvadorj J, Naiki Y, Tumurkhuu G, Hassan F, Islam S, Koide N, et al. Interleukin-10 inhibits tumor necrosis factor-α production in lipopolysaccharide-stimulated RAW 264.7 cells through reduced MyD88 expression. Innate Immun. (2008) 14:109–15. doi: 10.1177/1753425908089618
127. Chang J, Kunkel SL, Chang C-H. Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci USA. (2009) 106:18327–32. doi: 10.1073/pnas.0905815106
128. Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci USA. (2013) 110:11499–504. doi: 10.1073/pnas.1219852110
129. Sabat R, Grutz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev. (2010) 21:331–44. doi: 10.1016/j.cytogfr.2010.09.002
130. Jancalek R, Dubovy P, Svizenska I, Klusakova I. Bilateral changes of TNF-alpha and IL-10 protein in the lumbar and cervical dorsal root ganglia following a unilateral chronic constriction injury of the sciatic nerve. J Neuroinflammation. (2010) 7:11. doi: 10.1186/1742-2094-7-11
131. Jancalek R, Svizenska I, Klusakova I, Dubovy P. Bilateral changes of IL-10 protein in lumbar and cervical dorsal root ganglia following proximal and distal chronic constriction injury of peripheral nerve. Neurosci Lett. (2011) 501:86–91. doi: 10.1016/j.neulet.2011.06.052
132. Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, et al. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. (2012) 153:1091–106. doi: 10.1016/j.pain.2012.02.015
133. Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Kuhn MN, et al. Immunofluorescent spectral analysis reveals the intrathecal cannabinoid agonist, AM1241, produces spinal anti-inflammatory cytokine responses in neuropathic rats exhibiting relief from allodynia. Brain Behav. (2012) 2:155–77. doi: 10.1002/brb3.44
134. Khan J, Ramadan K, Korczeniewska O, Anwer MM, Benoliel R, Eliav E. Interleukin-10 levels in rat models of nerve damage and neuropathic pain. Neurosci Lett. (2015) 592:99–106. doi: 10.1016/j.neulet.2015.03.001
135. Milligan ED, Sloane EM, Langer SJ, Cruz PE, Chacur M, Spataro L, et al. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol Pain. (2005) 1:9. doi: 10.1186/1744-8069-1-9
136. Beutler AS, Reinhardt M. AAV for pain: steps towards clinical translation. Gene Ther. (2009) 16:461. doi: 10.1038/gt.2009.23
137. Milligan ED, Soderquist RG, Malone SM, Mahoney JH, Hughes TS, Langer SJ, et al. Intrathecal polymer-based interleukin-10 gene delivery for neuropathic pain. Neuron Glia Biol. (2006) 2:293–308. doi: 10.1017/S1740925X07000488
138. Sloane E, Ledeboer A, Seibert W, Coats B, Van Strien M, Maier SF, et al. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental multiple sclerosis: MOG-EAE behavioral and anatomical symptom treatment with cytokine gene therapy. Brain Behav Immun. (2009) 23:92–100. doi: 10.1016/j.bbi.2008.09.004
139. Grace PM, Loram LC, Christianson JP, Strand KA, Flyer-Adams JG, Penzkover KR, et al. Behavioral assessment of neuropathic pain, fatigue, and anxiety in experimental autoimmune encephalomyelitis (EAE) and attenuation by interleukin-10 gene therapy. Brain Behav Immun. (2017) 59:49–54. doi: 10.1016/j.bbi.2016.05.012
140. Soderquist RG, Milligan ED, Harrison JA, Chavez RA, Johnson KW, Watkins LR, et al. PEGylation of interleukin-10 for the mitigation of enhanced pain states. J Biomed Mater Res A. (2010) 93:1169–79. doi: 10.1002/jbm.a.32611
141. Oliveira AV, Rosa Da Costa AM, Silva GA. Non-viral strategies for ocular gene delivery. Mater Sci Eng C Mater Biol Appl. (2017) 77:1275–89. doi: 10.1016/j.msec.2017.04.068
