Whole Genome Shotgun Sequencing Detects Greater Lichen Fungal Diversity Than Amplicon-Based Methods in Environmental Samples
 Kyle Garrett Keepers1*
Kyle Garrett Keepers1*  Cloe S. Pogoda1
Cloe S. Pogoda1  Kristin H. White1 Carly R. Anderson Stewart1
Kristin H. White1 Carly R. Anderson Stewart1  Jordan R. Hoffman2 Ana Maria Ruiz2 Christy M. McCain1
Jordan R. Hoffman2 Ana Maria Ruiz2 Christy M. McCain1  James C. Lendemer2
James C. Lendemer2  Nolan Coburn Kane1 Erin A. Tripp1,3
Nolan Coburn Kane1 Erin A. Tripp1,3- 1Department of Ecology and Evolutionary Biology, University of Colorado, Boulder, CO, United States
- 2Institute of Systematic Botany, The New York Botanical Garden, New York, NY, United States
- 3Museum of Natural History, University of Colorado, Boulder, CO, United States
In this study we demonstrate the utility of whole genome shotgun (WGS) metagenomics in study organisms with small genomes to improve upon amplicon-based estimates of biodiversity and microbial diversity in environmental samples for the purpose of understanding ecological and evolutionary processes. We generated a database of full-length and near-full-length ribosomal DNA sequence complexes from 273 lichenized fungal species and used this database to facilitate fungal species identification in the southern Appalachian Mountains using low coverage WGS at higher resolution and without the biases of amplicon-based approaches. Using this new database and methods herein developed, we detected between 2.8 and 11 times as many species from lichen fungal propagules by aligning reads from WGS-sequenced environmental samples compared to a traditional amplicon-based approach. We then conducted complete taxonomic diversity inventories of the lichens in each one-hectare plot to assess overlap between standing taxonomic diversity and diversity detected based on propagules present in environmental samples (i.e., the “potential” of diversity). From the environmental samples, we detected 94 species not observed in organism-level sampling in these ecosystems with high confidence using both WGS and amplicon-based methods. This study highlights the utility of WGS sequence-based approaches in detecting hidden species diversity and demonstrates that amplicon-based methods likely miss important components of fungal diversity. We suggest that the adoption of this method will not only improve understanding of biotic constraints on the distributions of biodiversity but will also help to inform important environmental policy.
Introduction
Microbial diversity present in the environment is recognized increasingly for its important and varied roles in the health of ecosystems (Chen et al., 2018; Nottingham et al., 2018; Pike et al., 2018), particularly in the face of a changing climate (Cavicchioli et al., 2019). Unsurprisingly, a great deal of focus has been on quantifying biodiversity—the number, identity, and functions of species (Gotelli and Colwell, 2001; Faith, 2002; Barlow et al., 2007). It has also helped researchers more fully document the ranges of rare or endangered species through environmental DNA detection strategies (Olson et al., 2012; Thomsen et al., 2012; Spear et al., 2015). Further expanding the impacts of this relatively new field, microbial metagenomics has proven exceptionally useful toward informing remediation strategies of disturbed habitats such as ecologically sensitive biological soil crusts (BSC; Bowker, 2007; Steven et al., 2012). Failure to fully understand the microbial (biotic) community can thus dramatically limit understanding of what structures ecological interactions, species distributions, and environmental sustainability, all of which can, in turn, negatively impact informed conservation decision making (Guisan et al., 2013).
Increased accessibility and affordability of high throughput sequencing, has facilitated broad scale exploration of microbial community structure (Logares et al., 2012, 2014; Chen et al., 2017; Zhang et al., 2018). At present, the majority of broad-scale biotic diversity assessments employ primarily culture-independent, amplicon-based sequencing (Petrosino et al., 2009; Mande et al., 2012; Uyaguari-Diaz et al., 2016). This method relies on sufficiently variable, universally present regions of the genome, or “barcoding loci” (Hebert and Gregory, 2005; Kress and Erickson, 2008). Such loci must, first and foremost, be unique enough to yield distinctions between species present in a sample (Kolbert et al., 2004). A proliferation of bioinformatic pipelines developed for barcode sequencing has resulted in widespread capacity to analyze microbial diversity and community structure present in a variety of different environments, ranging from soil to the human body (e.g., QIIME; Caporaso et al., 2010; Kuczynski et al., 2012; Navas-Molina et al., 2013). These pipelines rely primarily on the 16S ribosomal DNA (rDNA) gene as a target for PCR amplification and subsequent sequencing to distinguish species (Winker and Woese, 1991; Kolbert et al., 2004; Petrosino et al., 2009). However, shortcomings of such amplicon-based approaches include moderate to extreme amplification bias (Acinas et al., 2005; Wang and Qian, 2009), thus effectively investigating only a fraction of total standing diversity.
Fungi decompose organic litter (Chapin et al., 2002; Osono, 2007), produce secondary compounds of tremendous importance to humans such as antibiotics (Keller et al., 2005), are used extensively in food production (e.g., bread, wine, beer; Campbell-Platt and Cook, 1989), and are common agricultural pests (e.g., Sclerotinia sclerotiorum; Amselem et al., 2011). Despite their immense ecological (Bever et al., 2001; Pitt and Hocking, 2009; Van Der Heijden and Horton, 2009) and economic importance (Sharma, 1989), fungal metagenomics has, on the whole, received less attention relative to bacterial metagenomics. For example, only 360 NCBI Bioprojects were archived for fungi using amplicon-based molecular barcodes compared to 5,121 Bioprojects available for studies of microbes using amplicon-based molecular barcodes (search conducted on June 21, 2019, in NCBI Bioproject Archive).
Lichens are a species-rich and evolutionarily heterogeneous assemblage of fungi that form obligate symbioses with a minimum of one primary photosynthetic partner, often in addition to other endolichenic fungi, algae, and bacteria (Ahmadjian and Jacobs, 1981; Seaward, 1997; Brodo et al., 2001; Papazi et al., 2015). Lichens are highly successful and ecologically important, as is evidenced by their abundance and diversity in terrestrial ecosystems around the world (Hawksworth, 1991). Along with bryophytes, cyanobacteria, and non-lichenized fungi, lichens are a crucial component of biological soil crust communities and function prominently in ecological restoration processes (Belnap, 2001; Belnap and Lange, 2001; Thompson et al., 2006; Bowker, 2007). In addition to their pivotal ecological contributions to such communities, lichenized fungi have relatively small metagenome sizes (Armaleo and May, 2009; Tripp et al., 2017), making them ideal targets for cost-effective genomics projects (Allen et al., 2018; Brigham et al., 2018; Funk et al., 2018; Pogoda et al., 2018, 2019). Given the above, lichens serve as an excellent system in which to explore the factors that constrain the establishment and development of obligate symbioses in nature, including those prevalent among soil crust communities. Such factors span the dynamics of propagule dispersal, the distribution and establishment of individual symbionts in the environment, and biotic interactions between extant symbionts in a given environment. To date, however, few studies have explored such avenues of research, but these have relied entirely on amplicon-based sequencing methods (Banchi et al., 2018; Eaton et al., 2018; but see, e.g., Tripp et al., 2017; Pizarro et al., 2019).
This shortage of fungal genomic resources broadly, and lichen genomic resources more specifically, makes it challenging to investigate key questions about lichen ecology, evolution, genetics, and physiology. Moreover, existing studies that have investigated fungal metagenomic communities, like bacteria, primarily relied on amplicon-based approaches. Given known complications arising from amplification bias (Acinas et al., 2005; Wang and Qian, 2009), one potential solution is to forego amplification of barcoding loci and instead utilize data from whole-genome shotgun sequencing (WGS). This method avoids classical PCR amplification biases but has been little employed to date, likely as a function of one to several challenges. In addition to increased costs of WGS relative to amplicon-based methods, the lack of developed, publicly available reference databases (e.g., complete or nearly complete rDNA complexes) as well as a paucity of bioinformatics pipelines have limited the utility of WGS as a primary tool with which to approach fungal and other microbial metagenomic research.
