
Exploring the Connection Between the Gut Microbiome and Parkinson’s Disease Symptom Progression and Pathology: Implications for Supplementary Treatment Options
Abstract
The contribution of the microbiota to induce gastrointestinal inflammation is hypothesized to be a key component of alpha-synuclein (aSyn) aggregation within the gastrointestinal (GI) tract in the pathological progression of Parkinson’s disease (PD). The function of the GI tract is governed by a system of neurons that form part of the enteric nervous system (ENS). The ENS hosts 100–500 million nerve cells within two thin layers lining the GI tract. The gut-brain axis (GBA) is the major communication pathway between the ENS and the central nervous system. It has become increasingly clear that the microbiota in the gut are key regulators of GBA function and help to maintain homeostasis in the immune and endocrine systems. The GBA may act as a possible etiological launching pad for the pathogenesis of age-related neurodegenerative diseases, such as PD, because of an imbalance in the gut microbiota. PD is a multi-faceted illness with multiple biological, immunological, and environmental factors contributing to its pathological progression. Interestingly, individuals with PD have an altered gut microbiota compared to healthy individuals. However, there is a lack of literature describing the relationship between microbiota composition in the gut and symptom progression in PD patients. This review article examines how the pathology and symptomology of PD may originate from dysregulated signaling in the ENS. We then discuss by targeting the imbalance within the gut microbiota such as prebiotics and probiotics, some of the prodromal symptoms might be alleviated, possibly curtailing the pathological spread of aSyn and ensuing debilitating motor symptoms.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting approximately 2–3% of people worldwide above the age of 65. With an increase in life expectancy and an aging population, PD has been determined to be the fastest growing neurological disorder [1]. PD affects males 1.5 times more than females with most cases arising in higher-income nations [1, 2]. Classified as a multi-faceted illness, there are several biological, immunological, and environmental factors that contribute to the pathological progression of this disease. Depending on these risk factors, the etiology of PD has been classically categorized as familial/genetic or sporadic/idiopathic. In this regard, the possibility that the factors contributing to PD exist on a spectrum with familial links that result in PD diagnosis at one end and purely environmental impacts at the other. Familial PD has a strong genetic component and is caused by specific gene mutations. However, since not all genes associated with PD give rise to Parkinsonian symptoms speaks to the role of environmental factors in the provocation of the illness. Further suggesting that sporadic/idiopathic PD may have multiple unknown contributing factors, or interactive effects of genetic vulnerability and environmental factors that make up the 95% majority of confirmed PD cases [3].
Whether the etiology of PD is familial or sporadic, cardinal symptoms among PD patients include resting tremor, bradykinesia, rigidity, postural instability, and stooped posture [4]. A diagnosis of PD is normally made only once these motor symptoms become apparent in an individual. Along with the emergence of the cardinal motor symptoms, it is also at this time that around 60–80% of dopaminergic neurons have degenerated in the substantia nigra (SN) and approximately 80% of dopamine levels are depleted in the nigrostriatal terminals [5, 6]. A key pathological hallmark associated with dopaminergic cell death is the intraneuronal misfolding and aggregation of the neuronal protein, alpha-synuclein (aSyn) [7].
aSyn is a 140 amino acid (aa) polypeptide encoded by the SNCA gene located at the q21-23 region of the long arms of chromosome 4 [8]. This protein is predominantly found in native monomeric forms at the presynaptic terminal in the brain and in various tissues such as the heart and the gastrointestinal (GI) tract [9, 10]. Although the function of aSyn remains elusive, the location of aSyn has hinted at its role in synaptic plasticity, vesicle packaging, and neurotransmitter release [11]. In 1997, two pivotal studies confirmed the involvement of aSyn in PD by identifying a missense point mutation of A53T in the SNCA gene that led to early-onset familial PD [12] and discovering that Lewy bodies (LBs) are mainly composed of aggregated forms of aSyn [13].
