 Milene Costa da Silva1,2,3,4,5
Milene Costa da Silva1,2,3,4,5 Michael O. Breckwoldt6,7
Michael O. Breckwoldt6,7 Francesca Vinchi1,2
Francesca Vinchi1,2 Margareta P. Correia4
Margareta P. Correia4 Ana Stojanovic4
Ana Stojanovic4 Carl Maximilian Thielmann1,2Michael Meister5,8Thomas Muley5,8Arne Warth5,9
Carl Maximilian Thielmann1,2Michael Meister5,8Thomas Muley5,8Arne Warth5,9 Michael Platten6,7Matthias W. Hentze2
Michael Platten6,7Matthias W. Hentze2 Adelheid Cerwenka4,10†
Adelheid Cerwenka4,10† Martina U. Muckenthaler1,2,5*†
Martina U. Muckenthaler1,2,5*†
- 1Department of Pediatric Oncology, Hematology and Immunology, University of Heidelberg, Heidelberg, Germany
- 2Molecular Medicine Partnership Unit (MMPU), Heidelberg University, European Molecular Biology Laboratory (EMBL), Heidelberg, Germany
- 3Graduate Program in Areas of Basic and Applied Biology (GABBA), Abel Salazar Biomedical Sciences Institute (ICBAS), University of Porto, Porto, Portugal
- 4Innate Immunity Group, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 5Translational Lung Research Center Heidelberg (TLRC), German Center for Lung Research (DZL), University of Heidelberg, Heidelberg, Germany
- 6Department of Neuroradiology, University Hospital Heidelberg, Heidelberg, Germany
- 7German Cancer Consortium, Clinical Cooperation Unit Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 8Translational Research Unit, Thoraxklinik at University Hospital Heidelberg, Heidelberg, Germany
- 9Institute of Pathology, University of Heidelberg, Heidelberg, Germany
- 10Division of Immunbiochemistry, Medical Faculty Mannheim, Heidelberg University, Heidelberg, Germany
Tumor-associated macrophages (TAMs) frequently help to sustain tumor growth and mediate immune suppression in the tumor microenvironment (TME). Here, we identified a subset of iron-loaded, pro-inflammatory TAMs localized in hemorrhagic areas of the TME. The occurrence of iron-loaded TAMs (iTAMs) correlated with reduced tumor size in patients with non-small cell lung cancer. Ex vivo experiments established that TAMs exposed to hemolytic red blood cells (RBCs) were converted into pro-inflammatory macrophages capable of directly killing tumor cells. This anti-tumor effect could also be elicited via iron oxide nanoparticles. When tested in vivo, tumors injected with such iron oxide nanoparticles led to significantly smaller tumor sizes compared to controls. These results identify hemolytic RBCs and iron as novel players in the TME that repolarize TAMs to exert direct anti-tumor effector function. Thus, the delivery of iron to TAMs emerges as a simple adjuvant therapeutic strategy to promote anti-cancer immune responses.
Introduction
The tumor microenvironment (TME) significantly influences tumor progression (1). It is characterized by high cellular complexity, including fibroblasts, stroma, and blood vessels, and infiltrates of immune cells. In several human cancers, tumor-associated macrophages (TAMs) are a major immune component of the TME (2–4). In particular, non-small cell lung cancer (NSCLC) was shown to have one of the highest TAMs densities when compared to other cancers, such as liver, ovary, breast, and prostate cancer (5). In general, macrophages display a high degree of functional plasticity, reflected by their capacity to integrate diverse signals from the microenvironment and to acquire distinct phenotypes (6–11).
In the TME, pro-inflammatory “M1 macrophages” counteract tumor growth either by activating adaptive immune responses or by directly killing tumor cells (12–15). By contrast, anti-inflammatory “M2 macrophages” sustain tumor cell growth (16) by promoting angiogenesis, matrix remodeling, and immune suppression (17, 18). Undesirably, most TAMs display the M2-like phenotype, sustaining tumor growth rather than supporting tumor elimination. Thus, reprogramming macrophages in the TME could represent a promising therapeutic strategy to improve anti-tumor activity (19).
Besides contributing to immune responses, macrophages play a critical role in the recycling of iron from red blood cells (RBCs). Macrophages located in the spleen and the liver engulf senescent RBCs and catabolize heme via the activity of heme oxygenases (HO-1 and HO-2) (20). Iron is either stored in ferritin or exported via ferroportin, the only known iron exporter (21, 22).
Recently, we discovered that anti-inflammatory macrophages shift toward the pro-inflammatory state after exposure to heme or iron (23). Additional studies supported the concept that iron can drive macrophages toward a pro-inflammatory phenotype (24, 25). These findings interconnect the dual functions of macrophages in iron handling and inflammation.
M1 macrophages are hallmarked by the production of reactive oxygen species (ROS) and pro-inflammatory cytokines, such as interleukin (IL)-1α/β, IL-6, tumor necrosis factor alpha (TNFα), and also by expression of inducible nitric oxide synthase (iNOS), cluster of differentiation (CD)86, major histocompatibility complex II, and CD14 (26, 27). They retain iron as a result of high levels of ferritin and low ferroportin expression (28, 29). By contrast, M2 macrophages produce anti-inflammatory cytokines, such as IL-10 and transforming growth factor beta (TGFβ), and are hallmarked by the expression of arginase 1, Ym1, and CD206 (26, 27). They express more ferroportin and less ferritin compared to M1 macrophages and display an “iron-recycling” phenotype (28, 29).
So far, the consequences of macrophage exposure to hemolytic RBCs were studied in hemolytic disease (e.g., sickle cell disease), where M1-like reprogramming by heme and iron aggravates tissue damage (23). We now explore the responses of TAMs to neoangiogenesis, which nourishes the tumor but also causes extravasation of RBCs and the release of heme and iron. We demonstrate that hemolysis in the TME reprograms TAMs to a pro-inflammatory phenotype, which shows an important role in inducing anti-tumor activity. Iron oxide nanoparticles can mimic these responses, suggesting a therapeutic strategy that can be exploited for (immunotherapy-based) anti-cancer approaches.
Materials and Methods
NSCLC Paraffin Slides
Paraffin slides were provided by the Lung Biobank Heidelberg, a member of the Biomaterial bank Heidelberg (BMBH) and the Biobank platform of the German Center for Lung Research (DZL). Tissue microarrays (TMAs) were provided by the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany), in accordance with the regulations of the tissue bank and the approval of the ethics committee of Heidelberg University. TMA classification was performed according to the sixth edition of the Tumor Node Metastasis staging system for NSCLC. Paraffin slides from 19 patients with NSCLC (Table 1) were analyzed under the microscope and divided into iron positive and iron negative according to the visible detection/absence of intracellular iron. Each paraffin slide included tumor center, invasive front and tumor periphery.
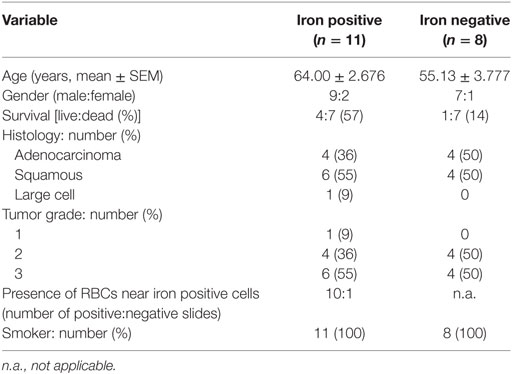
Table 1. Clinicopathological characterization of patients (n = 19) from histology slides of non-small cell lung cancer.
Tumor Suspensions
Human adenocarcinoma tumors were obtained from NSCLC patients (n = 4) who underwent resection for primary lung cancer at the Thoraxklinik of the University Hospital, Heidelberg, Germany. Fresh tumors (human and mouse) were mechanically dissociated and digested with DNAse (SIGMA) and Hyaluronidase (SIGMA). Tumor suspensions were strained using a 70 µm cell strainer (Becton Dickinson) and washed with PBS. For the isolation of human leukocytes, cell suspensions were layered over a density gradient solution (Biocoll Separating Solution, 1.077 g/ml, Biochrom AG, Germany) in a 1:1 volume ratio (450 g, 30 min, RT). Leukocytes were collected, washed twice (PBS) and resuspended (PBS, 4°C). Cells were further processed for magnetic isolation. For the preparation of mouse tumors, a gradient purification using a Lympholyte solution (Cederlane) was performed to remove RBCs and dead cells. Briefly, 7 ml of tumor suspension were added on top of 7 ml of Lympholyte solution and centrifuged (1,500 × g, 25 min, 20°C). The layer of live cells was removed and washed again in cold PBS. The cell pellet was resuspended in PBS (4°C) and kept on ice for the respective procedures.
Magnetic Isolation
Iron-loaded macrophages were isolated from cell suspensions as described in Ref. (30). Briefly, cell suspensions were resuspended in 5 ml of PBS and passed through an LS column attached to a magnetic board (Miltenyi Biotech). Columns were washed three times with 5 ml of PBS. Cells that were adherent to the column (magnetic fraction) were flushed with 5 ml PBS. Cells were either resuspended in PBS and centrifuged for cytospin preparations or lysed for RNA extraction.