In this study, we construct and then employ a new rDNA database spanning 273 species of lichenized fungi, built from a metagenomic survey of lichens in the southern Appalachian Mountain biodiversity hotspot, which is characterized by stark abiotic gradients and is home to over a thousand species of lichens (Dey, 1978; Brodo et al., 2001; Hodkinson, 2010; Lendemer et al., 2013; Tripp and Lendemer, 2019a,b; Tripp et al., in press). Coupled with development of a new bioinformatic pipeline, we identify lichen fungal symbionts present in WGS metagenomic environmental samples and then compare the efficacy of our approach against traditional ITS1-based amplicon sequencing of the same samples. We place our results in the broader framework of intensive biodiversity inventories of lichens at the same plots from which environmental samples were taken. Drawing on resulting data, we demonstrate that detection of symbionts using amplicon-free methods, here WGS, detects more species than amplicon-based methods. We introduce a new biodiversity metric, “Potential of Diversity” (hereafter PoD), which refers to the potential for species to occur at a given site in a given study area, regardless of whether the species is actually present at this site as determined by traditional taxonomic inventory. In the present study, PoD specifically refers to the ratio of lichen fungal symbionts detected on bare surfaces in the environment that would serve as biotic partners in subsequent lichen symbiosis (i.e., the potential lichens that could occur in a study plot based on presence of the required fungal symbiont). In this context, PoD is a useful metric in which to fully consider biotic constraints that limit establishment of obligate symbiotic organisms. We compare this metric to the number of lichens detected as established symbioses (i.e., the lichens that visibly occur at a plot, henceforth “TD” for taxonomic diversity).
Materials and Methods
Study Area, Field Plots and Field Sampling
This study was carried out in two one-hectare plots located in Great Smoky Mountains National Park, in the southern Appalachian Mountain Biodiversity Hotspot of eastern North America (Figure 1). The plots were selected to span the two extremes of a stark elevational (one high, one low) and ecological (bottomland hardwood vs. cloud-laden forest dominated by conifers) gradient in the region, so as to maximize the difference in extant lichen communities (i.e., minimize overlap) as well as potential environmental symbiont pools between the plots. The high-elevation (2,014 m) plot was located on the summit of Clingman's Dome in spruce-fir forest along the border of Swain County, North Carolina and Sevier County, Tennessee. The low-elevation (670 m) plot was located at White Oak Branch in mixed-hardwood forest above the north shore of Fontana Lake, in Swain County, North Carolina.
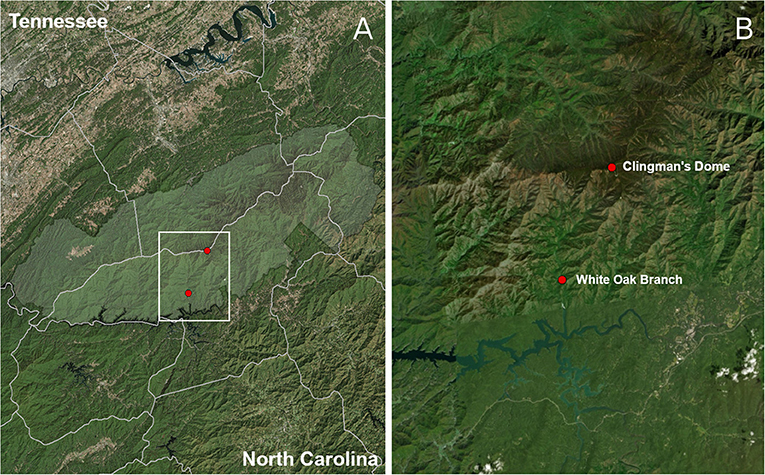
Figure 1. Plot map. Area shaded in light green (A) corresponds to Great Smoky Mountains National Park. White Oak Branch is the low-elevation sampling plot, at 670 m elevation, and Clingman's Dome, the highest peak in Tennessee, is at an elevation of 2,014 m (B).
Prior to environmental sampling, data pertaining to several ecological variables (e.g., tree DBH, woody plant inventory, habitat quality assessment, following Tripp et al., 2019) were recorded in order to delimit the plant communities and to ensure maximum difference in lichen communities (see above). Both plots were delimited to be uniform in vegetation type within a plot (i.e., not spanning more than one ecotone). In each plot, a full inventory of lichen species was conducted [carried out by JL, vouchers deposited in the herbaria of the New York Botanical Garden [NY] and University of Colorado, Boulder [COLO]]; methods following (Tripp et al., 2019). In each plot, 16 environmental samples were obtained by swabbing the surfaces of eight rocks and eight trees (yielding a total of 32 environmental samples) for 30 seconds with a sterile toothbrush (size, aspect, and identification were recorded for each rock and tree type; see below for additional details). In order to avoid sampling surfaces with artificially inflated propagule counts, such as spore deposits along river beds and bogs, we chose only vertically-oriented, bare surfaces of rocks and trees (i.e., free of any visible growth besides the bark of the tree, when applicable).
Thallus Collection
To facilitate direct comparison of amplicon-based sequencing to WGS-based sequencing of the 32 environmental samples, we first obtained lichen voucher specimens for species present throughout the Southern Appalachian Mountains (Table S1) as part of a system-wide investigation of drivers of lichen biodiversity and distributions in the region, including potential biotic constraints (e.g., presence of symbionts) such as those herein investigated. These samples were collected in order to build a new genomic reference database for lichens of the region (see below). Samples were collected and identified by JCL and EAT between December 2016 and January 2018. All lichen voucher specimens are deposited at NY and COLO (Table S1). Efforts were made to sample only single thalli for both macro- and microlichens. For macrolichens, ca. 1 × 1 cm of thallus was removed, targeting the margins and lobes. For microlichens, thallus was scraped from rock or tree substrates using a sterile razor blade. Samples were air dried in a laminar flow hood for 24 h then frozen at −20°C until transport to the University of Colorado for DNA extraction and subsequent sequencing.
Metagenomics Sampling Scheme
To quantify the number and identification of lichenized fungi present in the 32 environmental samples as well as assess differences between amplicon- vs. WGS-based sequencing approaches, we collected samples from eight rock plus eight tree surfaces within each hectare (n = 16 per plot). Substrates were randomly swabbed among those available, and only bare surfaces lacking visible bryophyte or lichen thalli were chosen. Standardized stencils of 10 × 10 cm were placed against the substrate and an individually packaged, sterile toothbrush was used to swab the surface for 30 s. The toothbrush containing the sample was then sealed in a sterile plastic bag. To process the samples, the bag was opened, the bristles were cut from the brush using a sterile blade, and a hole was cut into the corner of the bag using sterile scissors. The opened corner of the bag was placed into a sterile 1.5 mL microcentrifuge tube and the bristles were directly transferred into the tube. These samples were stored at −20°C until transport to the University of Colorado for subsequent extraction and sequencing.
DNA Extraction and Whole Genome Shotgun Sequencing
For both the 32 environmental samples as well as lichen species vouchered in order to build a new reference genomic database, dried samples were pulverized using tungsten carbide bearings in a Qiagen 96-well plate shaker. Genomic DNA (gDNA) was extracted from lichen thallus samples and toothbrush bristles using a Qiagen DNeasy 96 plant kit. Individual samples were transferred from 1.5 mL microcentrifuge tubes into 96 well plates used in the Qiagen kit. The manufacturer's protocol was modified to include a 10 min 65°C incubation step for ground material in lysis buffer as well as a 100% ethanol wash before final drying of the membrane prior to elution. Preliminary study found that these modifications improved lichen gDNA concentration and purity (Pogoda et al., 2018). Extracted samples were stored at −20°C prior to subsequent library preparation.
Whole genome shotgun sequencing was conducted on a total of 494 lichen thallus libraries [these collected throughout the southern Appalachian study area (Figure S1)] and 32 environmental samples on the Illumina NextSeq®. Each of the gDNA samples was prepared using the Nextera® XT DNA library prep kit, which is optimized for 1 ng of total input DNA. Each sample was uniquely tagged using the dual index adapters, Nextera® i5 and i7. Libraries prepared for sequencing on the NextSeq® utilized Illumina PhiX v.3 as a control and samples that passed QC were processed for paired-end 151 base pair reads on an Illumina NextSeq® sequencer at the University of Colorado's BioFrontiers Institute (Boulder, Colorado).