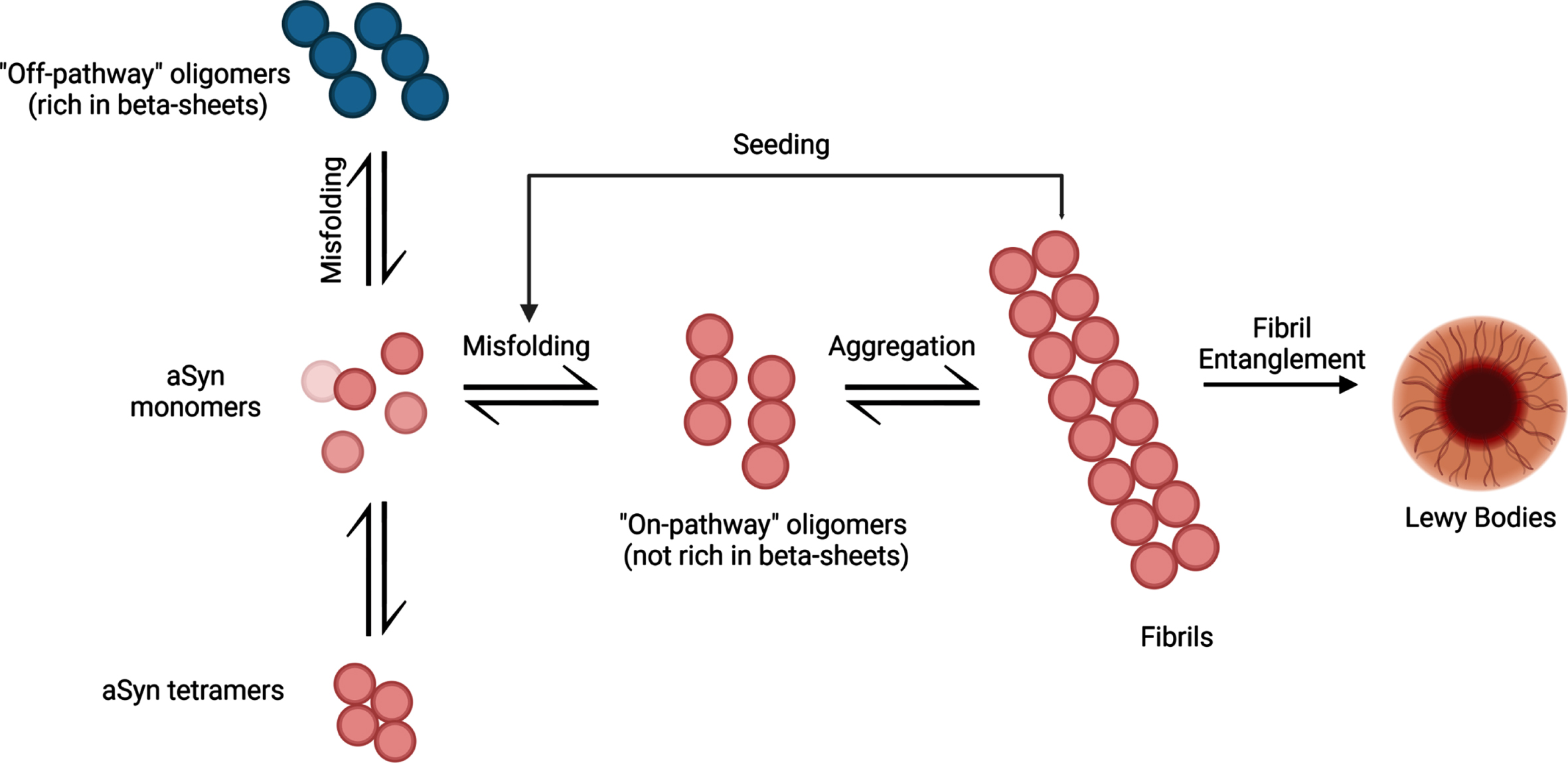
Initially, aSyn aggregation begins slowly as monomeric aSyn starts to misfold and clump into oligomeric structures. Protein degradation systems like the ubiquitin-proteasome system and autophagy-lysosomal pathways are present to combat the formation of aggregated aSyn [14]. Over time, the ability to degrade abnormal protein structures declines, causing increased levels of misfolded aSyn. This creates an imbalance between monomer and oligomer forms of aSyn, resulting in misfolded protein accumulation. As the aggregation continues to develop, oligomeric forms of aSyn develop into stable insoluble fibrils (Fig. 1). It is hypothesized that the fibrils may spread to neighboring cells in a prion-like manner and act as a template for monomeric aSyn proteins to misfold, propagating aSyn-associated neurodegeneration [15]. Altogether, examining the ability of pathogenic aSyn to spread to neighboring cells and induce aSyn-associated cellular death may help to uncover the trajectory of tissue-level damage and determine its relationship to the various symptom profiles associated with PD pathology.