142. Hu X, Shi YN, Zhang P, Miao M, Zhang T, Jiang B. d-Mannose: properties, production, and applications: an overview. Compr Rev Food Sci Food Saf. (2016) 15:773–85. doi: 10.1111/1541-4337.12211
143. Porru D, Parmigiani A, Tinelli C, Barletta D, Choussos D, Franco CD, et al. Oral D-mannose in recurrent urinary tract infections in women: a pilot study. J Clin Urol. (2014) 7:208–13. doi: 10.1177/2051415813518332
144. Martinez-Pomares L. The mannose receptor. J Leukoc Biol. (2012) 92:1177–86. doi: 10.1189/jlb.0512231
145. Taylor PR, Gordon S, Martinez-Pomares L. The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol. (2005) 26:104–10. doi: 10.1016/j.it.2004.12.001
146. Burudi EM, Riese S, Stahl PD, Regnier-Vigouroux A. Identification and functional characterization of the mannose receptor in astrocytes. Glia. (1999) 25:44–55. doi: 10.1002/(SICI)1098-1136(19990101)25:1<44::AID-GLIA5>3.0.CO;2-C
147. Burudi EM, Regnier-Vigouroux A. Regional and cellular expression of the mannose receptor in the post-natal developing mouse brain. Cell Tissue Res. (2001) 303:307–17. doi: 10.1007/s004410000311
148. Baetas-Da-Cruz W, Alves L, Pessolani MC, Barbosa HS, Regnier-Vigouroux A, Corte-Real S, et al. Schwann cells express the macrophage mannose receptor and MHC class II. Do they have a role in antigen presentation? J Peripher Nerv Syst. (2009) 14:84–92. doi: 10.1111/j.1529-8027.2009.00217.x
149. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
150. Fernandez N, Alonso S, Valera I, Vigo AG, Renedo M, Barbolla L, et al. Mannose-containing molecular patterns are strong inducers of cyclooxygenase-2 expression and prostaglandin E2 production in human macrophages. J Immunol. (2005) 174:8154–62. doi: 10.4049/jimmunol.174.12.8154
151. Lopez-Herrera A, Liu Y, Rugeles MT, He JJ. HIV-1 interaction with human mannose receptor (hMR) induces production of matrix metalloproteinase 2 (MMP-2) through hMR-mediated intracellular signaling in astrocytes. Biochim Biophys Acta. (2005) 1741:55–64. doi: 10.1016/j.bbadis.2004.12.001
152. Tachado SD, Zhang J, Zhu J, Patel N, Cushion M, Koziel H. Pneumocystis-mediated IL-8 release by macrophages requires coexpression of mannose receptors and TLR2. J Leukoc Biol. (2007) 81:205–11. doi: 10.1189/jlb.1005580
153. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
154. Chieppa M, Bianchi G, Doni A, Del Prete A, Sironi M, Laskarin G, et al. Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J Immunol. (2003) 171:4552–60. doi: 10.4049/jimmunol.171.9.4552
155. Üçeyler N, Valenza R, Stock M, Schedel R, Sprotte G, Sommer C. Reduced levels of antiinflammatory cytokines in patients with chronic widespread pain. Arthritis Rheum. (2006) 54:2656–64. doi: 10.1002/art.22026
156. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. (2008) 180:5771–7. doi: 10.4049/jimmunol.180.9.5771
157. Wilkerson JL, Milligan ED. The central role of glia in pathological pain and the potential of targeting the cannabinoid 2 receptor for pain relief. ISRN Anesthesiol. (2011) 2011:19. doi: 10.5402/2011/593894
158. Mechoulam R, Gaoni Y. A total synthesis of dl-Δ1-tetrahydrocannabinol, the active constituent of Hashish. J Am Chem Soc. (1965) 87:3273–5. doi: 10.1021/ja01092a065
159. Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures–a short review. Pharmacol Biochem Behav. (2008) 90:501–11. doi: 10.1016/j.pbb.2008.05.010
160. Guerrero-Alba R, Barragán-Iglesias P, González-Hernández A, Valdez-Moráles EE, Granados-Soto V, Condés-Lara M, et al. Some prospective alternatives for treating pain: the endocannabinoid system and its putative receptors GPR18 and GPR55. Front Pharmacol. (2019) 9:1496. doi: 10.3389/fphar.2018.01496
161. Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. (1995) 232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x
162. Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. (2003) 23:1398–405. doi: 10.1523/JNEUROSCI.23-04-01398.2003
163. Stempel AV, Stumpf A, Zhang H-Y, Özdogan T, Pannasch U, Theis A-K, et al. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron. (2016) 90:795–809. doi: 10.1016/j.neuron.2016.03.034
164. Turcotte C, Blanchet M-R, Laviolette M, Flamand N. The CB(2) receptor and its role as a regulator of inflammation. Cell Mol Life Sci. (2016) 73:4449–70. doi: 10.1007/s00018-016-2300-4
165. Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. (2007) 106:787–94. doi: 10.1097/01.anes.0000264765.33673.6c
166. Romero-Sandoval A, Nutile-Mcmenemy N, Deleo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. (2008) 108:722–34. doi: 10.1097/ALN.0b013e318167af74
167. Toth CC, Jedrzejewski NM, Ellis CL, Frey, WHII. Cannabinoid-mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol Pain. (2010) 6:16. doi: 10.1186/1744-8069-6-16
168. Rahn EJ, Makriyannis A, Hohmann AG. Activation of cannabinoid CB1 and CB2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. Br J Pharmacol. (2007) 152:765–77. doi: 10.1038/sj.bjp.0707333
169. Rahn EJ, Zvonok AM, Thakur GA, Khanolkar AD, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB2 receptors suppresses neuropathic nociception induced by treatment with the chemotherapeutic agent paclitaxel in rats. J Pharmacol Exp Ther. (2008) 327:584–91. doi: 10.1124/jpet.108.141994
170. Deng L, Guindon J, Vemuri VK, Thakur GA, White FA, Makriyannis A, et al. The maintenance of cisplatin- and paclitaxel-induced mechanical and cold allodynia is suppressed by cannabinoid CB2 receptor activation and independent of CXCR4 signaling in models of chemotherapy-induced peripheral neuropathy. Mol Pain. (2012) 8:71. doi: 10.1186/1744-8069-8-71
171. Rahn EJ, Deng L, Thakur GA, Vemuri K, Zvonok AM, Lai YY, et al. Prophylactic cannabinoid administration blocks the development of paclitaxel-induced neuropathic nociception during analgesic treatment and following cessation of drug delivery. Mol Pain. (2014) 10:27–27. doi: 10.1186/1744-8069-10-27
172. Rahn EJ, Hohmann AG. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. (2009) 6:713–37. doi: 10.1016/j.nurt.2009.08.002
173. Davis MP. Cannabinoids in pain management: CB1, CB2 and non-classic receptor ligands. Expert Opin Investig Drugs. (2014) 23:1123–40. doi: 10.1517/13543784.2014.918603
174. Donvito G, Nass SR, Wilkerson JL, Curry ZA, Schurman LD, Kinsey SG, et al. The endogenous cannabinoid system: a budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacology. (2018) 43:52–79. doi: 10.1038/npp.2017.204
175. Rahn EJ, Thakur GA, Wood JAT, Zvonok AM, Makriyannis A, Hohmann AG. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: antinociception without central nervous system side-effects. Pharmacol Biochem Behav. (2011) 98:493–502. doi: 10.1016/j.pbb.2011.02.024
176. Gantz I, Muraoka A, Yang YK, Samuelson LC, Zimmerman EM, Cook H, et al. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics. (1997) 42:462–6. doi: 10.1006/geno.1997.4752
177. Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, et al. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci USA. (2003) 100:4903–8. doi: 10.1073/pnas.0230374100