ITS1 Sequencing
To compare results of WGS-based sequencing to amplicon-based sequencing, amplification by PCR was performed on the 32 environmental samples using the ITS1-F (5'-CTTGGTCATTTAGAGGAAGTAA) (Gardes and Bruns, 1993) and ITS2 (5'-GCTGCGTTCTTCATCGATGC) (White et al., 1990) primers. Libraries were prepared for sequencing on the MiSeq® utilized Illumina PhiX v.3 as a control. Samples that passed QC were then processed for single end 251 base pair reads on the Illumina MiSeq® sequencer at the University of Texas' Genomics Sequencing and Analysis Facility (Austin, Texas).
Genome Assembly and Reference Genomic Database
Libraries were filtered with Trimmomatic-0.36 to trim adapters from reads, and with parameters “LEADING:3 TRAILING:3 MINLEN:100” (Bolger et al., 2014). Filtered reads were then assembled using SPAdes 3.9.0 with parameters “–careful -k 21,33,65,81” (Bankevich et al., 2012). This study utilized the whole nuclear ribosomal DNA (rDNA) complexes that were obtained from the de novo genome assembly of lichens that were collected as part of a broader study of lichen diversity in the southern Appalachian study region (Keepers et al., unpub. data). The rDNA complex is easily assembled due to its high copy number in the nuclear genome, and is long (i.e., >5,000 bp, in comparison to amplicon-based sequencing of ITS1, which is <500 bp), providing a larger target onto which sequenced reads may map. Moreover, the high-copy nature of the locus provides many opportunities per propagule to be counted in the downstream analyses. The rDNA complexes for each sample were identified by conducting a BLAST search of the rDNA complex of the trebouxioid algal photobiont from Cladonia uncialis against each of the assemblies. The algal rDNA was used in the search rather than a mycobiont sequence to avoid bias in BLAST hit length due to phylogenetic similarity. To identify complete or mostly complete rDNA complexes from the BLAST tables, contigs were required to have two or greater distinct hits and the span of these hits were required to be >1,000 bp in length. Sequences were parsed from the assemblies based on the nucleotide positions of the BLAST hits and oriented with the 18S in the 5' direction.
Phylogenetic Methods
Each putative rDNA contig parsed from the assemblies was BLAST searched against the NCBI non-redundant database. Any sequences whose best BLAST results mapped to non-lichenized fungi or to non-fungal species were excluded. To further vet the identities of the contigs in the database, sequences were aligned using MUSCLE aligner v3.8.31 (Edgar, 2004) for downstream phylogenetic analysis. The resulting preliminary alignment was trimmed to contain only highly homologous regions, then further trimmed to only sites for which >90% of taxa in the dataset contained sequence data. A best estimate phylogenetic tree was inferred under a model of GTR+I+Γ using MrBayes (Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003) with 10,000,000 MCMC generations and gaps treated as missing data. The first 25% trees were treated as burn-in and excluded from further analyses; eight runs were implemented and 32 chains were utilized. All other parameters were left at default values and the average standard deviation of split frequencies had converged to less than the recommended 0.01 by the end of the analysis. The trees from the posterior distribution were used to generate a majority consensus tree, upon which we mapped posterior probabilities.
This preliminary consensus tree was visualized using FigTree v.1.4.3, upon which we determined that 34 samples were likely misplaced based on disagreement with established large-scale phylogenic reconstructions of lichenized fungi (e.g., Miadlikowska et al., 2014). As such, these 34 samples were excluded from the alignment, which was then further pruned of duplicated taxon sequences such that the final matrix contained only one representative per species (and in each case, the longest rDNA contig was retained). The resulting final alignment included a total of 273 sequences and, after visual inspection following the same phylogenetic methods described above, was used as the reference database for subsequent queries of sequences obtained from sequencing of the environmental samples.
Read Mapping
The 251 bp amplicon reads were truncated to 151 bp to ensure comparability of amplicon-based sequences to WGS sequences. To exclude PCR primer sequence regions, amplicon reads were truncated to include only positions 50–200, representing the highly variable ITS1 sequence. Sequences from both the WGS and amplicon-based approaches were then aligned to the newly constructed reference rDNA database to generate short-read alignment maps. To ensure that reads uniquely mapped to a single locus in the database, several filtering steps were employed. First, only reads that mapped with a CIGAR score between 145 and 151 matches, corresponding to a mapping identity of between 96 and 100%, were retained. Second, only read-pairs for which both the left and right read mapped to the same contig were kept, and then only read-pairs for which both reads mapped with a SAM mapping quality of 31 or greater (out of a maximum of 60) were retained. Due to the highly conserved nature of portions of the coding regions of the ribosomal DNA complex, many reads belonging to species for which multiple congeners were represented in the database mapped almost equally well to multiple species. In these instances, the mapping score was bolstered by the estimated read separation. Thus, a higher SAM CIGAR score of 150 or 151 was required to retain reads that also mapped well to a member of the same genus.
Species Accumulation Curves
To facilitate comparison of species diversity and accumulation as assessed from environmental samples, species richness rarefaction curves for each of the 32 samples falling under four sampling regimes (i.e., eight each from high elevation rock, high elevation tree, low elevation rock, low elevation tree) were generated using the Diversity Stats calculator in EstimateS 9.1.0 (Colwell, 2013). Rarefaction curves were bootstrapped by randomizing the sample order 100 times.
Comparison of Environmental Sampling Methods and Expert-Based Inventory
To assess the congruence between the two metagenomic methods (i.e., WGS vs. amplicon) of detecting symbionts in environmental samples (potential of diversity, or “PoD”) as well as congruence of both methods to expert-based inventory (Coddington et al., 1991; Sørensen et al., 2012) of species found growing in each plot (taxonomic diversity, or “TD”), we pooled the taxa detected in all samples collected in each of the four sampling regimes (high rock, high tree, low rock, low tree) to produce a single list of species detected through a given method) and calculated Jaccard indices in each inventory. These were derived from a presence/absence matrix that consisted of the species present in the rDNA reference database that were found at each of the two elevation extremes via (1) the expert-based lichen biodiversity inventory, (2) the lichenized fungi detected in environmental samples using amplicon-based sequencing, and (3) the lichenized fungi detected in environmental samples using WGS sequencing. Similarity between the lichens detected with the different methods is reported below as JTreatment,Treatment (e.g., JWGS,Vouchered is similarity of lichenized fungi detected by WGS of environmental samples compared to that detected by expert-based inventory).
Results
Ribosomal DNA Database
The final reference genomic database of lichen rDNA contained complete or nearly complete ribosomal DNA complexes (NTS, ETS, 18S, ITS1, 5.8S, ITS2, 26/28S) for 273 unique species of lichenized fungi within Pezizomycotina, representing 25 orders and 57 families. The database spanned 1,770,139 bp of sequence, with an average contig length of 6,484 bp.
Detection of Lichenized Fungi Using Expert, WGS, and Amplicon-Based Inventory
Taxonomic diversity (TD: the number of vouchered species) of lichens found growing in the study plots totaled 57 species in the high-elevation plot and 83 species in the low-elevation plot, yielding a total of 136 species in both (n = 4 species occurred in both). Of these 136 species, 78 (~60%) were represented in the rDNA database generated in this study and thus could potentially be matched to sequences from the environmental samples. Note that the additional species in the database were vouchered from other plots in the study area and were used to ascertain whether this method detected species that were not vouchered in the expert-based field inventory.