Fig. 1
The aggregation process of aSyn protein. aSyn protein is normally found in natively unfolded monomeric structures but can also be found in α-helical tetramers when binding to lipid compounds. Aggregation of misfolded aSyn protein can either form oligomers that are rich in β-sheet formations (off-pathway) or oligomers that are not rich in β-sheet formations (on-pathway). Oligomers that are not rich in β-sheet conformations aggregate into fibrils, ultimately leading to the formation of LBs. These pathologic aSyn fibrils can act as a template for aSyn monomers and induce misfolding, creating a cycle of misfolding and aggregation of aSyn. Created with BioRender.com.
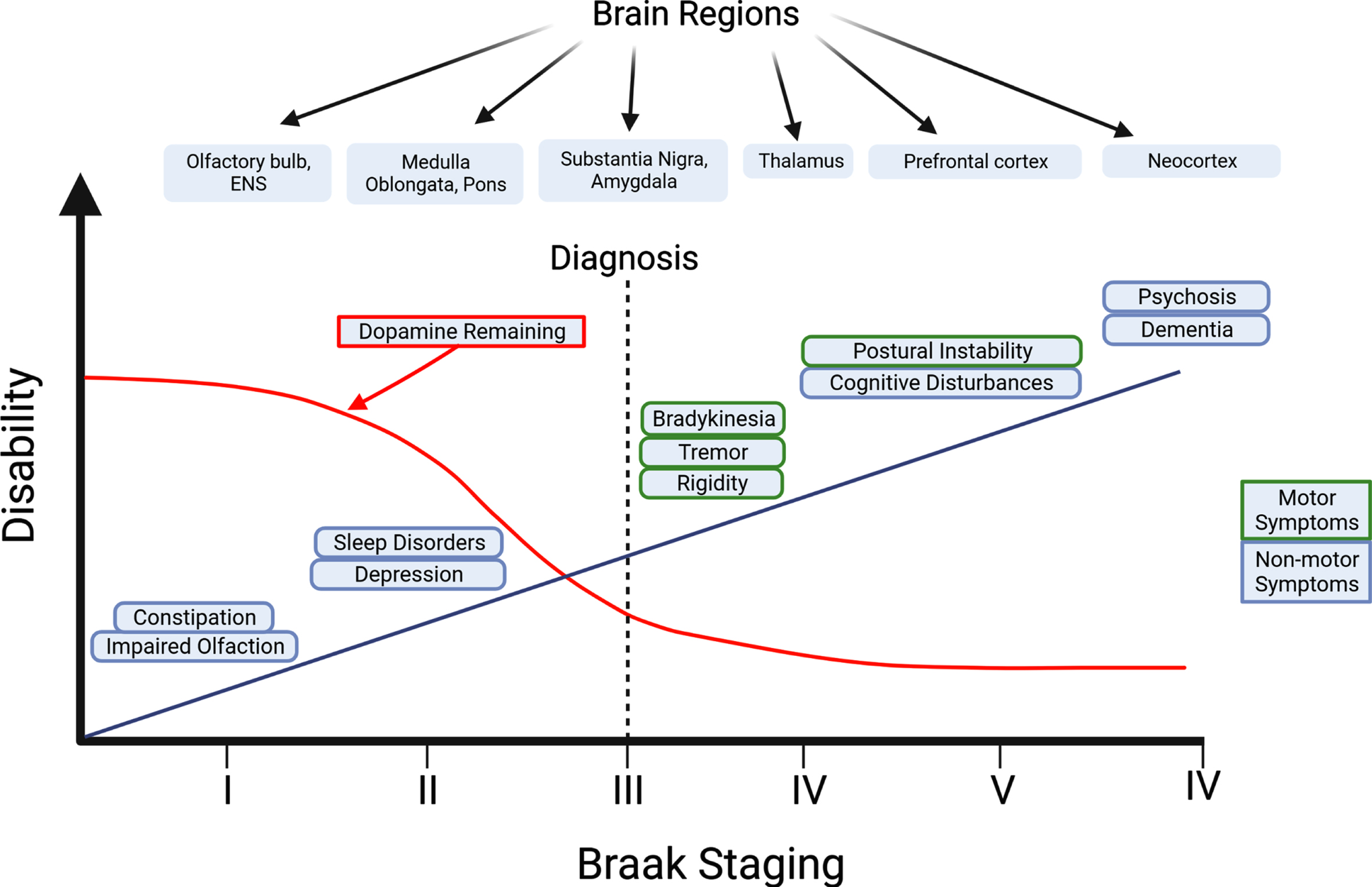
Although the diagnosis of PD is dependent on the emergence of motor symptoms, prodromal non-motor symptoms often start before motor symptoms emerge (Fig. 2) [16]. These non-motor symptoms range from cognitive and behavioral symptoms, to sleep disorders, and gastrointestinal issues, with gastrointestinal symptoms affecting 80% of PD [17]. Because gastrointestinal issues are common in PD and arise years prior to motor symptoms, viewing changes in gastrointestinal function may prove to be a biomarker in the early identification and treatment of PD [18, 19].