178. Burstein SH, Mcquain CA, Ross AH, Salmonsen RA, Zurier RE. Resolution of inflammation by N-arachidonoylglycine. J Cell Biochem. (2011) 112:3227–33. doi: 10.1002/jcb.23245
179. Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med. (2015) 212:1203–17. doi: 10.1084/jem.20150225
180. Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. (2008) 139:225–36. doi: 10.1016/j.pain.2008.04.006
181. Marichal-Cancino BA, Fajardo-Valdez A, Ruiz-Contreras AE, Mendez-Díaz M, Prospero-García O. Advances in the physiology of GPR55 in the central nervous system. Curr Neuropharmacol. (2017) 15:771–8. doi: 10.2174/1570159X14666160729155441
182. Akiyama H, Mcgeer PL. Brain microglia constitutively express beta-2 integrins. J Neuroimmunol. (1990) 30:81–93. doi: 10.1016/0165-5728(90)90055-R
183. Moneta ME, Gehrmann J, Topper R, Banati RB, Kreutzberg GW. Cell adhesion molecule expression in the regenerating rat facial nucleus. J Neuroimmunol. (1993) 45:203–6. doi: 10.1016/0165-5728(93)90181-W
184. Evans R, Patzak I, Svensson L, De Filippo K, Jones K, Mcdowall A, et al. Integrins in immunity. J Cell Sci. (2009) 122:215–25. doi: 10.1242/jcs.019117
185. Evans BJ, Mcdowall A, Taylor PC, Hogg N, Haskard DO, Landis RC. Shedding of lymphocyte function–associated antigen-1 (LFA-1) in a human inflammatory response. Blood. (2006) 107:3593–9. doi: 10.1182/blood-2005-09-3695
186. Emoto M, Emoto Y, Brinkmann V, Miyamoto M, Yoshizawa I, Stäber M, et al. Increased resistance of LFA-1-deficient mice to lipopolysaccharide-induced shock/liver injury in the presence of TNF-α and IL-12 is mediated by IL-10: a novel role for LFA-1 in the regulation of the proinflammatory and anti-inflammatory cytokine balance. J Immunol. (2003) 171:584–93. doi: 10.4049/jimmunol.171.2.584
187. Wang JG, Collinge M, Ramgolam V, Ayalon O, Fan XC, Pardi R, et al. LFA-1-Dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J Immunol. (2006) 176:2105–13. doi: 10.4049/jimmunol.176.4.2105
188. Suzuki J-I, Yamasaki S, Wu J, Koretzky GA, Saito T. The actin cloud induced by LFA-1–mediated outside-in signals lowers the threshold for T-cell activation. Blood. (2007) 109:168–75. doi: 10.1182/blood-2005-12-020164
189. Wang Y, Kai H, Chang F, Shibata K, Tahara-Hanaoka S, Honda S-I, et al. A critical role of LFA-1 in the development of Th17 cells and induction of experimental autoimmune encephalomyelytis. Biochem Biophys Res Commun. (2007) 353:857–62. doi: 10.1016/j.bbrc.2006.12.104
190. Lam N, Ornatowski W, Alberti LB, Wilkerson JL, Moezzi D, Sun MS, et al. The role of leukocyte accumulation and diminished spinal IL-10 expression in chronic neuropathy. Program No. 166.12. In: 2013 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience (2013).
191. Verma NK, Kelleher D. Not just an adhesion molecule: LFA-1 contact tunes the T lymphocyte program. J Immunol. (2017) 199:1213–21. doi: 10.4049/jimmunol.1700495
192. Noor S, Sanchez JJ, Vanderwall AG, Sun MS, Maxwell JR, Davies S, et al. Prenatal alcohol exposure potentiates chronic neuropathic pain, spinal glial and immune cell activation and alters sciatic nerve and DRG cytokine levels. Brain Behav Immun. (2017) 61:80–95. doi: 10.1016/j.bbi.2016.12.016
193. Noor S, Sanchez JJ, Pervin Z, Sanchez JE, Sun MS, Epler L, et al. Neuropathic pain susceptibility in prenatal alcohol exposed (PAE) females is mediated by the proinflammatory actions of lymphocyte function-associated antigen (LFA)-1 on immune and glial cells. Program No. 666.18. In: 2018 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience (2018).