Mapping of paired-end WGS reads to the rDNA reference database resulted in the detection of a total of 94 lichenized fungi present in both plots: 43 species from the high-elevation plot and 71 from the low-elevation plot (Table 1). Conversely, using the traditional amplicon-based approach of mapping ITS1 reads to the rDNA reference database resulted in the detection of a total of 34 lichenized fungi present in both plots: 21 species from the high-elevation plot and 18 species from the low-elevation plot (Table 1). Species detection accumulated consistently faster by sample using WGS in all four sampling regimes (Figure 2).Of the 57 species vouchered during the inventory of the high-elevation plot, 13 were detected using both sequencing methods although only seven of these species represented the same species between the two plots (Figure 3; Table 1). Conversely, of the 83 species that were vouchered during the inventory of the low-elevation plot, 17 were detected using either sequencing method, although only 4 were detected by both (Figure 3; Table 1).

Table 1. Inventory of species of lichenized fungi collected at the high-elevation Clingman's Dome and low-elevation White Oak Branch plots.
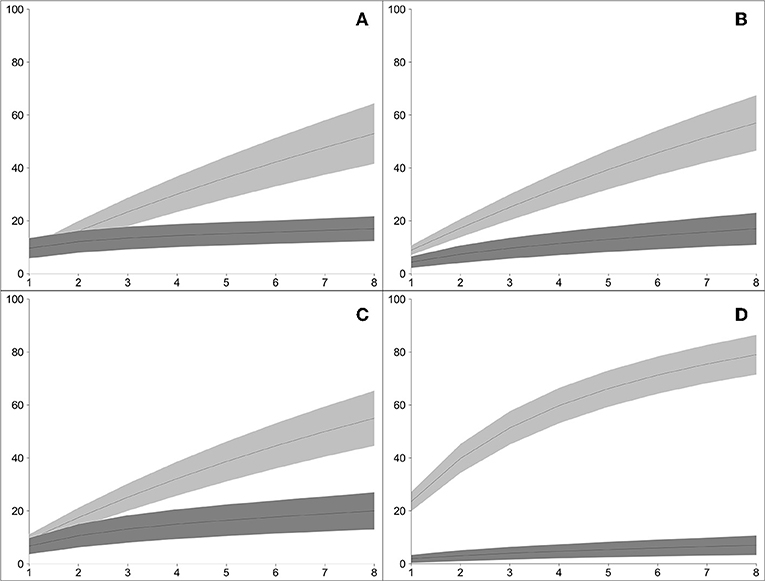
Figure 2. Bootstrapped rarefaction curves of the accumulation of species of lichenized fungi, comparing WGS-based rDNA approach (light gray bands) vs. amplicon-based ITS1 approach (dark gray bands), in the four sampling regimes in two plots: (A) high-elevation rock samples; (B) low-elevation rock samples; (C) high-elevation tree samples; (D) low-elevation tree sample (x-axis: number of environmental samples; y-axis: number of species of lichenized fungi detected). In all four sampling regimes, the WGS-based approached developed in this study detected greater species diversity than the amplicon-based approach.
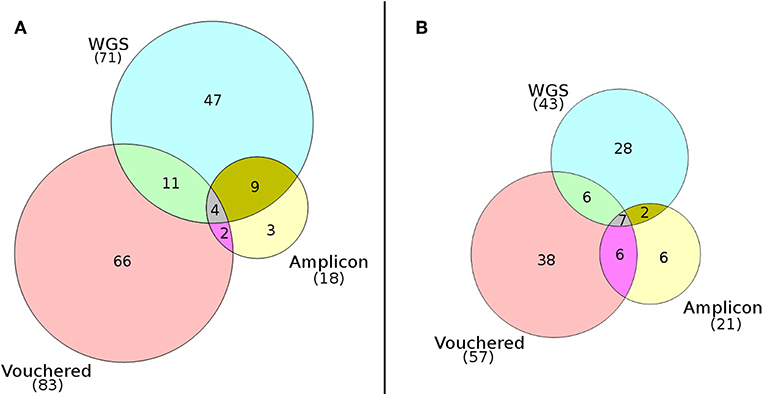
Figure 3. Venn diagrams comparing the numbers of species of lichenized fungi detected at (A) the low-elevation White Oak Branch plot and (B) the high-elevation summit of Clingman's Dome. Diagrams show an overall low overlap of detected species using the WGS approach (PoD), the amplicon approach (PoD), and the expert-based inventory approach (TD).
The similarity between the community of lichens detected growing at the plots via expert-based inventory vs. the pools of lichen fungal symbionts species detected in environmental samples using WGS or amplicon sequencing are reported as Jaccard indices in Table 2. The similarity values for the assemblages of lichenized fungi detected in the environment at the high-elevation plot compared to those vouchered in the expert-based inventory (JWGS,Vouchered = 0.148, JAMP,Vouchered = 0.200) were roughly twice as high as their respective indices at the low-elevation plot (JWGS,Vouchered = 0.108, JAMP,Vouchered = 0.063). This result conveys a greater congruence between the PoD inventory based on WGS sequencing to the TD inventory, than the PoD inventory based on amplicon sequencing to the TD inventory.
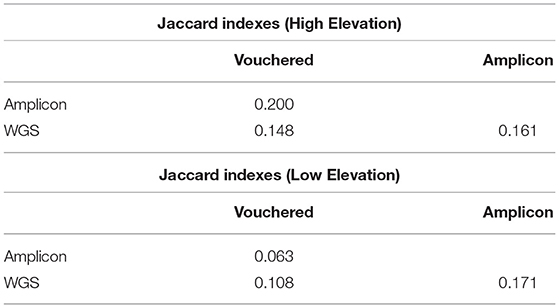
Table 2. Comparison of the congruence between the species detected by either method and the vouchered species at each plot.
Discussion
Our results demonstrate that, in comparison to amplicon sequencing, the WGS approach detects a greater number of species that exist within an environment as propagules. When analyzed in tandem with data from expert-based taxonomic inventories of the same locations, there is an added capability to compare the number and community composition of extant, fully formed symbioses that occur in nature, to those that could potentially occur based on the pool of symbionts in the environment. In turn, this PoD-to-TD comparison helps unravel biotic constraints on the distributions of biodiversity. The estimation of species counts and taxonomic diversity using WGS metagenome sequencing has been conceptually validated in planktonic microbial communities (Poretsky et al., 2014). However, to our knowledge, the application of this method to macro-eukaryotes is only just emerging (Donovan et al., 2018). This study provides a substantial increase in the number of rDNA sequences from lichenized fungi now made available for future research. At the time of writing (April 2019), there were 779 complete (>3,000 bp) ribosomal DNA sequences within the Pezizomycotina (if “18S,” “complete,” and either “26S” or “28S” are required in the search) publicly available on GenBank. Accounting for the 49 complete lichenized fungal ribosomal DNA sequences that had already been submitted from the database curated here, our additional 224 new sequences represent a 29% increase in genomic resources for this locus for lichens.
Our workflow presented herein adds to the growing toolset of molecular contributions to biodiversity science. We have demonstrated the utility of using WGS metagenomic libraries to estimate the pool of available symbionts that are present in an environment. Given the expected ongoing decreases in sequencing cost (Schuster, 2007), we anticipate that WGS will be readily adopted in many study systems and organisms.
Understanding the Distributions of Lichens and Their Propagules
At both sampling plots (high-elevation and low-elevation), species were detected based on environmental (PoD) sampling that were not present based on TD sampling. In other words, lichen mycobionts were detected in the environmental samples but were not detected by the inventory of lichens growing at the site. There are two plausible explanations for these results. First, and likely applicable to most such instances, species detected in the environmental samples but not in the inventories were present only as propagules derived from other locations. It has been shown that the gametes of sexually reproducing lichens can disperse several kilometers (Ronnås et al., 2017), and even clonal propagules are capable of dispersing over a kilometer (Gjerde et al., 2015; Eaton et al., 2018), although these studies suggest that such long-distance dispersal events are rare relative to the total reproductive propagule output of any given individual lichen thallus. It is thus not unexpected that the bank of lichen propagules on a given surface could include representatives of a more diverse pool of lichen species, these derived from a broader geographic neighborhood, regardless of the suitability of the substrate for colonization by those species. Indeed, similar patterns of have been recovered from amplicon sequencing of lichen propagules from indoor dust samples in the United States (Tripp et al., 2016).