Fig. 2
Progression of non-motor and motor symptoms in Parkinson’s disease. In the Braak Model of PD, it is hypothesized that the origin of PD originates in the PNS such as the olfactory bulbs and the ENS (Stage I). Non-motor symptoms can start to appear months or years prior to the onset of motor symptoms. Transmission of pathogenic forms of aSyn is thought to spread throughout the body in a prion-like manner, where aggregated aSyn can travel from the ENS to the brain via the vagus nerve. The diagnosis of PD is made once motor symptoms become apparent (Stage III). Long-term progression of PD causes a decrease in dopamine levels in the brain and an increase in the severity of motor and non-motor symptoms such as postural instability and cognitive disturbances, resulting in a dramatic decline in the quality of life of PD patients. Created with BioRender.com.
BRAAK STAGING
First described in 2003, the “Braak’s hypothesis” suggests PD pathology follows a six-stage sequence during disease development that may start in the peripheral nervous system (PNS) [20]. This model is further expanded by the “dual-hit” hypothesis, as it suggests that PD may originate from environmental toxins such as air pollutants and viruses that enter through the nasal and/or GI route before spreading towards the brain via the vagus nerve [21]. Alternatively, in other patients with PD, it also has been hypothesized to originate in the central nervous system (CNS) and affect the amygdala first before spreading to the SN and PNS [22]. In this “CNS-first” model, possible sites of LB pathology may originate in the amygdala or in off-regions such as the olfactory bulb [23]. In addition, there are key differences between the “PNS-first” and the “CNS-first” PD phenotypes. Primarily, in the “PNS-first” model there is a strong association in patients that experience REM sleep behavior disorders (RBD) in the prodromal phase, whereas in the “CNS-first” model, there is a lack of RBDs. Interestingly, despite nigrostriatal dopaminergic dysfunction occurring after PNS damage in the “PNS-first” model compared to dopaminergic dysfunction occurring before PNS involvement in the “CNS-first” model, motor symptom progression is faster in the “PNS-first” model [23]. While it is probable that there are different subtypes of PD as suggested by Borghammer & Van Den Berge [22], the “PNS-first” model continues to be used as the gold standard for PD progression since most PD-positive patients show this pattern of pathology [22]. When looking at the biopsies of PD patients, all patients showed signs of LB pathology in the esophagus, suggesting that LB formations could arise outside the CNS [24]. Further autopsy reports showed LB pathology in other ENS areas like the stomach, colon, and rectum [25]. To explore the consequences of LB pathology in the ENS, preformed aSyn fibrils (PFFs) were injected into the gastrointestinal tract of wild-type mice. Within days, phosphorylated-aSyn pathology was observed in both the ENS and the dorsal motor nucleus of the vagus nerve (DMV) before reaching the substantia nigra pars compacta (SNpc) [26, 27]. Taken together, these pieces of evidence help support Braak’s hypothesis of PD pathology originating in the PNS before reaching the brain via aSyn prion-like mechanisms.
THE ENTERIC NERVOUS SYSTEM’S ROLE IN PD
Referred to as the “second brain”, the enteric nervous system (ENS) contains 100–500 million neurons, making it the largest collection of neurons outside the CNS [28, 29]. These neurons are held within two thin layers lining the GI tract from the esophagus to the rectum and share similar chemical coding, structure, and function as neurons within the CNS [30]. The ENS consists of two networks of cells: the submucosal plexus, which regulates local functioning such as absorption and secretion, and the myenteric plexus, that is more involved in muscle movement including gut motility [28, 29]. Despite the ENS functioning independently from the CNS, frequent communication occurs between the two systems via the gut-brain axis (GBA). The GBA is a bidirectional communication pathway between the brain and the gut, and consists of crosstalk between the neuronal, hormonal, and immune systems [31]. The GBA is composed of multiple components including the CNS, the autonomic nervous system (ANS), the ENS, and the hypothalamic-pituitary-adrenal (HPA) axis [32]. Neuronal inputs from the ANS and the ENS innervate the vagus nerve and travel up to the lower brainstem regions. From here, information is passed on to higher order information processing neural networks (e.g., emotional, cognitive) of the brain [30]. While the GBA is a bidirectional pathway, most connections come from afferent projections from the gut and are heavily regulated by the gut microbiome [33].