194. Sanchez JJ, Sanchez JE, Noor S, Ruffaner-Hanson CD, Davies S, Wagner CR, et al. Targeting the β2-integrin LFA-1, reduces adverse neuroimmune actions in neuropathic susceptibility caused by prenatal alcohol exposure. Acta Neuropathol Commun. (2019) 7:54. doi: 10.1186/s40478-019-0701-y
195. Doverty M, White JM, Somogyi AA, Bochner F, Ali R, Ling W. Hyperalgesic responses in methadone maintenance patients. Pain. (2001) 90:91–6. doi: 10.1016/S0304-3959(00)00391-2
196. Vanderah TW, Suenaga NMH, Ossipov MH, Malan TP, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. (2001) 21:279–86. doi: 10.1523/JNEUROSCI.21-01-00279.2001
197. Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. (2005) 80:319–24. doi: 10.1002/bip.20254
198. Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. (2006) 104:570–87. doi: 10.1097/00000542-200603000-00025
199. Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain. (2009) 10:316–22. doi: 10.1016/j.jpain.2008.10.003
200. Hay JL, Kaboutari J, White JM, Salem A, Irvine R. Model of methadone-induced hyperalgesia in rats and effect of memantine. Eur J Pharmacol. (2010) 626:229–33. doi: 10.1016/j.ejphar.2009.09.056
201. Roeckel L-A, Le Coz G-M, Gavériaux-Ruff C, Simonin F. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience. (2016) 338:160–82. doi: 10.1016/j.neuroscience.2016.06.029
202. Lacagnina MJ, Watkins LR, Grace PM. Toll-like receptors and their role in persistent pain. Pharmacol Ther. (2018) 184:145–58. doi: 10.1016/j.pharmthera.2017.10.006
203. Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. (2010) 24:83–95. doi: 10.1016/j.bbi.2009.08.004
204. Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, et al. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience. (2010) 165:569–83. doi: 10.1016/j.neuroscience.2009.10.011
205. Ferrini F, Trang T, Mattioli TAM, Laffray S, Del'guidice T, Lorenzo LE, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl? homeostasis. Nat Neurosci. (2013) 16:183–92. doi: 10.1038/nn.3295
206. Grace PM, Ramos KM, Rodgers KM, Wang X, Hutchinson MR, Lewis MT, et al. Activation of adult rat CNS endothelial cells by opioid-induced toll-like receptor 4 (TLR4) signaling induces proinflammatory, biochemical, morphological, and behavioral sequelae. Neuroscience. (2014) 280:299–317. doi: 10.1016/j.neuroscience.2014.09.020
207. Johnson JL, Rolan PE, Johnson ME, Bobrovskaya L, Williams DB, Johnson K, et al. Codeine-induced hyperalgesia and allodynia: investigating the role of glial activation. Transl Psychiatry. (2014) 4:e482. doi: 10.1038/tp.2014.121
208. Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, et al. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci USA. (2016) 113:E3441–50. doi: 10.1073/pnas.1602070113
209. Raghavendra V, Rutkowski MD, Deleo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. (2002) 22:9980–9. doi: 10.1523/JNEUROSCI.22-22-09980.2002
210. Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. (2004) 24:7353–65. doi: 10.1523/JNEUROSCI.1850-04.2004
211. Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. (2008) 22:1178–89. doi: 10.1016/j.bbi.2008.05.004
212. Tumati S, Largent-Milnes TM, Keresztes A, Ren J, Roeske WR, Vanderah TW, et al. Repeated morphine treatment-mediated hyperalgesia, allodynia and spinal glial activation are blocked by co-administration of a selective cannabinoid receptor type-2 agonist. J Neuroimmunol. (2012) 244:23–31. doi: 10.1016/j.jneuroim.2011.12.021
213. Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur J Neurosci. (2008) 28:20–9. doi: 10.1111/j.1460-9568.2008.06321.x
214. Lewis SS, Loram LC, Hutchinson MR, Li C-M, Zhang Y, Maier SF, et al. (+)-Naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. J Pain. (2012) 13:498–506. doi: 10.1016/j.jpain.2012.02.005
Keywords: cytokines, chemokines, gene therapy, chronic pain, IL-10, IL-4, TNF-α, CCL2
Citation: Vanderwall AG and Milligan ED (2019) Cytokines in Pain: Harnessing Endogenous Anti-Inflammatory Signaling for Improved Pain Management. Front. Immunol. 10:3009. doi: 10.3389/fimmu.2019.03009
Received: 25 September 2019; Accepted: 09 December 2019;
Published: 23 December 2019.
Edited by:
Waldiceu A. Verri, State University of Londrina, BrazilReviewed by:
Sebastien Talbot, Université de Montréal, CanadaNiels Eijkelkamp, University Medical Center Utrecht, Netherlands
Copyright © 2019 Vanderwall and Milligan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arden G. Vanderwall, avanderwall@salud.unm.edu; Erin D. Milligan, emilligan@salud.unm.edu