Tumor Model
Female and male [C57BL/6N and Slc40a1C326S/C326S (31)] mice were used at 8- to 10-weeks of age. Experiments were approved by “Regierungspräsidium Karlsruhe” under the project number G267/12. Lewis lung carcinoma (LLC) cells were injected (1 × 106 in 100 µl PBS) subcutaneously into the flanks of mice. When indicated, LLC cells were co-injected with cross-linked iron oxide (CLIO)-FITC nanoparticles (8 mg of iron/kg of mouse), or only in PBS. Tumor size was assessed by caliper measurements at the indicated time points and volumes were calculated using the following formula: V = 1/2(length [mm] × width [mm]2) as previously described (32). Mice were sacrificed at the indicated time points. Mice that developed ulcers or necrotic tumors were sacrificed and not considered for the experiments. Blood was removed directly from the heart by cardiac puncture. Subcutaneous tumors were resected and dissected carefully to avoid tissue damage and bleedings induced during animal preparation. Tumors were transferred to PBS on ice and tumor weight (g) was measured on a scale. Tumors were processed for cytospin, FACS analysis and FACS sorting, snap frozen until further analysis or fixed in formalin for immunohistochemistry and histological analysis.
Dissection of Hemorrhagic Areas from LLC Tumors
After careful resection, LLC tumors were washed in PBS and placed in a petry dish under a stereo microscope (Olympus SZ51). Tumors were evaluated for the presence of hemorrhagic areas. Hemorrhagic (H) areas were distinguished from non-hemorrhagic (NH) areas by a strong red coloration. With the help of tweezers and scalpel, H and NH areas from the same tumor were dissected, separated, and stored accordingly for further processing. As validation and quality control for the dissection procedure, heme was quantified.
Histology and Immunohistology
Single cell suspensions (200 µl) were centrifuged (500 r.p.m, 5 min) in a Cytospin Cytocentrifuge (Thermo Scientific). Tissues were fixed for 24 h in 10% neutral buffered formalin (Sigma-Aldrich), dehydrated, and embedded in paraffin. Tissue sections (3–5 µm) were stained for iron using Accustain Iron Stain No. HT20 (Sigma-Aldrich) following manufacturer’s instructions. When indicated, Perls’ blue staining was further enhanced using the DAB peroxidase substrate kit SK-4100 (Vector Labs). Quantification of iron staining was performed using the Image Pro-Premier 3D software. The software calculated the area of pixels corresponding to blue staining (iron staining). For immunostaining, cytospin samples were fixed and permeabilized in ice-cold acetone for 5 min, washed in PBS and treated with H2O2 to block endogenous peroxidase. For immunohistochemistry, sections were treated for 10 min with 3% H2O2 (Sigma-Aldrich) and subjected to microwave-mediated antigen retrieval using the Citraplus reagent (Biogenex). Immunostaining was performed according to the instructions of the Vectastain ABC mouse, rat, and rabbit kits (Vector Labs). Anti-mouse ferroportin staining was performed using MTP11-A rabbit polyclonal antibody (Anti-Mouse Metal Transporter Protein1/Ferroportin (MTP1/IREG1/Fpn) from Alpha Diagnostics); rabbit IgG was used as isotype control for the ferroportin staining. Anti-human CD68 staining was performed using the monoclonal mouse anti-human PG-M1 clone (DAKO). Tissue slides were developed using the Vector AEC substrate (Vector Labs), rinsed with distilled water, counterstained with hematoxylin, washed in PBS, and mounted using the VectaMount AQ mounting medium (Vector Labs). Images were acquired with a Ni-E Nikon microscope.
Preparation of In Vitro TAMs
Bone marrow cells were flushed from tibia and femur using ice-cold HBSS and filtered through a 70 µm cell strainer. Cells were seeded at a density of 700,000 cells/ml in equal volumes of conditioned media (CM) from LLC cells (or DMEM used as control) and complete RPMI1640-Glutamax medium (Life Technologies) [supplemented with 10% of heat-inactivated FBS (Thermo Scientific), 1% penicillin/streptomycin (Sigma-Aldrich) and 10 ng/ml M-CSF (Sigma-Aldrich)]. For 12 well plates, cells were incubated with 0.5 ml of CM plus 0.5 ml of complete RPMI. For 6 well plates, cells were incubated with 1 ml of CM plus 1 ml of complete RPMI. After 4 days, the medium was removed and centrifuged (1,500 r.p.m for 5 min) to remove cells in suspension. Macrophages were incubated in the same media with additional CM for 24 h (0.5 ml for 12 well plates and 1 ml for 6 well plates). For each independent experiment, BMDM were prepared from three different mice. At least three independent experiments were performed for each figure panel.
RBC Preparation
Red blood cell aging was performed as described (33). Mouse blood was collected on EDTA tubes, washed with PBS (3×), and resuspended in Hepes buffer (10 mM Hepes, 140 mM NaCl, BSA 0.1%, pH 7.4). Cells kept at 4°C overnight were considered non-aged RBCs. For in vitro RBCs aging (aRBC), cells (1 × 108 cells/ml in Hepes buffer) were incubated overnight at 30°C with 2.5 mM calcium and 0.5 mM of the ionophore A23187 (Calbiochem). Before incubation with TAMs, both RBCs fractions were washed twice (PBS, 1,500 r.p.m, 5 min) and resuspended in RPMI.
Statistical Analysis
Data are shown as mean ± SEM, and the number of mice (n) is indicated. Statistical analyses were performed using Prism v.6 (GraphPad). Comparisons between two groups were performed with two-sided Welch t-tests, and among three or more than three groups with one-way ANOVA, followed by Bonferroni post-test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 are indicated.
Results
Iron Accumulates in a Subset of TAMs in Human NSCLC
To explore whether iron is detectable in the TME, Perls’ staining was performed on human NSCLC pathological specimen (n = 19 samples). Iron positive staining was detected in 11 cases while 8 samples were iron negative. Table 1 summarizes the available clinical information from these patients grouped by the iron staining results. While cancer cells were negative for iron staining in all samples, some infiltrating cells were clearly iron positive. Interestingly, iron positive cells accumulated in the vicinity of RBCs (10/11 patients) (Figure 1A; Table 1). In addition, iron staining strongly overlapped with positive immune staining for CD68, a macrophage marker (Figure 1B). To further analyze if iron accumulates in TAMs, we isolated leukocytes from fresh tumor tissue (human lung adenocarcinoma) (Table 2) and separated them by exposure to a magnetic field according to their iron content. Cells retained in the magnetic fraction (due to their high iron content) stained positive for iron while cells in the flow through were negative for iron (Figure 1C). Cells within the magnetic fraction were strongly CD68 positive demonstrating that TAMs accumulate iron in lung adenocarcinoma. In addition the macrophage population in the flow through was iron negative (Figure 1C). To explore whether iron-loaded TAMs (iTAMs) are associated with specific areas in the tumor, we performed iron staining in TMA from 116 patients with NSCLC. From each patient, three areas of the original histology block were represented: normal lung, tumor center, and the invasion front (Figure 1D). 38 of 116 patient samples stained positive for iron in the tumor center and/or the invasion front (Table 3). Quantification of iron staining revealed that signals are significantly higher in the invasion front and tumor center when compared to normal lung (Figure 1E). We next analyzed if iron content correlates with tumor size (length in centimeters). Even if it represents a relatively crude clinical parameter, the tumor size of patients scored as iron positive was significantly smaller compared to iron negative tumors (Figure 1F) independently of the histological subtype (Figure S1A in Supplementary Material) and tumor grade (Figure S1B in Supplementary Material). Taken together these data demonstrate that iron accumulates in a subset of TAMs that localize in the vicinity of RBCs in invasive areas of the tumor. Furthermore, the presence of iTAMs correlates with smaller tumor size.