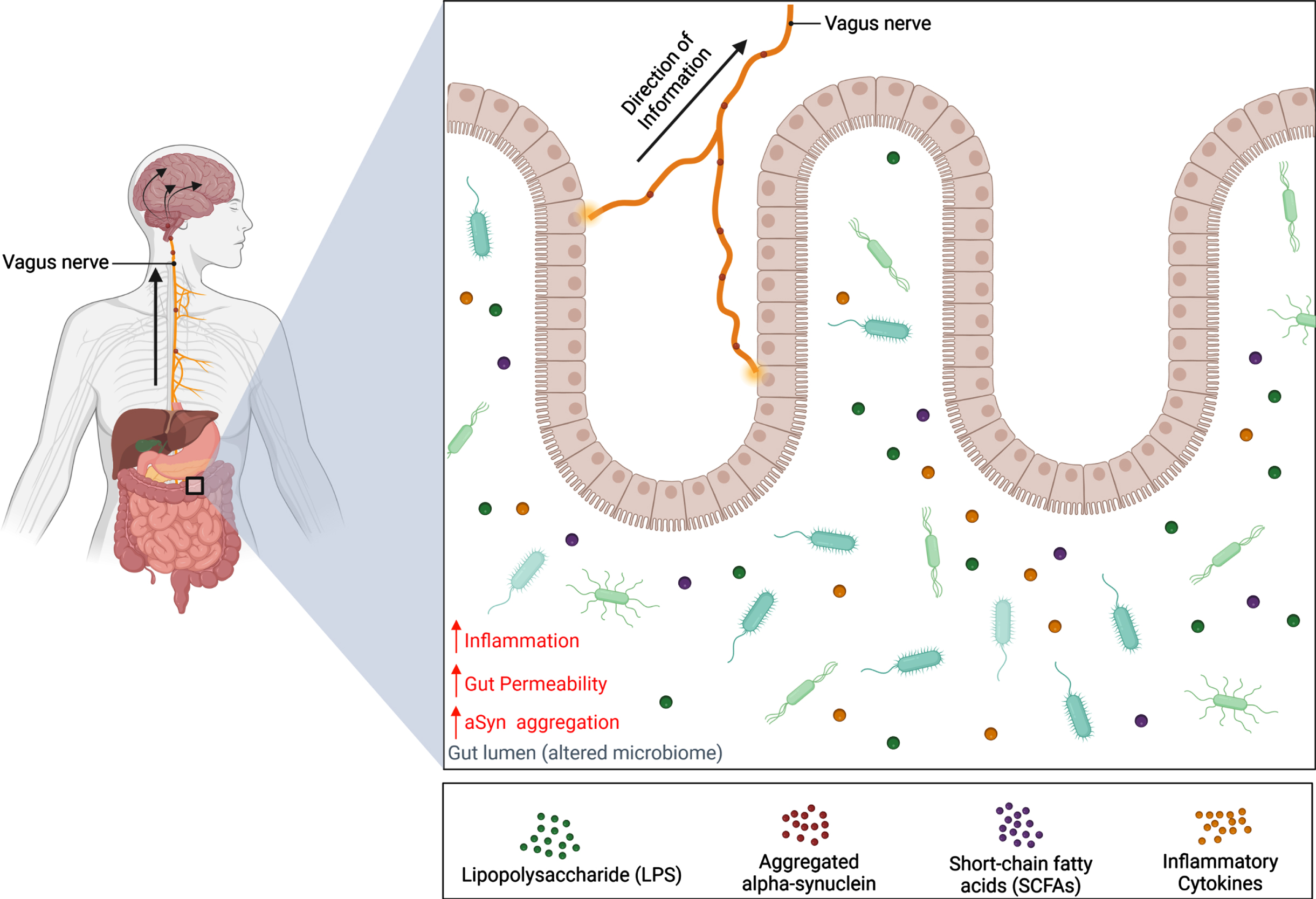
The gut microbiome consists of a balance between bacteria, viruses, fungi, archaea, and many unicellular eukaryotes belonging to hundreds of different species [34, 35]. With a diverse microbiota profile, the gut microbiome influences the ENS in numerous physiological processes that include digestion, nutrient absorption, development, and metabolism [34, 36]. As microbiota are sensitive to changes in the local environment, their abundance levels and function can be altered by various factors such as vaginal birth, diet, genetics, environmental exposures, and antibiotic use [37]. Consequently, this may cause a shift in gut microbial composition towards an overabundance of pathogenic bacteria that are capable of releasing endotoxins that induce inflammation and compromise intestinal wall integrity, thereby leading to numerous diseases like IBS and inflammatory bowel disease (Fig. 3) [38–40].