The presence of a lichenized fungal propagule alone represents only one element of the total biotic community needed for a lichen species to grow into a mature individual, with the presence or absence of other algal, bacterial, and or other fungal species potentially representing constraints to development of a given lichen thallus (see Tripp et al., 2019). Other constraints include abiotic factors such as elevational or precipitation regimes selected for by different lichen species. One example of this phenomenon in our dataset is Bulbothrix scortella, which was detected only in WGS sequencing of the environmental samples from the high-elevation plot. Bulbothrix scortella is a subtropical species that occurs only at low-elevations in the southern Appalachian Mountains (Hale, 1976; Lendemer et al., 2013). As such, the detection of the species with only WGS sequencing likely reflects the dispersal of a lichenized fungal propagule to the high-elevation plot (from nearby low-elevation habitats), where it is unlikely to become established and undergo further development owing to abiotic and/or biotic constraints.
A second and less likely explanation for our results is failure to detect the presence of a lichen using expert-based TD inventories. Lichens are widely recognized as microhabitat specialists with many species present as small or spatially restricted populations in a given geographic area (Peck et al., 2004; Belinchón et al., 2015; Boch et al., 2016; Dymytrova et al., 2016). Examples from our own work have demonstrated that in some instances, inventories of the same area by more than one collector will yield <50% overlap in the cohorts of documented species (Lendemer et al., 2016). In the present study, Loxospora elatina is a species known to have a distribution restricted to temperate boreal forests (i.e., high elevation habitats in the southern Appalachian Mountains) in North America and Europe (Tønsberg, 1992; Lendemer, 2013). This species was detected by both sequencing methods in the high-elevation plot, but not found in the TD inventory. Thus, it is possible that in a limited number of instances, the species detected in the environmental samples but not in TD inventories were present in low abundance and not detected. However, our expert-based inventories for the broader research project under which the current study falls (i.e., investigating drivers of diversity and distributions of lichens in the southern Appalachian Mountains) target exhaustive sampling until full vouchering of the entire pool of species diversity has been accomplished (Tripp et al., 2019).
Although replicate sampling of additional high- and low-elevation plots was not carried out in this study, we nonetheless detected clear differences in the cohorts of species between the two elevational extremes with all methods herein employed. High- and low-elevations in the southern Appalachians host different lichen communities that share in common only a small number of species (Lendemer and Tripp, 2008, Allen and Lendemer, 2016; Muscavitch and Lendemer, 2016; Lendemer et al., 2017; Tripp and Lendemer, 2019a,b; Tripp et al., 2019). Thus, differences between the inventories of the two plots are to be expected. Some of the differences in the cohorts of species detected through environmental sampling may reflect the overall dispersal limitation of lichen propagules that leads the propagule pool to be dominated by locally occurring species. We hypothesize that at higher elevations one would expect the detection of fewer “long distance dispersal” events such as is described for B. scortella above, when compared to lower elevations. The ability of a propagule to disperse is influenced in part by gravity (Ronnås et al., 2017), and thus propagules will face an uphill battle in dispersing upward through passive wind dispersal.
Benefits and Limitations of WGS vs. Amplicon Sequencing
Our analyses detected approximately four times as many species using the WGS approach compared to the amplicon-based method in the low-elevation plot, and twice as many in the high-elevation plot. The higher number of species detected by WGS affords a stricter threshold for what counts as a detected species in the SAM file (e.g., by only counting species as detected if they occur at least a certain number of times throughout the SAM file, or by requiring a higher SAM mapping quality score before counting the species as detected).
The community present in a metagenomics sample contains the DNA of species that are represented by many cells in some cases, and potentially by a single cell in other cases. Such stark differences in input quantity from different species in the sample leads to potential issues for both WGS and amplicon environmental sequencing methods. While both methods rely on low abundance DNA being extracted in sufficient quality and quantity to be detected in downstream analysis, amplicon-based metagenomics suffers from difficulties in primer design, including issues with the use of universal primers (Acinas et al., 2005; Wang and Qian, 2009). Moreover, unequal (biased) amplification is of wide concern in skewing the distribution of amplicons in the PCR product (Acinas et al., 2005; Sipos et al., 2010). Conversely, one potential issue with WGS metagenomics is that for species with low abundance in the sample, the probability of sampling that species in the prepared library will scale inversely with the proportion of the prepared library that is sequenced. For these reasons, the cohorts of species detected in the community may differ minimally to substantially between the two methods.
One complication of our approach is the presence of group I introns (DePriest and Been, 1992; DePriest, 1993; Gargas et al., 1995) in the coding sequences (18S, 5.8S, and 28S) of many lichenized fungal ribosomal complexes. These introns may be horizontally transferred between species (Hibbett, 1996; Fitzpatrick, 2012; Roy and Irimia, 2012). Thus, it is possible that a read originating from a group I intron in the rDNA complex of a species that is not present in our dataset may appear to map to the database to a species that recently received a horizontal transfer of that intron. However, this will occur very rarely, and even less-so as the database improves, for several reasons. First, our heuristics for read filtering are fairly strict, requiring read-pairs to uniquely map with high percent-identity to members of the database. Unless the intron transferred very recently, the rapid evolution of such introns (Dujon, 1989; Gargas et al., 1995; Roy and Irimia, 2012) would prevent the false detection of a species within the database. Moreover, these introns may facilitate a higher rate of species detection, due to their rapid evolution relative to the coding regions that are too conserved to uniquely identify species in many cases.
Moving forward, we aim to implement the approach presented here to measure the congruence between the observed TD of lichens in the southern Appalachians and the PoD measured in the propagule bank, as one step forward in understanding potential biotic constraints on the distributions of lichen biodiversity. The environmental covariates that predict this congruence may enable the development of cost-effective conservation measures in ecologically sensitive keystone systems such as biological soil crusts (Eldridge, 2000; Belnap, 2003; Belnap and Lange, 2013). We propose that the WGS method presented here should be added to the toolset used by molecular taxonomists in pursuit of conservation and restoration efforts, in addition to facilitating more general understanding on what structures the distributions of species. The datasets and bioinformatic resources presented here represent an important set of new tools and approaches that can be used to address a broad range of questions.
Data Availability Statement
The database consisting of the complete (or nearly complete) ribosomal DNA complexes of 273 lichen mycobionts is available at Zenodo record #3256645. The WGS and amplicon metagenomics libraries are published on the SRA database, SUB5873533.
Author Contributions
KK conducted the research, analyzed the data, and interpreted the data. KK, CP, ET, and JL wrote the paper. ET, JL, CM, and NK conceptualized the project and secured and managed funding. JL, ET, KK, KW, CA, and JH collected and vouchered samples. Processing, curation, and archiving of voucher specimens and field data was managed by AR. KK, KW, CP, and CA performed the extractions and library preps. NK helped develop the bioinformatics pipeline and edited the paper.
Funding
This work was supported by a grant from the National Science Foundation's Dimensions of Biodiversity Program [award #1542639 [University of Colorado] and award #1432629 [New York Botanical Garden]], as well as the National Science Foundation's IGERT award #1144807 for the IQBiology program at University of Colorado's BioFrontiers Institute.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the reviewers for their constructive improvements to this manuscript. Publication of this article was funded by the University of Colorado Boulder Libraries Open Access Fund.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00484/full#supplementary-material
Figure S1. The southern Appalachian study area spans the states of Alabama, Georgia, Tennessee, South Carolina, North Carolina, Virginia, and West Virginia.