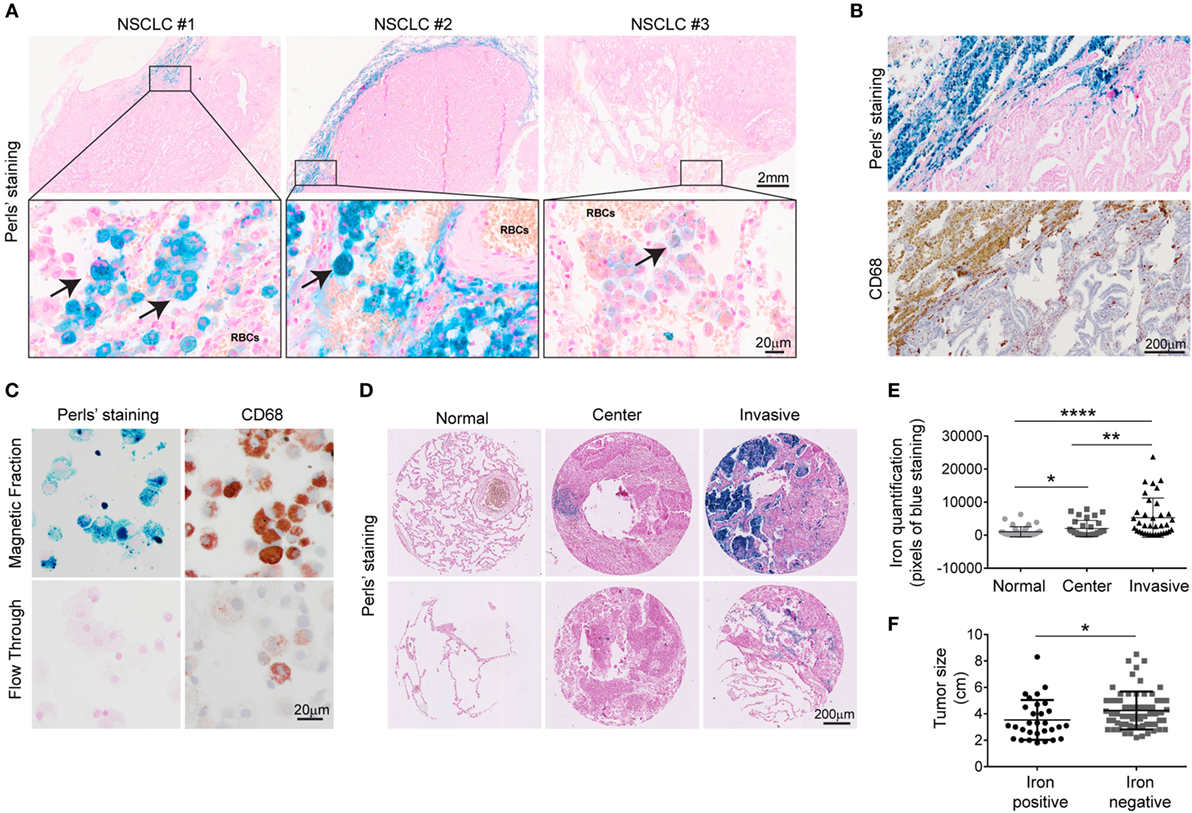
Figure 1. Tumor-associated macrophages associated with invasive margin accumulate iron and correlate with smaller tumor size. (A) Representative examples of three different patients with non-small cell lung cancer (NSCLC). Arrows indicate iron-positive cells (blue staining). Red blood cells (RBCs) are identified by morphology. (B,C) Representative examples of Perls’ staining and anti-CD68 immunostaining in lung adenocarcinoma (B) and tumor-associated leukocytes after magnetic isolation (C), blue staining indicates iron and red staining represents CD68 positive cells (representative of four patients). (D) Representative Perls’ staining in normal lung, tumor center and invasive front in lung squamous cell carcinoma (upper panel) and lung adenocarcinoma (lower panel). (E) Quantification of Perls’ staining in normal lung, center and invasion front of NSCLC. Results are shown as area of pixels corresponding to blue staining (n = 38). (F) Comparison of tumor size in a cohort of NSCLC patients divided by iron content: iron positive (n = 29) and iron negative (n = 65). Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.

Table 2. Clinicopathological characterization of patients (n = 4) of non-small cell lung carcinoma (fresh tumors).
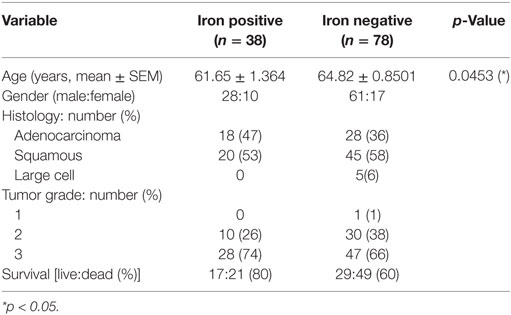
Table 3. Clinicopathological characterization of patients (n = 116) from tissue microarrays (TMAs) of non-small cell lung carcinoma.
iTAMs Show Increased Expression of Markers for Iron Import and Decreased Expression of the Iron Export Protein Ferroportin
To understand the molecular mechanism(s) of how iron accumulates in TAMs, we used the LLC mouse model, a widely used syngeneic model for NSCLC (34). Consistent with our findings in human NSCLC, infiltrating cells in the proximity of RBCs stained positive for iron (Figure 2A), whereas cancer cells were negative for iron staining. We next isolated TAMs from LLC tumors and sorted for the surface markers CD11b+/Gr-1−/F4/80+ (35) by flow cytometry (see gating strategy in Figure S2A in Supplementary Material). We further separated them into iron-spared (i(-)TAMs) and iron-loaded TAMs (iTAMs) by magnetic isolation (Figure 2B). iTAMs express elevated mRNA levels of Cd163 [the scavenger receptor for hemoglobin and haptoglobin-hemoglobin complexes (36), and Hmox1 (the inducible isoform of heme oxygenase responsible for heme degradation)], while mRNA expression of ferroportin (Fpn), was low and similar to iron-spared TAMs (Figure 2C). At the protein level, ferroportin was not detectable in TAMS, but in splenic macrophages that were analyzed as a control (Figure S3A in Supplementary Material). Ferroportin is internalized and degraded by the binding of the hepatic iron-hormone hepcidin. Inflammation increases hepcidin levels and decreases ferroportin cell surface expression causing iron retention in macrophages (37–42). Tumor-bearing mice neither showed elevated serum levels of the inflammatory cytokines IL-6 and IL-1β, known activators of hepcidin expression (43, 44) (Figure S3B in Supplementary Material) nor increased hepatic hepcidin mRNA levels (Hamp1) (Figure S3C in Supplementary Material). Consistently, the expression of ferroportin was detectable in cell types contributing to systemic iron supplies, such as splenic macrophages, Kupffer cells and enterocytes (Figure S3D in Supplementary Material). Taken together these data suggest that iron retention in TAMs does not depend on a hepcidin-dependent decrease of ferroportin expression. Consistently, TAMs (CD11b+/Gr-1−/F4/80+) isolated from LLC tumors of Slc40a1C326S mice, which express a ferroportin allele with a point mutation (C326S) that causes resistance to hepcidin-binding (31), did not express ferroportin protein in most TAMs (Figure 2D) pointing toward a hepcidin-independent downregulation of ferroportin. We conclude that iTAMs are hallmarked by a phenotype of hemoglobin recycling and iron retention.
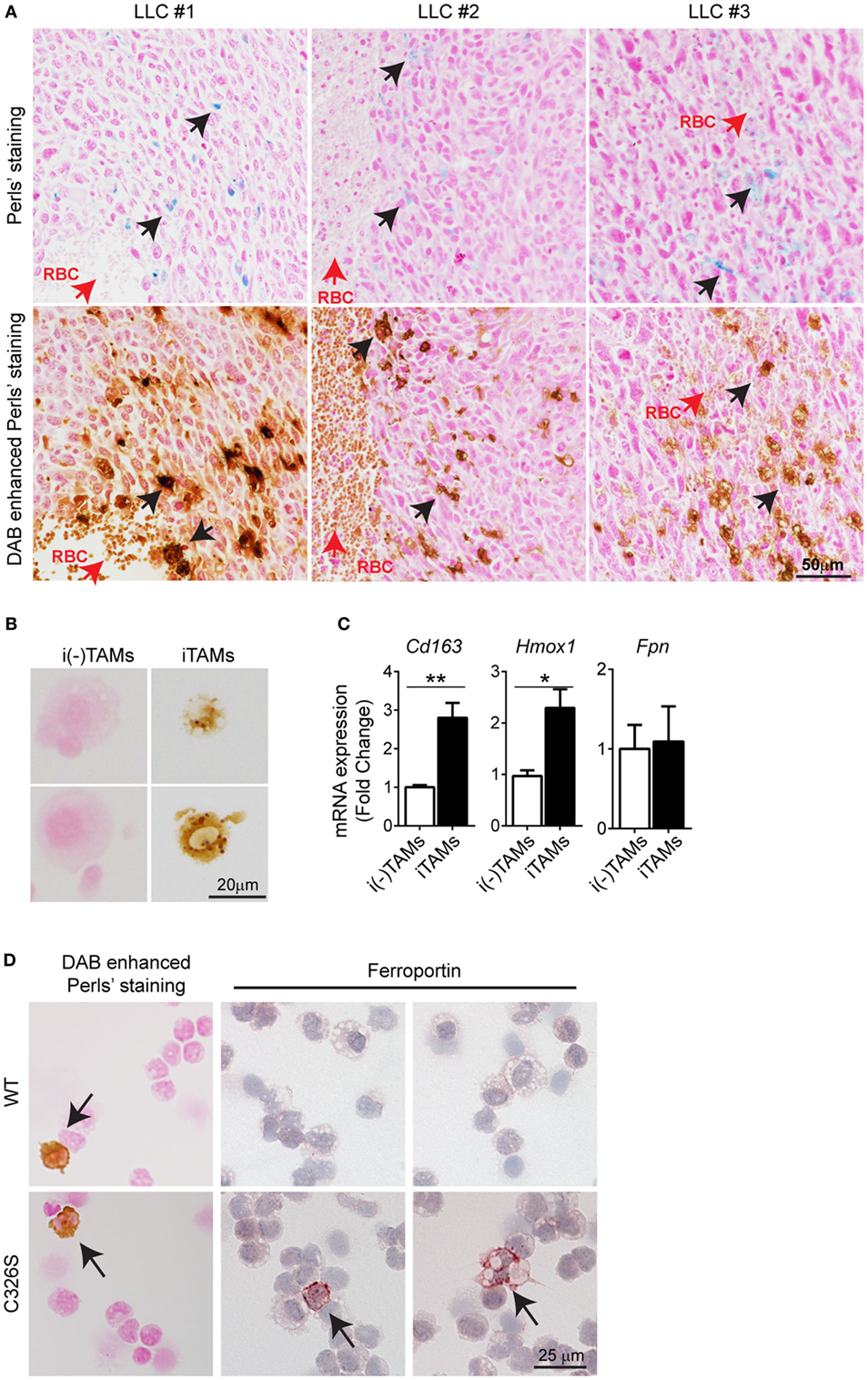
Figure 2. Iron-loaded tumor-associated macrophages (TAMs) from Lewis lung carcinoma (LLC) tumors localize near sites of red blood cells (RBCs) extravasation. (A) Consecutive slides with Perls’ staining of three different LLC tumors (upper panel) and DAB enhanced Perls’ staining (lower panel). Black arrows indicate iron-loaded TAMs and red arrows indicate RBCs. (B) DAB enhanced Perls’ staining of sorted iron-spared (i(-)TAMs) and iron-loaded (iTAMs) TAMs after magnetic isolation. (C) mRNA expression of Cd163, Hmox1, and Fpn in iron-spared (i(-)TAMs) and iron-loaded (iTAMs) TAMs determined by quantitative RT-PCR (three independent experiments, each experiment with TAMs pooled from eight mice). (D) DAB enhanced Perls’ staining and anti-ferroportin staining in TAMs sorted from LLC tumors of WT and Slc40a1C326S mice (C326S). Images are representative of 4 mice and arrows indicate TAMs positive for iron and ferroportin staining respectively. All mRNA levels were normalized to Rpl19 mRNA expression and all tissues were collected 15 days after LLC inoculation. Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Hemorrhagic Areas in LLC Tumors Show Increased Inflammation