Fig. 3
Schematic representation of the gut-brain axis in Parkinson’s disease. Alternations of the microbiota composition can be caused by various factors such as genetics and environmental risks. Dysbiosis of the microbiome can contribute to PD pathology in the gut before spreading to the brain via the vagus nerve. This results in a decrease in abundance of beneficial bacteria and a reduction of their metabolites, SCFAs, which have been shown to lower inflammation and gut permeability. Meanwhile, the abundance of harmful bacteria increases, along with the release of endotoxins like LPS. It is hypothesized that these endotoxins can increase inflammatory cytokine expression and cause the gut to become permeable for bacteria and endotoxins to enter the CNS and induce aSyn misfolding. Consequently, aSyn pathology can develop in the gut and travel up the vagus nerve where pathogenic aSyn can spread to brain regions in a prion-like manner. Created with BioRender.com.
GUT MICROBIOTA ALTERATIONS IN PD
The GI symptoms that PD patients experience may be caused by changes in the composition of the GI microbiome. In studies that investigate the composition of a patient’s microbiome, there are two techniques available: 16 S rRNA gene-based amplicon sequencing and whole-genome shotgun metagenomic sequencing. The former technique extracts the DNA from all cells in the area of interest and targets the 16 S rRNA gene portion of the bacterial genome and amplifies it using PCR for microbial composition. Whereas whole-genome shotgun sequencing takes the DNA from cells and breaks it up into small fragments and amplifies by means of PCR. Next, a DNA library is created from these DNA fragments. By using computer software, these fragments are joined together from their overlapping sequences to create a larger strand of DNA, eventually forming a whole genome sequence [41]. While both techniques identify the composition of microbial communities, inconsistent findings may arise when comparing studies that use different sequencing approaches [42]. Given that 16 S rRNA sequencing is more affordable it is more commonly used, although output using this technique limits its findings to genus-level. In contrast, WGS is known for being more expensive, yet it can provide more details pertaining to the species-specific microbial compositions [41].
Overall, several studies have suggested that distinct differences in gut microbiota exist among PD patients compared to healthy controls [43–48]. For example, in a recent meta-analysis, the microbial genera Lactobacillus, Akkermansia, and Bifidobacterium were increased in PD patients, while the microbial Lachnospiraceae family, and Faecalibacterium genus were reduced [43]. Interrelationships between different PD phenotypes and abundance of key species of bacteria have also been found as levels of gram-negative bacteria, Enterobacteriaceae, were higher in PD patients with postural instability and gait difficulty compared to tremor-dominant PD patients [45] while PD duration and disease severity were positively correlated with Bacteroidetes and Proteobacteria phylum [44].
Altered microbial composition in the gut can also modify the number of by-products that bacteria produce. Bacterial metabolites known as short-chain fatty acids (SCFAs) are created by bacteria that break down fiber and resistant starch from the diet. The main SCFAs produced are acetate, propionate, and butyrate [49]. Although SCFAs levels are reduced in PD patients, the role of SCFA in PD remains controversial [50, 51]. Numerous beneficial functions have been associated with SCFAs as they help regulate intestinal wall integrity, curb inflammation, and reduce blood-brain barrier (BBB) permeability [51, 52]. However, in an animal study conducted by Sampson et al. [50], oral administration of SCFAs to germ-free mice accelerated aSyn pathology and exacerbated inflammation mediated by activated microglia, suggesting that an extreme imbalance in SCFAs could have negative outcomes for disease.