References
Acinas, S. G., Sarma-Rupavtarm, R., Klepac-Ceraj, V., and Polz, M. F. (2005). PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71, 8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005
Ahmadjian, V., and Jacobs, J. B. (1981). Relationship between fungus and alga in the lichen Cladonia cristatella tuck. Nature 289, 169–172. doi: 10.1038/289169a0
Allen, J. L., and Lendemer, J. C. (2016). Climate change impacts on endemic, high-elevation lichens in a biodiversity hotspot. Biodivers. Conserv. 25, 555–568. doi: 10.1007/s10531-016-1071-4
Allen, J. L., McKenzie, S. K., Sleith, R. S., and Alter, S. E. (2018). First genome-wide analysis of the endangered, endemic lichen Cetradonia linearis reveals isolation by distance and strong population structure. Am. J. Bot. 105, 1556–1567. doi: 10.1101/237164
Amselem, J., Cuomo, C. A., Van Kan, J. A., Viaud, M., Benito, E. P., Couloux, A., et al. (2011). Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 7:e1002230. doi: 10.1371/journal.pgen.1002230
Armaleo, D., and May, S. (2009). Sizing the fungal and algal genomes of the lichen Cladonia grayi through quantitative PCR. Symbiosis 49:43. doi: 10.1007/s13199-009-0012-3
Banchi, E., Ametrano, C. G., Stanković, D., Verardo, P., Moretti, O., Gabrielli, F., et al. (2018). DNA metabarcoding uncovers fungal diversity of mixed airborne samples in Italy. PLoS ONE 13:e0194489. doi: 10.1371/journal.pone.0194489
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comp. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barlow, J., Gardner, T. A., Araujo, I. S., Ávila-Pires, T. C., Bonaldo, A. B., Costa, J. E., et al. (2007). Quantifying the biodiversity value of tropical primary, secondary, and plantation forests. Proc. Natl. Acad. Sci. U.S.A. 104, 18555–18560. doi: 10.1073/pnas.0703333104
Belinchón, R., Yahr, R., and Ellis, C. J. (2015). Interactions among species with contrasting dispersal modes explain distributions for epiphytic lichens. Ecography 38, 762–768. doi: 10.1111/ecog.01258
Belnap, J. (2001). “Comparative structure of physical and biological soil crusts,” in Biological Soil Crusts: Structure, function, and Management (Berlin, Heidelberg: Springer) 177–191. doi: 10.1007/978-3-642-56475-8_15
Belnap, J. (2003). The world at your feet: desert biological soil crusts. Front. Ecol. Environ. 1, 181–189. doi: 10.1890/1540-9295(2003)001[0181:TWAYFD]2.0.CO;2
Belnap, J., and Lange, O. L. (2001). “Structure and functioning of biological soil crusts: a synthesis,” in Biological Soil Crusts: Structure, Function, and Management (New York, NY; Berlin; Heidelberg: Springer), 471–479. doi: 10.1007/978-3-642-56475-8_33
Belnap, J., and Lange, O. L., (Eds.). (2013). Biological Soil Crusts: Structure, Function, and Management, Vol. 150. Springer Science & Business Media.
Bever, J. D., Schultz, P. A., Pringle, A., and Morton, J. B. (2001). Arbuscular mycorrhizal fungi: more diverse than meets the eye, and the ecological tale of why: the high diversity of ecologically distinct species of arbuscular mycorrhizal fungi within a single community has broad implications for plant ecology. AIBS Bull. 51, 923–931. doi: 10.1641/0006-3568(2001)051[0923:AMFMDT]2.0.CO;2
Boch, S., Prati, D., Schöning, I., and Fischer, M. (2016). Lichen species richness is highest in non-intensively used grasslands promoting suitable microhabitats and low vascular plant competition. Biodivers. Conserv. 25, 225–238. doi: 10.1007/s10531-015-1037-y
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowker, M. A. (2007). Biological soil crust rehabilitation in theory and practice: an underexploited opportunity. Restor. Ecol. 15, 13–23. doi: 10.1111/j.1526-100X.2006.00185.x
Brigham, L. M., Allende, L. M., Shipley, B. R., Boyd, K. C., Higgins, T. J., Kelly, N., et al. (2018). Genomic insights into the mitochondria of 11 eastern North American species of Cladonia. Mitochondrial DNA Part B 3, 508–512. doi: 10.1080/23802359.2018.1463827
Brodo, I. M., Sharnoff, S. D., and Sharnoff, S. (2001). Lichens of North America. New Haven; London: Yale University Press.
Campbell-Platt, G., and Cook, P. E. (1989). Fungi in the production of foods and food ingredients. J. Appl. Bacteriol. 67, 117s−131s. doi: 10.1111/j.1365-2672.1989.tb03776.x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335. doi: 10.1038/nmeth.f.303
Cavicchioli, R., Ripple, W. J., Timmis, K. N., Azam, F., Bakken, L. R., Baylis, M., et al. (2019). Scientists' warning to humanity: microorganisms and climate change. Nat. Rev. Microbiol. 1:569–86. doi: 10.1038/s41579-019-0222-5
Chapin, F. S., Matson, P. A., and Mooney, H. A. (2002). Terrestrial Decomposition. New York, NY: Springer New York, 151–175.
Chen, W., Pan, Y., Yu, L., Yang, J., and Zhang, W. (2017). Patterns and processes in marine microeukaryotic community biogeography from Xiamen coastal waters and intertidal sediments, southeast China. Front. Microbiol. 8:1912. doi: 10.3389/fmicb.2017.01912
Chen, Z. M., Chang, W. H., Zheng, A. J., Zhang, S., Cai, H. Y., and Liu, G. H. (2018). Comparison of gut microbial diversity in beijing oil and arbor acres chickens. Rev. Brasil. Ciênc. Avícol. 20, 37–44. doi: 10.1590/1806-9061-2017-0549
Coddington, J. A., Griswold, C. E., Silva Davila, D., Penaranda, E., and Larcher, S. F. (1991). “Designing and testing sampling protocols to estimate biodiversity in tropical ecosystems,” in The Unity of Evolutionary Biology: Proceedings of the Fourth International Congress of Systematic and Evolutionary Biology, ed E. C. Dudley (Portland, OR: Dioscorides Press), 44–60.
Colwell, R. K. (2013). EstimateS: Statistical Estimation of Species Richness and Shared Species From Samples. Version 9. Available online at: http://purl.oclc.org/estimates.
DePriest, P. T. (1993). Small subunit rDNA variation in a population of lichen fungi due to optional group-I introns. Gene 134, 67–74. doi: 10.1016/0378-1119(93)90175-3
DePriest, P. T., and Been, M. D. (1992). Numerous group I introns with variable distributions in the ribosomal DNA of a lichen fungus. J. Mol. Biol. 228, 315–321. doi: 10.1016/0022-2836(92)90819-6
Dey, J. P. (1978). Fruticose and foliose lichens of the high-mountain areas of the southern Appalachians. Bryologist 81, 1–93. doi: 10.2307/3242271
Donovan, P. D., Gonzalez, G., Higgins, D. G., Butler, G., and Ito, K. (2018). Identification of fungi in shotgun metagenomics datasets. PLoS ONE 13:e0192898. doi: 10.1371/journal.pone.0192898
Dujon, B. (1989). Group I introns as mobile genetic elements: facts and mechanistic speculations—a review. Gene 82, 91–114. doi: 10.1016/0378-1119(89)90034-6
Dymytrova, L., Stofer, S., Ginzler, C., Breiner, F. T., and Scheidegger, C. (2016). Forest-structure data improve distribution models of threatened habitat specialists: implications for conservation of epiphytic lichens in forest landscapes. Biol. Conserv. 196, 31–38. doi: 10.1016/j.biocon.2016.01.030
Eaton, S., Zuniga, C., Czyzewski, J., Ellis, C. J., Genney, D., Haydon, D., et al. (2018). A method for the direct detection of airborne dispersal in lichens. Mol. Ecol. Resour. 18, 240–250. doi: 10.1111/1755-0998.12731
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eldridge, D. (2000). Ecology and management of biological soil crusts: recent developments and future challenges. Bryologist 103, 742–747. doi: 10.1639/0007-2745(2000)103[0742:EAMOBS]2.0.CO;2
Faith, D. P. (2002). Quantifying biodiversity: a phylogenetic perspective. Conserv. Biol. 16, 248–252. doi: 10.1046/j.1523-1739.2002.00503.x
Fitzpatrick, D. A. (2012). Horizontal gene transfer in fungi. FEMS Microbiol. Lett. 329, 1–8. doi: 10.1111/j.1574-6968.2011.02465.x
Funk, E. R., Adams, A. N., Spotten, S. M., Van Hove, R. A., Whittington, K. T., Keepers, K. G., et al. (2018). The complete mitochondrial genomes of five lichenized fungi in the genus Usnea (Ascomycota: Parmeliaceae). Mitochondrial DNA Part B 3, 305–308. doi: 10.1080/23802359.2018.1445485
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Gargas, A., DePriest, P. T., and Taylor, J. W. (1995). Positions of multiple insertions in SSU rDNA of lichen-forming fungi. Mol. Biol. Evol. 12, 208–218.