We observed that iTAMs co-localize with RBCs in the TME. We next quantified tumor micro-bleedings in response to LLC tumor growth by high field magnetic resonance imaging (MRI). Micro-bleedings were detectable at day 7 after LLC inoculation and increased significantly in number with tumor progression (Figures 3A,B). In addition, dynamic contrast-enhanced (DCE) imaging showed that tumor vessels were more permeable than those within muscle tissue (Figure 3C), and thus more fragile and leaky, leading to the occurrence of micro-bleedings within tumors. Heme, a product of RBC degradation, promotes inflammation by activating macrophages, neutrophils, and endothelial cells (23, 45). We next dissected hemorrhagic areas (H) and NH areas from the same tumor (Figure 3D) and analyzed their properties. Hemorrhagic areas showed increased heme levels and Hmox1 mRNA expression (Figures 3E,F), as well as elevated Cd163 levels (Figure 3F), consistent with the accumulation of iTAMs in areas of RBC extravasation. In addition, the percentage of Gr-1+ cells (gated as CD11b+/Gr-1+) was increased (Figure 3G). The marker Gr-1 (Ly-6C/Ly-6G) is expressed in neutrophils and granulocytes and myeloid-derived suppressor cells (35). Interestingly, expression levels of chemokines known for their neutrophil and myeloid cell chemoattractant activity, KC (Cxcl1) and MIP-2 (Cxcl2) were increased in hemorrhagic compared to NH areas (Figure 3H). In addition, expression of M-CSF (Csf1) and GM-CSF (Csf2) that drive macrophage differentiation were also elevated (Figure 3I). In hemorrhagic areas TAMs (CD11b+/Gr-1-/F4/80+) expressed less CD206, an M2 polarization marker, suggesting a shift of macrophages toward a pro-inflammatory phenotype (Figure 3J). Consistently, mRNA expression of the M1 markers Nos2 and Il-6 were increased in H areas (Figure 3K). In summary, we show that hemorrhagic areas occur due to RBC extravasation from permeable vessels in the TME and are characterized by an infiltration of leukocytes, the accumulation of CD206low iTAMs, as well as increased inflammation.
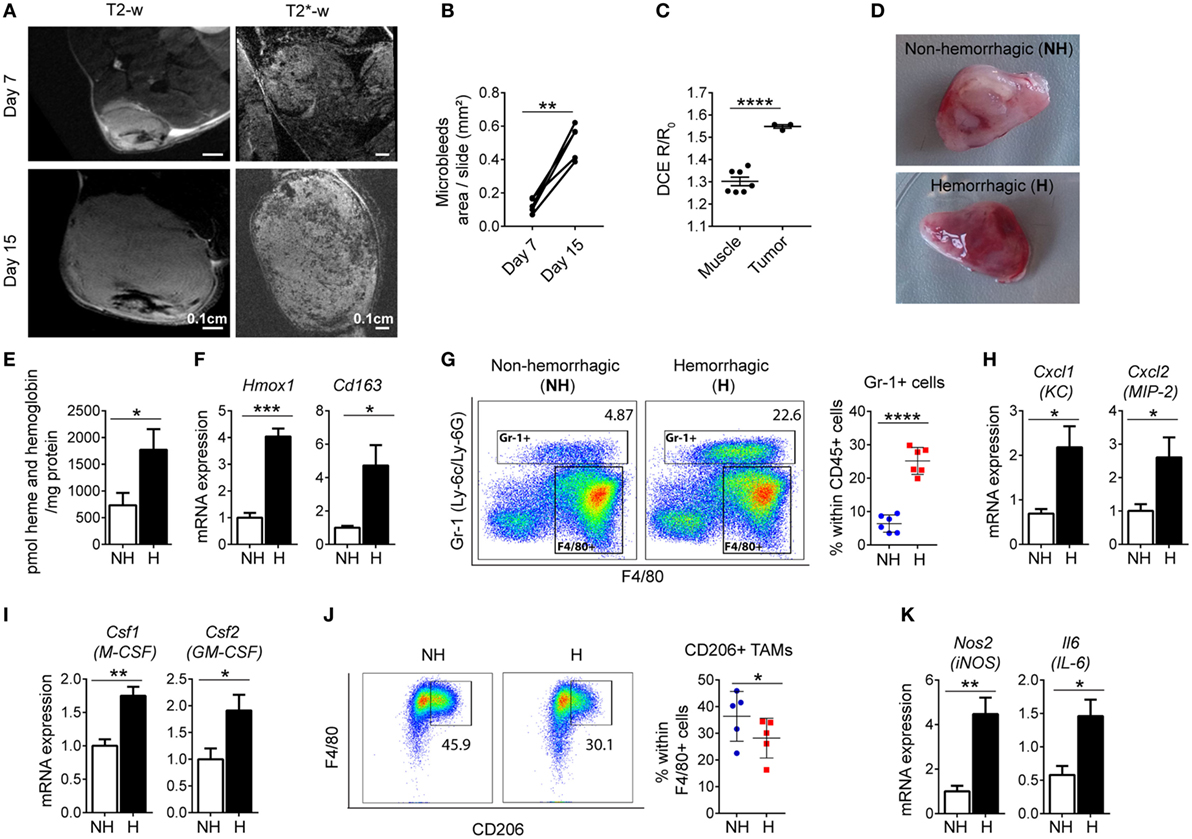
Figure 3. Hemorrhagic areas from Lewis lung carcinoma (LLC) tumors show increased inflammation. (A) Magnetic resonance imaging of LLC tumors, 7 and 15 days after LLC inoculation with T2-w sequence and T2*-w gradient echo sequence (representative of n = 8). (B) Quantification of micro-bleeds by T2*-w gradient echo sequence (n = 5). (C) Dynamic contrast-enhanced imaging in muscle (n = 7) and in tumor tissue (n = 3) at day 7 after LLC inoculation. (D) Co-existence of non-hemorrhagic (NH) area and hemorrhagic (H) area in a LLC tumor. (E) Heme and hemoglobin quantification in NH and H areas (n = 6). (F) Hmox1 and Cd163 mRNA expression determined by quantitative RT-PCR in total lysates of NH and H areas (n = 6). (G) Representative plots and quantification of Gr-1+ cells in NH and H areas (n = 6). (H,I) mRNA expression of Cxcl1 and Cxcl2, (H) and Csf1 and Csf2 (I) determined by quantitative RT-PCR of total lysates of NH and H areas (n = 6). (J) Representative flow cytometry plots of F4/80+ CD206+ TAMs and quantification in NH and H areas (n = 5). (K) mRNA expression of Nos2 and Il6 determined by quantitative RT-PCR of total lysates of NH and H areas (n = 6). All mRNA levels were normalized to Rpl19 mRNA expression and expressed as fold change relative to NH. All tumors were collected 15 days after LLC inoculation. Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Hemolytic RBCs Induce Pro-inflammatory Responses in TAMs
To further understand the impact of RBC extravasation and degradation in TAMs, we next established an in vitro system by differentiating isolated bone marrow cells into TAMs. We incubated cells isolated from the bone marrow with (CM) from LLC cells for 4 days, and analyzed adherent cells that differentiated into macrophages (CD11cneg/Gr-1neg/CD11bpos/F4/80pos) (Figure 4A). We observed that macrophages differentiated with CM polarized toward an anti-inflammatory, tumor-tolerant phenotype showing increased expression of Arginase 1, Ccl2, and Vegf (Figure 4A). We next mimicked conditions in hemorrhagic areas of the TME by adding RBCs to macrophages. In order to mimic senescent hemolytic RBCs (e.g., as would be expected to occur in the prooxidant inflammatory TME) we prepared aged RBC by treating RBC overnight with calcium. In vitro TAMs accumulated iron when incubated with either aged RBCs (aRBC) (to mimic senescent/hemolytic RBCs) or non-aged RBCs (Figure 4B). Heme (heme and hemoglobin) concentration was significantly higher in the supernatant of macrophages treated with aged RBCs (Figure 4C). Additionally, heme was also detected in the supernatant on non-aged RBC, suggesting that inflammation may induce RBC breakage. Aged RBCs exposed to CM from LLC cells changed shape and size (higher FSC-A) and were more prone to hemolysis when compared to control media (Figure S4A–C in Supplementary Material). Furthermore, mRNA expression of Hmox1 and Spi-c, markers for “iron recycling macrophages” was increased in macrophages treated with both RBCs sources (Figure 4D). Similar to observations in hemorrhagic areas of the tumor, the presence of RBCs increased mRNA expression of Cxcl1 (KC), Cxcl2 (MIP-2), and Csf1 (M-CSF) (Figure 4E), suggesting that the activation of macrophages by heme/iron may trigger recruitment of myeloid cells. We further observed that in the presence of aged RBCs, pro-inflammatory markers (CD86, Il6, Nos2, and Tnfa) were increased, while the M2 markers remained unchanged (Arginase 1) or were decreased (CD206, Ym1, and Il10) (Figures 4F,G). Our results show that the responses of macrophages to RBCs in the in vitro model mirrored those in the hemorrhagic areas of the TME, with iron accumulation in macrophages (iTAMs) and a shift toward a pro-inflammatory phenotype.