Due to the microbiome in the gut being extremely heterogeneous, it may be possible that any alterations in the gut may affect the abundance of several bacterial populations. These changes may cascade to extreme boundaries such as changes in SCFA production. Thus, this review seeks to provide insight for how altered gut microbiota composition may influence various mechanisms that propagate PD symptomatology.
THE IMPACT OF ALTERED GUT MICROBIOTA COMPOSITION ON PD SYMPTOMOLOGY
Inflammation
One contributing factor to gut dysfunction is inflammation, which is also a major component of PD pathogenesis [53]. Recent studies suggest that systemic inflammation in both the CNS and PNS contributes to promoting PD pathology via the chronic release of pro-inflammatory cytokines and the proliferation of immune cells stemming from alterations in gut functioning [54]. As described above, the local environment that surrounds the gut is heavily influenced by the microbiota. Due to alterations in gut composition, the number of by-products that the bacteria produce and release into the gut’s local environment may affect PD pathogenesis. A meta-analysis determined that bacteria belonging to the families Lachnospiraceae and Ruminococcaceae were reduced in PD patients. These families are known producers of SCFAs like butyrate and similar reductions in their levels have been found in other inflammatory disorders [43]. Other meta-analysis studies have found similar results with decreased levels of Firmicutes belonging to the genera Roseburia and Faecalibacterium, both of which are SCFA-producing bacteria [55]. Bacteria from the genus Faecalibacterium are some of the most abundant found in the healthy human gut. They play a vital role in regulating gut health as anti-inflammatory bacteria by balancing immune-related factors and regulating microbiome metabolism [56]. In an in vitro study, Sokol et al. [57] showed that peripheral blood mononuclear cells treated with Faecalibacterium prausnitzii bacteria isolated from human stool samples produced the highest levels of anti-inflammatory cytokines (e.g., IL-10) and the lowest levels of pro-inflammatory cytokines (e.g., IL-12 and IFN-γ) when compared to other commensal bacterial species. Since alterations in the abundance of Faecalibacterium bacteria have been implicated in other gastrointestinal disorders, it may be no surprise that similar changes are observed in PD patients, which likely can contribute to the dysregulated immune-related mechanisms in the gut of these individuals [56].
Other bacteria, such as Escherichia coli (E. coli), have been shown to be increased in PD patients. One study determined that the overabundance of E. coli bacteria in the intestinal mucosa increased colon permeability in PD patients and was associated with several diseases marked by inflammation of the gut including Crohn’s disease and ulcerative colitis [58, 59]. In addition, the increase in the abundance of certain gram-negative bacteria like E. coli, which can contain amyloid curli proteins on the cell surface, may trigger PD pathology [60]. A study conducted by Sampson et al. [60] reported that aSyn overexpressing (ASO) mutant mice displayed worsening motor and GI dysfunction when these mice were colonized with curli-producing E. coli. Further investigation showed that these mice had enhanced aSyn aggregation and inflammatory markers like IL-6 and TNF-α in the colon. In a similar study, Chen et al. [61] observed increased levels of TNF and IL-6 in several brain regions including in the striatum, SN, and the hippocampus in rats that were orally administered curli-producing E. coli bacteria.
Helicobacter pylori (H. pylori), a gram-negative bacterium, has also been shown to have a higher prevalence in PD patients. Although the mechanisms of H. pylori in PD remain unclear, individuals that are H. pylori-positive were at a 59% increased risk of developing PD [62]. When compared to H. pylori-negative PD patients, H. pylori-positive individuals experienced a longer disease duration overall and required higher doses of levodopa for treatment of motor symptoms [63]. Studies have shown that H. pylori can infect the gut by attaching to gastric epithelial cells and releasing proteins like vacuolating cytotoxin A, a toxin that can cause rapid tissue damage and apoptosis [64, 65]. Consequently, these events lead to the activation of the immune system by increasing inflammatory cytokines such as IL-8 and IL-1β [65, 66]. In addition, H. pylori can also contain lipopolysaccharides (LPS) on the cell surface that act as endotoxins, and trigger inflammation and apoptosis within the gut [67, 68].
Mood
Of the non-motor symptoms that arise in PD, depression and anxiety have been commonly observed. While PD patients report a significant decrease in the quality of life (QoL) after being diagnosed [69], it was also reported that anxiety and depression-like symptoms can be experienced before motor symptoms [70]. While the mechanisms for these mood disorders are not fully established, several factors, such as inflammation and neurotransmitter imbalance, have been shown to contribute to the onset of these non-motor symptoms [71, 72].