Gjerde, I., Blom, H. H., Heegaard, E., and Sætersdal, M. (2015). Lichen colonization patterns show minor effects of dispersal distance at landscape scale. Ecography 38, 939–948. doi: 10.1111/ecog.01047
Gotelli, N. J., and Colwell, R. K. (2001). Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 4, 379–391. doi: 10.1046/j.1461-0248.2001.00230.x
Guisan, A., Tingley, R., Baumgartner, J. B., Naujokaitis-Lewis, I., Sutcliffe, P. R., Tulloch, A. I., et al. (2013). Predicting species distributions for conservation decisions. Ecol. Lett. 16, 1424–1435. doi: 10.1111/ele.12189
Hale, M. E. Jr. (1976). A monograph of the lichen genus Bulbothrix Hale (Parmeliaceae). Smithsonian Contrib. Bot. 32, 1–29. doi: 10.5479/si.0081024X.32
Hawksworth, D. L. (1991). The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol. Res. 95, 641–655. doi: 10.1016/S0953-7562(09)80810-1
Hebert, P. D., and Gregory, T. R. (2005). The promise of DNA barcoding for taxonomy. Syst. Biol. 54, 852–859. doi: 10.1080/10635150500354886
Hibbett, D. S. (1996). Phylogenetic evidence for horizontal transmission of group I introns in the nuclear ribosomal DNA of mushroom-forming fungi. Mol. Biol. Evol. 13, 903–917. doi: 10.1093/oxfordjournals.molbev.a025658
Hodkinson, B. P. (2010). A first assessment of lichen diversity for one of North America's ‘biodiversity hotspots’ in the southern Appalachians of Virginia. Castanea 75, 126–133. doi: 10.2179/09-033.1
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogeny. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Keller, N. P., Turner, G., and Bennett, J. W. (2005). Fungal secondary metabolism—from biochemistry to genomics. Nat. Rev. Microbiol. 3:937. doi: 10.1038/nrmicro1286
Kolbert, C. P., Rys, P. N., Hopkins, M., Lynch, D. T., Germer, J. J., O'Sullivan, C. E., et al. (2004). “16S ribosomal DNA sequence analysis for identification of bacteria in a clinical microbiology laboratory” in Molecular Microbiology: Diagnostic Principles and Practice (Washington, DC: ASM Press), 361–377.
Kress, W. J., and Erickson, D. L. (2008). DNA barcodes: genes, genomics, and bioinformatics. Proc. Natl. Sci. U.S.A. 105, 2761–2762. doi: 10.1073/pnas.0800476105
Kuczynski, J., Stombaugh, J., Walters, W. A., González, A., Caporaso, J. G., and Knight, R. (2012). Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protocols Microbiol. 27, 1E−5. doi: 10.1002/9780471729259.mc01e05s27
Lendemer, J. C. (2013). Two new sterile species of loxospora (Sarrameanaceae: Lichenized Ascomycetes) from the mid-atlantic coastal plain. J. North Carol. Acad. Sci. 129, 71–81. doi: 10.7572/2167-5880-129.3.71
Lendemer, J. C., Anderson Stewart, C. R., Besal, B., Goldsmith, J., Griffith, H., Hoffman, J. R., et al. (2017). The lichens and allied fungi of mount mitchell state park, North Carolina: a first checklist with comprehensive keys and comparison to historical data. Castanea 82, 69–97. doi: 10.2179/17-126
Lendemer, J. C., Harris, R. C., and Ruiz, A. M. (2016). A review of the lichens of the dare regional biodiversity hotspot in the mid-atlantic coastal plain of North Carolina, eastern North America. Castanea 81, 1–77. doi: 10.2179/15-073R2
Lendemer, J. C., Harris, R. C., and Tripp, E. A. (2013). The lichens and allied fungi of Great Smoky Mountains National Park. Mem. N. Y. Botan. Garden 104, 1–152.
Lendemer, J. C., and Tripp, E. A. (2008). Contributions to the lichen flora of North Carolina: a preliminary checklist of the lichens of Gorges State Park. The Bryologist 111, 57–67. doi: 10.1639/0007-2745(2008)111[57:CTTLFO]2.0.CO;2
Logares, R., Haverkamp, T. H., Kumar, S., Lanzén, A., Nederbragt, A. J., Quince, C., et al. (2012). Environmental microbiology through the lens of high-throughput DNA sequencing: synopsis of current platforms and bioinformatics approaches. J. Microbiol. Methods 91, 106–113. doi: 10.1016/j.mimet.2012.07.017
Logares, R., Sunagawa, S., Salazar, G., Cornejo-Castillo, F. M., Ferrera, I., Sarmento, H., et al. (2014). Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 16, 2659–2671. doi: 10.1111/1462-2920.12250
Mande, S. S., Mohammed, M. H., and Ghosh, T. S. (2012). Classification of metagenomic sequences: methods and challenges. Brief. Bioinformatics 13, 669–681. doi: 10.1093/bib/bbs054
Miadlikowska, J., Kauff, F., Högnabba, F., Oliver, J. C., Molnár, K., Fraker, E., et al. (2014). A multigene phylogenetic synthesis for the class Lecanoromycetes (Ascomycota): 1307 fungi representing 1139 infrageneric taxa, 317 genera and 66 families. Mol. Phylogenet. Evol. 79, 132–168. doi: 10.1016/j.ympev.2014.04.003
Muscavitch, Z. M., and Lendemer, J. C. (2016). A new species of Acanthothecis (Ostropales), highlights subtropical floristic elements of the southern Appalachian lichen biota in eastern North America. Bryologist 119, 350–360. doi: 10.1639/0007-2745-119.4.350
Navas-Molina, J. A., Peralta-Sánchez, J. M., González, A., McMurdie, P. J., Vázquez-Baeza, Y., Xu, Z., et al. (2013). “Advancing our understanding of the human microbiome using QIIME,” in Methods in Enzymology, Vol 531 (Cambridge, MA: Academic Press), 371–444. doi: 10.1016/B978-0-12-407863-5.00019-8
Nottingham, A. T., Fierer, N., Turner, B. L., Whitaker, J., Ostle, N. J., McNamara, N. P., et al. (2018). Microbes follow Humboldt: temperature drives plant and soil microbial diversity patterns from the Amazon to the Andes. Ecology 99, 2455–2466. doi: 10.1002/ecy.2482
Olson, Z. H., Briggler, J. T., and Williams, R. N. (2012). An eDNA approach to detect eastern hellbenders (Cryptobranchus a. alleganiensis) using samples of water. Wildlife Res. 39, 629–636. doi: 10.1071/WR12114
Osono, T. (2007). Ecology of ligninolytic fungi associated with leaf litter decomposition. Ecol. Res. 22, 955–974. doi: 10.1007/s11284-007-0390-z
Papazi, A., Kastanaki, E., Pirintsos, S., and Kotzabasis, K. (2015). Lichen symbiosis: nature's high yielding machines for induced hydrogen production. PLoS ONE 10:e0121325. doi: 10.1371/journal.pone.0121325
Peck, J. E., Grabner, J., Ladd, D., and Larsen, D. R. (2004). Microhabitat affinities of Missouri Ozarks lichens. Bryologist 107, 47–61. doi: 10.1639/0007-2745(2004)107[47:MAOMOL]2.0.CO;2
Petrosino, J. F., Highlander, S., Luna, R. A., Gibbs, R. A., and Versalovic, J. (2009). Metagenomic pyrosequencing and microbial identification. Clin. Chem. 55, 856–866. doi: 10.1373/clinchem.2008.107565
Pike, L. J., Viciani, E., and Kumar, N. (2018). Genome watch: microbial diversity knows no borders. Nat. Rev. Microbiol. 16:66. doi: 10.1038/nrmicro.2017.166