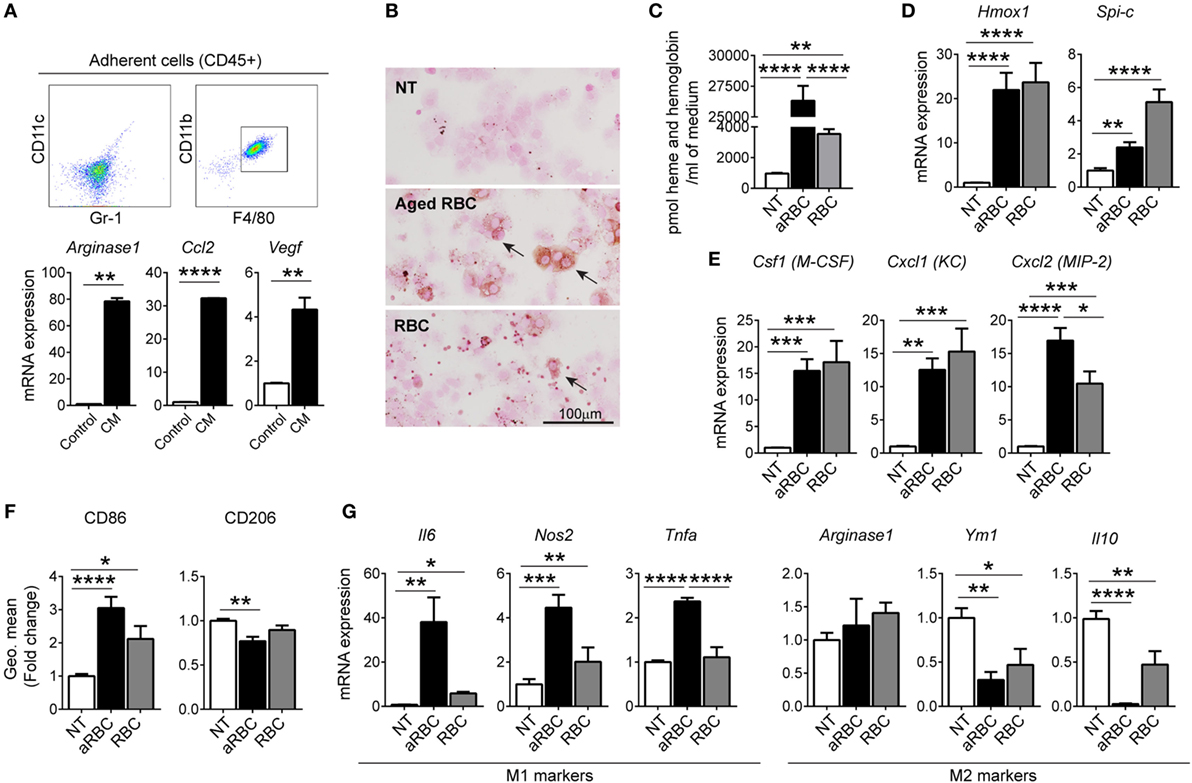
Figure 4. Hemolytic red blood cells (RBCs) shift tumor-associated macrophages (TAMs) polarization toward an M1-like phenotype. (A) Representative flow cytometry plots of BMDM differentiated with conditioned media (CM) from Lewis lung carcinoma (LLC) cells (in vitro TAMs) and mRNA expression of Arginase 1, Ccl2, and Vegf compared to control BMDM. (B) DAB enhanced Perls’ staining (indicated by arrows) of in vitro TAMs, non-treated (NT), treated with aged RBCs (aRBC) or RBCs (RBC) (representative of n = 3). (C) Heme and hemoglobin quantification in the supernatant of in vitro TAMs, NT, treated with aged RBCs (aRBC) or RBCs (RBC). (D,E) mRNA expression of Hmox1 and Spi-c (D) and Csf1, Cxcl1, and Cxcl2 (E) in in vitro TAMs, NT, treated with aged RBCs (aRBC) or RBCs (RBC), determined by quantitative RT-PCR. (F) Quantification of CD86 and CD206 expression by flow cytometry in in vitro TAMs NT, treated with aged RBCs (aRBC) or RBCs (RBC), results are shown as geometric mean fold change to NT samples (n = 9). (G) mRNA expression of M1 markers: Il6, Nos2 and Tnfa and M2 markers: Arginase 1, Ym1, and Il10 in in vitro TAMs NT, treated with aged RBCs (aRBC) or RBCs (RBC). All cultures were analyzed 24 h after the respective treatment. All mRNA levels were determined by quantitative RT-PCR and normalized to Rpl19 mRNA expression [shown as fold change to NT samples (n = 9)]. Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
TAMs Exposed to Hemolytic RBCs Promote Tumor Cell Death
Pro-inflammatory macrophages in the TME can promote tumor cell death by producing ROS (46, 47). To test the effect of iron loading on tumor cell killing in vitro, we co-cultured macrophages exposed to hemolytic RBCs with LLC cells. This co-culture strongly reduced LLC cell viability (Figure 5A, gating strategy shown in Figure S2B in Supplementary Material); macrophages showed increased ROS levels (Figure 5B) and a shift toward pro-inflammatory phenotype characterized by the decreased expression of CD206 and increased expression of CD86 (Figure 5C). We next performed TUNEL staining and DAB enhanced Perls’ staining of consecutive slides of LLC tumors. Consistent with our findings in cultured cells, apoptotic areas co-localize with hemorrhagic areas and with the presence of iTAMs in the TME of LLC tumors (Figure 5D). Our findings demonstrate that macrophage iron loading in the TME correlates with and leads to a pro-inflammatory phenotype and anti-tumor activity. Thus, increasing the population of iTAMs in the TME emerged as a promising new therapeutical option to counteract tumor growth.
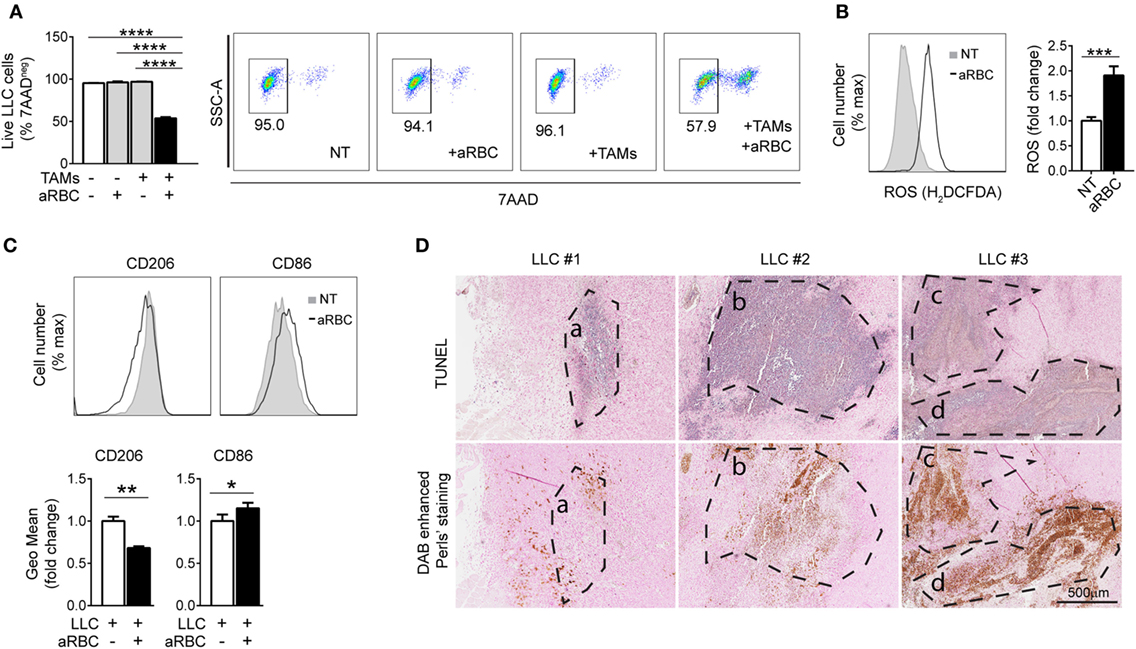
Figure 5. Macrophages exposed to hemolytic red blood cells (RBCs) promote tumor cell death. (A) Viability of Lewis lung carcinoma (LLC) cells co-cultured with in vitro tumor-associated macrophages (TAMs) and aged RBCs (aRBC) measured by flow cytometry. Results are shown as% of 7AAD negative cells (representative plots of LLC stained with 7AAD). (B) Representative plots and reactive oxygen species (ROS) quantification by flow cytometry in in vitro TAMs co-cultured with LLC cells and non-treated (NT) or treated with aged RBCs (aRBC). Results are shown as fold change to NT samples (n = 6). (C) Quantification of CD206 and CD86 expression by flow cytometry in in vitro TAMs co-cultured with LLC cells and aged RBCs (aRBC). Results are shown as geometric mean fold change to in vitro TAMs+LLC (white bar) (n = 9). All cultures were analyzed 24 h after the respective treatment. (D) Consecutive slides of LLC, showing four different areas overlapping (A–D) of tumors (upper panel) with TUNEL staining for apoptosis (purple staining) and (lower panel) DAB enhanced Perls’ staining (brown staining represents iTAMs and RBCs). Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Phagocytosis of Iron Nanoparticles by TAMs Inhibits Tumor Growth
We tested CLIO nanoparticles as a strategy to deliver iron to macrophages without provision of this growth factor to tumor cells. CLIO nanoparticles are used for in vivo imaging by magnetic resonance and are specifically ingested by phagocytic cells such as TAMs, rather than neighboring cell types such as tumor cells or other leukocytes (48). Similar to TAMs exposed to RBCs, macrophages accumulated CLIO nanoparticles (Figure 6A) which induced decreased expression of CD206 (Figures 6A,B). We next tested whether TAMs treated with CLIO displayed anti-tumor activity in vivo and in vitro. Incubation of LLC cells with CLIO-treated TAMs significantly reduced tumor cell viability (Figure 6C), correlating with a decrease in the expression of CD206 in TAMs (Figure 6D). We injected mice with LLC cells and analyzed tumor growth with or without co-injection of CLIO. 15 days after injection, tumors from CLIO-co-injected mice were significantly smaller than control tumors (Figure 6E). The CLIO nanoparticles accumulated in TAMs (Figure 6F), which were predominantly localized in the periphery of the tumor and near the invasive front (Figure 6G). The presence of iTAMs was also detected in NT samples, near areas of RBCs extravasation (Figure 6G). In accordance, we observed a moderate reduction of CD206 expression and increased CD86 expression (Figure 6H), together with a significant increase of the CD8/CD4 T-cell ratio (Figure 6I). These data show that the injection of iron nanoparticles limits tumor growth in the LLC cell mouse model, translating the in vitro effect observed with hemolytic RBCs. Our data suggest that iron nanoparticles injection could be developed as a therapeutic strategy to inhibit tumor growth via TAMs reprogramming.
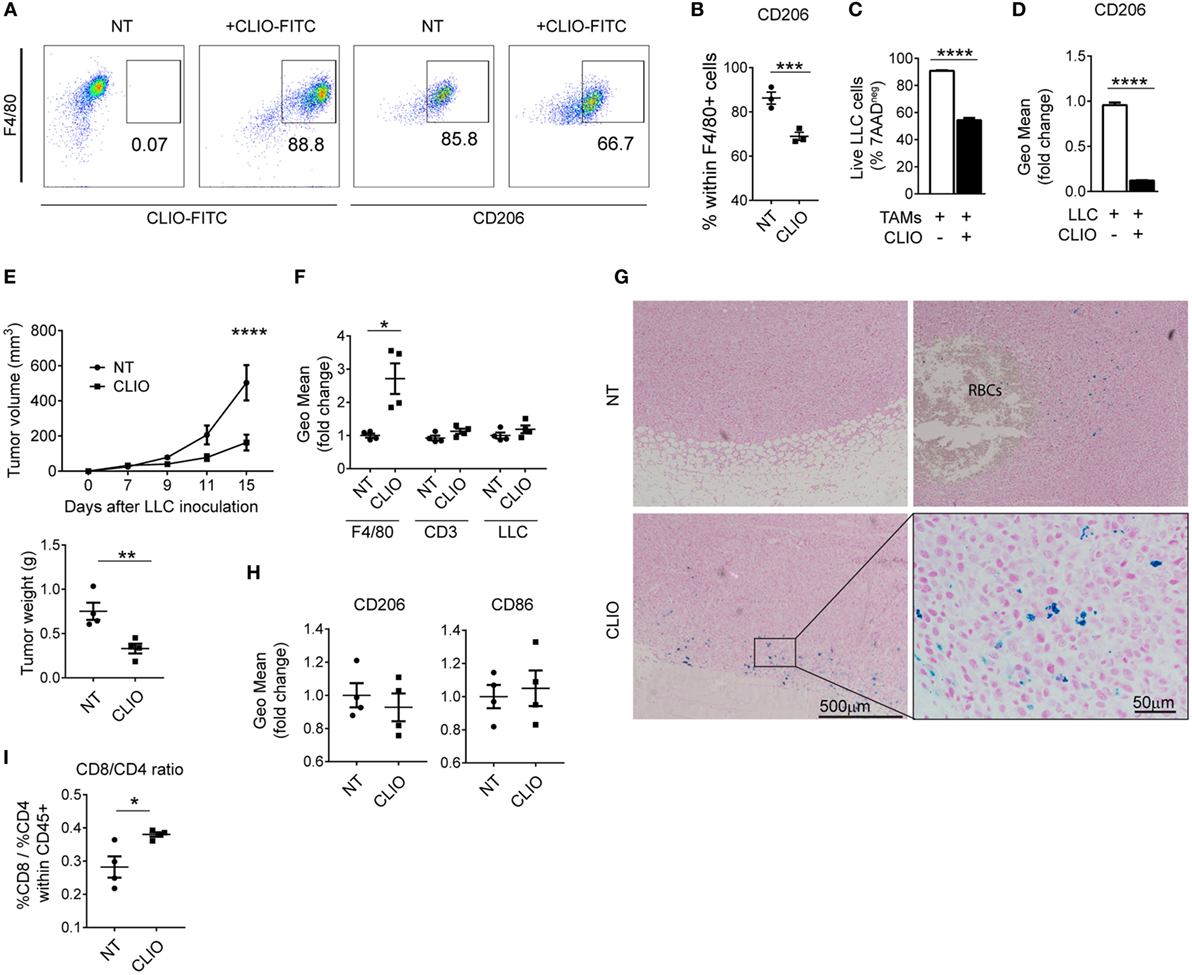
Figure 6. Iron nanoparticles accumulate in tumor-associated macrophages (TAMs) and delay tumor growth. (A) Representative plots of in vitro TAMs analyzed for the uptake of cross-linked iron oxide (CLIO)-FITC nanoparticles and expression of CD206. (B) Percentage of CD206 positive cells in in vitro TAMs (F4/80+), non-treated (NT) or incubated with CLIO-FITC nanoparticles. (C) Viability of Lewis lung carcinoma (LLC) cells measured by flow cytometry, in the presence of in vitro TAMs, CLIO-FITC at 48 h. Results are shown as% of 7AAD negative cells. (D) Expression of CD206 measured by flow cytometry in in vitro TAMs in the presence of LLC cells and incubated with CLIO-FITC, as geometric mean fold change (Geo Mean) to control (TAMs+LLC) at 48 h. (E) Tumor volume of sc LLC tumors NT or co-injected with CLIO-FITC nanoparticles and tumor weight at 15 days after LLC inoculation. (F) CLIO-FITC uptake in TAMs (F4/80), CD3+ and LLC cells measured by flow cytometry. Results are shown as geometric mean fold change (Geo Mean) compared to cells from NT tumors. (G) Representative Perls’ staining of LLC tumors NT or co-injected with CLIO-FITC nanoparticles (CLIO). RBCs are indicated in the NT sample. Blue staining represents iron-loaded TAMs. (H) Quantification of CD86 and CD206 expression by flow cytometry in F4/80+ cells in tumors NT or co-injected with CLIO-FITC nanoparticles (CLIO) results are shown as geometric mean fold change to NT samples (n = 9). (I) Ratio of the% of CD8/CD4 cells within CD45+ cells in tumors non-treated (NT) or co-injected with CLIO-FITC nanoparticles (CLIO). All tumors were collected 15 days after LLC inoculation. Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Discussion
In most malignancies, anti-inflammatory TAMs are detected in the TME which frequently correlates with poor prognosis (2, 49–53). Although TAMs could have the ability to eliminate tumor cells, they rather display a tumor supportive phenotype in most tumors, promoting angiogenesis and exerting immune suppressive functions (Figure 7A) (52). Thus, converting macrophages from a pro-tumoral to anti-tumoral phenotype is relevant for anti-cancer therapy. So far, iron was seen as an essential nutrient for tumor cell growth (54) but its contribution to immune responses in the TME remained unexplored.