Pitt, J. I., and Hocking, A. D. (2009). “The ecology of fungal food spoilage,” in Fungi and Food Spoilage (Boston, MA: Springer), 3–9. doi: 10.1007/978-0-387-92207-2_2
Pizarro, D., Dal Grande, F., Leavit, S. D., Dyer, P. S., Schmitt, I., Crespo, A., et al. (2019). Whole-genome sequence data uncover widespread heterothallism in the largest group of lichen-forming fungi. Genome Biol. Evol. 11, 721–730. doi: 10.1093/gbe/evz027
Pogoda, C. S., Keepers, K. G., Lendemer, J. C., Kane, N. C., and Tripp, E. A. (2018). Reductions in complexity of mitochondrial genomes in lichen-forming fungi shed light on genome architecture of obligate symbioses. Mol. Ecol. 27, 1155–1169. doi: 10.1111/mec.14519
Pogoda, C. S., Keepers, K. G., Nadiadi, A. Y., Bailey, D. W., Lendemer, J. C., Tripp, E. A., et al. (2019). Genome streamlining via complete loss of introns has occurred multiple times in lichenized fungal mitochondria. Ecol. Evol. 9, 4245–4263. doi: 10.1002/ece3.5056
Poretsky, R., Rodriguez, -R. L. M., Luo, C., Tsementzi, D., and Konstantinidis, K. T. (2014). Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 9:e93827. doi: 10.1371/journal.pone.0093827
Ronnås, C., Werth, S., Ronnås, C., Werth, S., Ovaskainen, O., Varkonyi, G., et al. (2017). Discovery of long-distance gamete dispersal in a lichen-forming ascomycete. New Phytologist 216, 216–226. doi: 10.1111/nph.14714
Ronquist, F., and Huelsenbeck, J. P. (2003). MRBAYES 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Roy, S. W., and Irimia, M. (2012). Genome evolution: where do new introns come from? Curr. Biol. 22, R529–R531. doi: 10.1016/j.cub.2012.05.017
Schuster, S. C. (2007). Next-generation sequencing transforms today's biology. Nat. Methods 5:16. doi: 10.1038/nmeth1156
Seaward, M. R. D. (1997). Major impacts made by lichens in biodeterioration processes. Int. Biodeterior. Biodegr. 40, 269–273. doi: 10.1016/S0964-8305(97)00056-5
Sharma, H. S. (1989). Economic importance of thermophilous fungi. Appl. Microbiol. Biotechnol. 31, 1–10. doi: 10.1007/BF00252517
Sipos, R., Székely, A., Révész, S., and Márialigeti, K. (2010). Addressing PCR biases in environmental microbiology studies. Methods Mol. Biol. 599, 37–58. doi: 10.1007/978-1-60761-439-5_3
Sørensen, L. L., Coddington, J. A., and Scharf, N. (2012). Inventorying and estimating subcanopy spider diversity using semiquantitative sampling methods in an afromontane forest. Pest Manage. Sampl. 31, 319–330. doi: 10.1603/0046-225X-31.2.319
Spear, S. F., Groves, J. D., Williams, L. A., and Waits, L. P. (2015). Using environmental DNA methods to improve detectability in a hellbender (Cryptobranchus alleganiensis) monitoring program. Biol. Conserv. 183, 38–45. doi: 10.1016/j.biocon.2014.11.016
Steven, B., Gallegos-Graves, L. V., Starkenburg, S. R., Chain, P. S., and Kuske, C. R. (2012). Targeted and shotgun metagenomic approaches provide different descriptions of dryland soil microbial communities in a manipulated field study. Environ. Microbiol. Rep. 4, 248–256. doi: 10.1111/j.1758-2229.2012.00328.x
Thompson, W. A., Eldridge, D. J., and Bonser, S. P. (2006). Structure of biological soil crust communities in Callitris glaucophylla woodlands of New South Wales, Australia. J. Veg. Sci. 17, 271–280. doi: 10.1111/j.1654-1103.2006.tb02446.x
Thomsen, P. F., Kielgast, J., Iversen, L. L., Møller, P. R., Rasmussen, M., and Willerslev, E. (2012). Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE 7:e41732. doi: 10.1371/journal.pone.0041732
Tønsberg, T. (1992). The sorediate and isidiate, corticolous, crustose lichens in Norway. Sommerfeltia 14, 1–331.
Tripp, E. A., and Lendemer, J. C. (2019a). Highlights from 10 years of lichenological research in Great Smoky Mountains National Park: celebrating the United States National Park Service centennial. Syst. Bot. 44, 943–980. doi: 10.1600/036364419X15710776741332
Tripp, E. A., and Lendemer, J. C. (2019b). Field Guide to the Lichens of Great Smoky Mountains National Park. Tennessee: University of Tennessee Press, Knoxville.
Tripp, E. A., Lendemer, J. C., Barberán, A., Dunn, R. R., and Fierer, N. (2016). Biodiversity gradients in obligate symbiotic organisms: exploring the diversity and traits of lichen propagules across the United States. J. Biogeogr. 43, 1667–1678. doi: 10.1111/jbi.12746
Tripp, E. A., Lendemer, J. C., and McCain, C. M. (2019). Habitat quality and disturbance drive lichen species richness in a temperate biodiversity hotspot. Oecologia 190, 445–457. doi: 10.1007/s00442-019-04413-0
Tripp, E. A., Zhuang, Y., and Lendemer, J. C. (2017). A review of existing whole genome data suggests lichen mycelia may be haploid or diploid. Bryologist 120, 302–310. doi: 10.1639/0007-2745-120.3.302
Uyaguari-Diaz, M. I., Chan, M., Chaban, B. L., Croxen, M. A., Finke, J. F., Hill, J. E., et al. (2016). A comprehensive method for amplicon-based and metagenomic characterization of viruses, bacteria, and eukaryotes in freshwater samples. Microbiome 4:20. doi: 10.1186/s40168-016-0166-1
Van Der Heijden, M. G., and Horton, T. R. (2009). Socialism in soil? The importance of mycorrhizal fungal networks for facilitation in natural ecosystems. J. Ecol. 97, 1139–1150. doi: 10.1111/j.1365-2745.2009.01570.x
Wang, Y., and Qian, P. Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 4:e7401. doi: 10.1371/journal.pone.0007401
White, T. J., Bruns, T., Lee, S. J. W. T., and Taylor, J. W. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. 18, 315–322. doi: 10.1016/B978-0-12-372180-8.50042-1
Winker, S., and Woese, C. R. (1991). A definition of the domains Archaea, Bacteria and Eucarya in terms of small subunit ribosomal RNA characteristics. Syst. Appl. Microbiol. 14, 305–310. doi: 10.1016/S0723-2020(11)80303-6
Keywords: metagenomics, ribosomal RNA, sequence database, biological soil crusts, lichen, potential of diversity, taxonomic diversity
Citation: Keepers KG, Pogoda CS, White KH, Anderson Stewart CR, Hoffman JR, Ruiz AM, McCain CM, Lendemer JC, Kane NC and Tripp EA (2019) Whole Genome Shotgun Sequencing Detects Greater Lichen Fungal Diversity Than Amplicon-Based Methods in Environmental Samples. Front. Ecol. Evol. 7:484. doi: 10.3389/fevo.2019.00484
Received: 10 July 2019; Accepted: 28 November 2019;
Published: 13 December 2019.
Edited by:
Maik Veste, Brandenburg University of Technology Cottbus-Senftenberg, GermanyReviewed by:
Martin Grube, University of Graz, AustriaRobert Friedman, University of South Carolina, United States
Copyright © 2019 Keepers, Pogoda, White, Anderson Stewart, Hoffman, Ruiz, McCain, Lendemer, Kane and Tripp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyle Garrett Keepers, kylekeepers@gmail.com