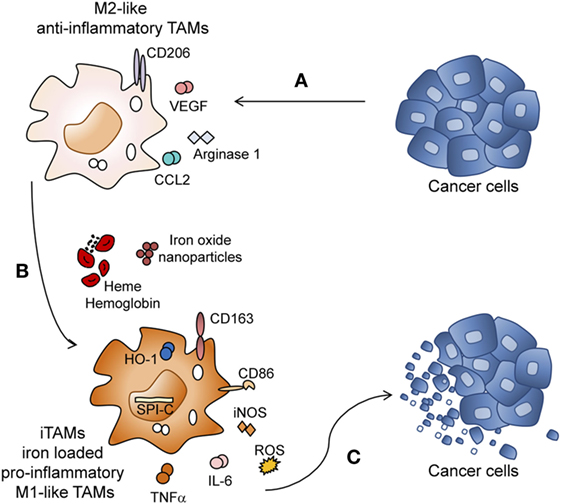
Figure 7. Heme and iron shift the polarization of tumor-associated macrophages (TAMs) toward a pro-inflammatory phenotype. (A) Lewis lung carcinoma (LLC) tumor cells promote M2 polarization of TAMs by inducing the expression of CD206, Arginase 1, Ccl2, and Vegf. (B) M2 macrophages exposed to the degradation products of hemolytic RBCs (heme, hemoglobin, and iron) or iron nanoparticles are reprogrammed to iron-loaded TAMs (iTAMs), with an M1-like inflammatory phenotype (increased production of Tnfa, Il6 and reactive oxygen species (ROS) and increased expression of Nos2 (iNOS), CD86, Cd163, Spi-c, and Hmox1). (C) iTAMs show tumoricidal activity by decreasing the viability of LLC cells.
Here, we discover a novel role of RBCs, heme and iron in the TME, which shapes the immune response. Hemolytic RBCs that extravasate from vessels during neoangiogenesis have the capacity to reprogram M2-like TAMs into pro-inflammatory (M1-like) TAMs with an ability to kill tumor cells. iTAMs show low expression of the iron exporter ferroportin, suggesting an inability to provide iron as a growth factor to the tumor. Of note, ferroportin expression is decreased at the mRNA level independent of hepcidin activity. iTAMs are further hallmarked by the expression of CD86high, CD206low, Cd163high, Hmox1high, show a pro-inflammatory phenotype associated with the production of ROS and pro-inflammatory cytokines (TNFα and IL-6), and are capable of killing tumor cells (Figures 7B–C). Interestingly, the same characteristics of this subset of macrophages can be elicited by applying exogenous iron sources, such as iron oxide nanoparticles.
Our findings in the TME are reminiscent of observations in hemolytic disease where heme and iron polarize splenic and liver macrophages toward an M1-like phenotype, contributing to inflammation and tissues damage (23). In accordance with our findings, a pro-inflammatory response of macrophages to heme and iron was further described in the wound-healing process, in the injured spinal cord and in hemophilic mice (24, 25, 55). Furthermore, free heme sensitizes hepatocytes to TNF-α and oxidative stress-induced cell death (56). Here, we show that hemolytic RBCs do not directly promote tumor cell death but require the M1-like pro-inflammatory activation of TAMs, which release pro-inflammatory cytokines and ROS (Figures 5A,B).
Non-small cell lung cancer patients with detectable iron in the TME have significant smaller tumors (Figure 1F). This supports the idea that exposure of TAMs to hemolytic RBCs and subsequent iron retention, which promotes their inflammatory phenotype, impacts on tumor cell viability (28, 57). In fact, the presence of M1 macrophages in several tumor entities such as NSCLC has been associated with better patient survival (58–61). By contrast, in breast cancer, TAMs are characterized by an “iron-donor” phenotype and ferroportin expression is detectable (62). These differences in the TAM phenotypes are likely explained by the cytokine/chemokine composition of the tumor niche. Key players may be M-CSF (Csf1) and GM-CSF (Csf2), which are implicated in the control of ferroportin expression (63, 64). GM-CSF suppresses ferroportin mRNA expression (64), thus the ratio of GM-CSF/M-CSF in the tumor tissue may explain ferroportin expression levels in macrophages. The composition of the TME, including the presence of RBCs directly affects macrophage polarization as well as the expression of iron genes. We studied lung cancer as a model, but we believe that this concept can be further extended to other tumor entities. Future studies will aim to understand how RBCs in the TME of other tumor subtypes affects TAM function. This knowledge will be important to design anti-tumor therapies.
Dependent on the pathophysiological context iron-induced M1 pro-inflammatory macrophage polarization can be detrimental or beneficial, thus representing an interesting target for therapy. In hemolytic disorders, heme and iron-dependent M1-like macrophage polarization contributes to inflammation and tissue damage, an effect which can be prevented by applying heme and iron scavengers (23). In the context of the TME, iron-mediated reprogramming of pro-tumoral M2-like TAMs into anti-tumoral M1-like TAMs is desirable to prevent tumor growth. We expect that this knowledge can be applied therapeutically by delivering iron to TAMs.
The “natural” population of iTAMs appears only in late stages of tumor development, when the angiogenic switch occurs and tumors are already well established. In this scenario, the number of iTAMs may be insufficient to exert a sufficiently large, protective anti-tumor response. Nevertheless, we observed that tumors with iTAMs are of significantly smaller size (Figure 1F), suggesting that iTAMs may impact on tumor growth. Our observations motivated us to use iron nanoparticles as a “specific” means to deliver iron to TAMs in early stages of tumor development. Application of iron nanoparticles to diminish tumor growth served as a proof-of-concept for the therapeutic potential of such an approach in vitro and in a mouse tumor model. Recently, ferumoxytol, another type of iron nanoparticle, was shown to reduce growth of subcutaneous adenocarcinoma and to prevent the development of liver metastasis by promoting a pro-inflammatory type of macrophages (65). We propose that the iron moiety of the nanoparticle plays a key role in determining TAM polarization and anti-tumoral activity. We suggest that the administration of iron formulations at early stages of tumor development will contribute to the inhibition of tumor growth, representing as a promising strategy for cancer therapy. Increasing the physiological population of TAMs via iron/heme delivery is expected to produce only mild adverse effects for cancer patients. However, the development of “iron sources” for targeted delivery to the TME is required. We expect that such a therapeutic approach will be of benefit in combination with current therapies, such as immunotherapy, to improve anti-cancer responses. Immune check point inhibitors, such as drugs that block programmed cell death-1 (PD-1) were shown to activate the immune system and trigger anti-tumor activity. These drugs are currently being applied to treat NSCLC (66, 67). PD-1 is expressed in the surface of macrophages and binds to the receptor PD-L1 on the surface of T cells, reducing cytokine production and suppressing T-cell proliferation (68). As a combination approach, increasing the population of iTAMs, together with the use of monoclonal antibodies or drugs that block T-cell inhibition, is expected to improve the immune response against cancer. T-cell activation requires the interaction of CD28 expressed on the T-cell surface with CD80 or CD86 expressed by macrophages (69). Since iTAMs express high levels of CD86, the amplification of the iTAM population may boost T-cell activation and suppress tumor growth. Consistently, we observe an increase of the CD8 cytotoxic T cells/CD4 helper T cells ratio in tumors treated with iron nanoparticles (Figure 6I).
In conclusion, we defined a novel pro-inflammatory niche within the TME. This niche contains hemorrhagic areas, where RBCs release heme and iron which is subsequently taken up by TAMs. Heme and iron differentiate M2-like TAMs into a M1-proinflammatory phenotype capable of reducing tumor growth. We further provide a proof-of-concept that iron nanoparticle treatment of TAMs reduces tumor growth. Our findings have potential to be further explored for translation into clinical applications to improve cancer therapy.
Ethics Statement
Paraffin slides were provided by the Lung Biobank Heidelberg, a member of the Biomaterial bank Heidelberg (BMBH) and the Biobank platform of the German Center for Lung Research (DZL). Tissue microarrays (TMAs) were provided by the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany), in accordance with the regulations of the tissue bank and the approval of the ethics committee of Heidelberg University. Animal Experiments were approved by “Regierungspräsidium Karlsruhe” Germany, under the project number G267/12.
Author Contributions
MCS designed the project, performed the experiments and wrote the manuscript; MOB performed the MRI experiments and gave advice on nanoparticle experiments; FV provided advice for experiments, critical discussion and wrote the manuscript; MPC, AS and CMT helped with some experiments; MM, TM and AW provided human histology slides and patient data; MP provided critical reading and discussion; MWH provided critical reading and wrote the manuscript; AC and MUM designed and supervised the project and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Liz Meister (Thoraxklinik at University Hospital Heidelberg) for helping with the procedures to isolate cells from human tumors; Dr. Margareta Müller and Dr. Sabine Hensler (Furtwangen University, Furtwangen) for sharing the knowledge with co-culture experiments; Dr. Hellmut Augustin for critical discussions; Andreas Simmelbauer for helping with the quantification of Perls’ staining; Prof. Dr. Ursula Klingmüller and Dr. Katharina Robichon for support in the Luminex experiment. We specially thank Maria de Sousa for critical reading and advice. We further thank R. Weissleder (MGH, Boston) for the kind gift of cross-linked iron oxide nanoparticles and Manuel Fischer for help with MRI experiments. We thank the Institute of Pathology, University of Heidelberg, for histological processing of tissue samples. We acknowledge the financial support of the Deutsche Forschungsgemeinschaft and Ruprecht-Karls-Universität Heidelberg within the funding programme Open Access Publishing.
Funding
This work was supported by Fundação para a Ciência e Tecnologia by means of a PhD fellowship SFRH/BD/51290/2010 awarded to MCS; “Stiftungen und Preise 2012” awarded by Faculty of Medicine, Heidelberg; Deutsches Zentrum für Lungenforschung (DZL) with the reference TP CF-1.1 and by the Deutsche Forschungsgemeinschaft (SFB1038). MOB was supported by a Physician-Scientist Fellowship of the Medical Faculty, University of Heidelberg, the Hoffmann–Klose Foundation (University of Heidelberg) and the Novartis Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01479/full#supplementary-material.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
2. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol (2009) 86:1065–73. doi:10.1189/jlb.0609385
3. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today (1992) 13:265–70. doi:10.1016/0167-5699(92)90008-U
4. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
5. Jung KY, Cho SW, Kim YA, Kim D, Oh BC, Park DJ, et al. Cancers with higher density of tumor-associated macrophages were associated with poor survival rates. J Pathol Transl Med (2015) 49:318–24. doi:10.4132/jptm.2015.06.01
6. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
7. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
8. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
9. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi:10.1016/j.immuni.2014.01.006
10. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643
11. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi:10.1038/nature12034
12. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331:1612–6. doi:10.1126/science.1198443
13. Mills CD, Shearer J, Evans R, Caldwell MD. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol (1992) 149:2709–14.
14. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27:462–72. doi:10.1016/j.ccell.2015.02.015
15. O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med (2012) 209:1869–82. doi:10.1084/jem.20112738
16. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18:349–55. doi:10.1016/j.semcancer.2008.03.004
17. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi:10.1016/S1471-4906(02)02302-5
18. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi:10.1038/nrc1256
19. Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, et al. Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol (2016) 18:790–802. doi:10.1038/ncb3371
20. Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A (1997) 94:10919–24. doi:10.1073/pnas.94.20.10919
21. Korolnek T, Hamza I. Macrophages and iron trafficking at the birth and death of red cells. Blood (2015) 125:2893–7. doi:10.1182/blood-2014-12-567776
22. Soares MP, Hamza I. Macrophages and iron metabolism. Immunity (2016) 44:492–504. doi:10.1016/j.immuni.2016.02.016
23. Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood (2016) 127:473–86. doi:10.1182/blood-2015-08-663245
24. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest (2011) 121:985–97. doi:10.1172/JCI44490
25. Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron (2014) 83:1098–116. doi:10.1016/j.neuron.2014.07.027
26. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
27. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi:10.1146/annurev-physiol-022516-034339
28. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol (2010) 40:824–35. doi:10.1002/eji.200939889
29. Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica (2010) 95:1814–22. doi:10.3324/haematol.2010.023879
30. Franken L, Klein M, Spasova M, Elsukova A, Wiedwald U, Welz M, et al. Splenic red pulp macrophages are intrinsically superparamagnetic and contaminate magnetic cell isolates. Sci Rep (2015) 5:12940. doi:10.1038/srep12940
31. Altamura S, Kessler R, Grone HJ, Gretz N, Hentze MW, Galy B, et al. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab (2014) 20:359–67. doi:10.1016/j.cmet.2014.07.007
32. D’Amico L, Mahajan S, Capietto AH, Yang Z, Zamani A, Ricci B, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med (2016) 213:827–40. doi:10.1084/jem.20150950
33. Delaby C, Rondeau C, Pouzet C, Willemetz A, Pilard N, Desjardins M, et al. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS One (2012) 7:e42199. doi:10.1371/journal.pone.0042199
34. Kellar A, Egan C, Morris D. Preclinical murine models for lung cancer: clinical trial applications. Biomed Res Int (2015) 2015:621324. doi:10.1155/2015/621324
35. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
36. Schaer DJ, Schaer CA, Buehler PW, Boykins RA, Schoedon G, Alayash AI, et al. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood (2006) 107:373–80. doi:10.1182/blood-2005-03-1014
37. Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal (2014) 26:2539–50. doi:10.1016/j.cellsig.2014.07.029
38. Pinnix ZK, Miller LD, Wang W, D’Agostino R Jr, Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med (2010) 2:43ra56. doi:10.1126/scisignal.3001127
39. Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res (2015) 75:2254–63. doi:10.1158/0008-5472.CAN-14-2465
40. Liu XB, Nguyen NB, Marquess KD, Yang F, Haile DJ. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis (2005) 35:47–56. doi:10.1016/j.bcmd.2005.04.006
41. Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem (2002) 277:39786–91. doi:10.1074/jbc.M201485200
42. Guida C, Altamura S, Klein FA, Galy B, Boutros M, Ulmer AJ, et al. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood (2015) 125:2265–75. doi:10.1182/blood-2014-08-595256
43. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest (2004) 113:1271–6. doi:10.1172/JCI200420945
44. Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci U S A (2005) 102:1906–10. doi:10.1073/pnas.0409808102
45. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol (2014) 5:115. doi:10.3389/fphar.2014.00115
46. Tan HY, Wang N, Li S, Hong M, Wang X, Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longev (2016) 2016:2795090. doi:10.1155/2016/2795090
47. Mills CD, Lenz LL, Harris RA. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res (2016) 76:513–6. doi:10.1158/0008-5472.CAN-15-1737
48. Leimgruber A, Berger C, Cortez-Retamozo V, Etzrodt M, Newton AP, Waterman P, et al. Behavior of endogenous tumor-associated macrophages assessed in vivo using a functionalized nanoparticle. Neoplasia (2009) 11:459–468,452 p following 468. doi:10.1593/neo.09356
49. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
50. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
51. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
52. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell (2006) 124:263–6. doi:10.1016/j.cell.2006.01.007
53. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol (2008) 66:1–9. doi:10.1016/j.critrevonc.2007.07.004
54. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer (2013) 13:342–55. doi:10.1038/nrc3495
55. Nieuwenhuizen L, Schutgens RE, Coeleveld K, Mastbergen SC, Roosendaal G, Biesma DH, et al. Hemarthrosis in hemophilic mice results in alterations in M1-M2 monocyte/macrophage polarization. Thromb Res (2014) 133:390–5. doi:10.1016/j.thromres.2013.10.039
56. Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med (2010) 2:51ra71. doi:10.1126/scitranslmed.3001118
57. Recalcati S, Locati M, Gammella E, Invernizzi P, Cairo G. Iron levels in polarized macrophages: regulation of immunity and autoimmunity. Autoimmun Rev (2012) 11:883–9. doi:10.1016/j.autrev.2012.03.003
58. Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J (2009) 33:118–26. doi:10.1183/09031936.00065708
59. Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer (2010) 10:112. doi:10.1186/1471-2407-10-112
60. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res (2012) 4:376–89. https://www.ncbi.nlm.nih.gov/pubmed/23145206
61. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi:10.1038/nrclinonc.2016.217
62. Marques O, Porto G, Rema A, Faria F, Cruz Paula A, Gomez-Lazaro M, et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer (2016) 16:187. doi:10.1186/s12885-016-2228-y
63. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med (2016) 22:945–51. doi:10.1038/nm.4146
64. Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology (2010) 215:788–95. doi:10.1016/j.imbio.2010.05.020
65. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol (2016) 11:986–94. doi:10.1038/nnano.2016.168
66. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med (2015) 373:1627–39. doi:10.1056/NEJMoa1507643
67. Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol (2015) 16:257–65. doi:10.1016/S1470-2045(15)70054-9
68. Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol (2015) 27:39–46. doi:10.1093/intimm/dxu095
Keywords: tumor-associated macrophages, macrophage polarization, hemolytic red blood cells, heme, iron, non-small cell lung cancer, iron nanoparticles, anti-tumor activity
Citation: Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, Meister M, Muley T, Warth A, Platten M, Hentze MW, Cerwenka A and Muckenthaler MU (2017) Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front. Immunol. 8:1479. doi: 10.3389/fimmu.2017.01479
Received: 15 September 2017; Accepted: 23 October 2017;
Published: 08 November 2017
Edited by:
Jason Roszik, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Fabian Benencia, Ohio University, United StatesAnna Karolina Kozlowska, Poznan University of Medical Sciences, Poland
Jörg Wischhusen, University of Würzburg, Germany
Copyright: © 2017 Costa da Silva, Breckwoldt, Vinchi, Correia, Stojanovic, Thielmann, Meister, Muley, Warth, Platten, Hentze, Cerwenka and Muckenthaler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martina U. Muckenthaler, martina.muckenthaler@med.uni-heidelberg.de
†These authors have contributed equally to this work.