Anxiety is a severe type of mental illness and is the most prominent psychiatric feature of PD patients as it affects more than half of those with this disease [69]. While anxiety is characterized by chronic overactivation of the HPA axis, human studies have suggested that acute stress can increase intestinal permeability via mast cell activation [73]. Paired with an inflammatory state caused by altered gut microbiota composition, the intestinal and BBB permeability becomes severely weakened. Accordingly, LPS released from gram-negative bacteria such as E. coli can travel up the vagus nerve, bypass the BBB, and over-activate the HPA axis, leading to anxiety-like behaviors [74]. Numerous mechanisms have been proposed for how LPS affects the HPA axis. First, LPS can activate the HPA axis through means of cytokine expression and activation of toll-like receptor (TLR) 4 [75]. Second, LPS can stimulate the release of corticosteroid-releasing hormone (CRH), which then results in the upregulation of ACTH leading to the secretion of cortisol [76, 77]. Lastly, LPS can directly influence cortisol by activating adrenal cells through cyclooxygenase-dependent mechanisms [78]. Demonstrated in vivo, intraperitoneal injection of LPS was administered in mice that were then assessed for behavioral outcomes. Mice that received a LPS injection had increased plasma corticosterone levels and displayed more anxiety-like behaviors as more time was spent in the closed arms of the elevated plus maze, as well as the outer limits of the open field test [79].
Although it is not surprising that individuals with PD report a decrease in QoL, it may be that altered gut microbiota composition could play a major role in modulating one aspect of QoL like depressive behaviors via impaired neurotransmission. While changes in monoamine activity are not the sole cause of depression, they are a critical component that underlies depressive-like symptoms. As previously mentioned, the ENS shares similar chemical signaling as the CNS. More than 90% of serotonin and other neurotransmitters such as gamma-aminobutyric acid (GABA) and dopamine are produced by gut microbes [80–82]. While serotonin that is synthesized outside the CNS cannot cross the BBB, the serotonin precursor, tryptophan, can. Considered an essential amino acid, tryptophan cannot be produced within the body and therefore must be consumed through the diet. Once in the gut, tryptophan can be delivered to the brain for serotonin production. However, there are multiple mechanisms in the GI tract that may take up the tryptophan supply and metabolize it into other metabolites such as indole metabolites and metabolites in the kynurenine pathway [83]. With an abundance of gram-negative bacteria such as E. coli, tryptophan is converted into indole formations, oxindole and isatin, with tryptophanase enzymes [84]. When these indole metabolites were introduced in germ-free rats by colonizing E. coli in the gut, the rats demonstrated helplessness behavior in a tail suspension test, indicative of depressive-like behaviors, and increased anxiety-like behaviors on the open-field and elevated plus maze tests [84]. Furthermore, the inflammatory environment that is created in the gut by altered microbe composition can synthesize more tryptophan into kynurenine metabolites. One such kynurenine metabolite, called quinolinic acid, is neurotoxic and can cause cellular loss and contribute to PD-associated neurodegeneration [85, 86].
In addition to disturbances in serotonin production, altered gut microbiota composition may also impact dopamine production. As gram-negative bacteria abundance increases, beneficial bacteria, such as Prevotellaceae, can decrease. This negative change in bacteria has been linked to a decrease in a gut hormone called ghrelin in PD patients [45]. In an animal study that induced dopamine cell loss with MPTP, treatment with ghrelin promoted a neuroprotective effect on dopaminergic neurons by inhibiting the accumulation and phosphorylation of aSyn, promoting autophagy, and inhibiting endoplasmic reticulum-mediated apoptosis [87, 88]. Similarly, lower levels of SCFA-producing bacteria have been linked to dopamine deficiencies and depressive-like behaviors [89, 90]. The administration of SCFAs (acetate, propionate, and butyrate) has been shown to promote anti-depressive effects in animal models [91]. Specifically, butyrate can act as a histone deacetylase and bind onto G protein-coupled receptors, FA3 receptors, and prevent dopaminergic cell death and lead to increased striatal dopamine levels [92, 93].
Alpha-synuclein protein aggregation
Based on the “PNS-first” hypothesis, aSyn aggregation has been hypothesized to originate in the gut before traveling to the brain via prion-like transmission [15]. Studies have suggested that levels of aSyn aggregation can be influenced indirectly by the gut microbiome [94]. Kim et al. [67] induced human recombinant aSyn into forming fibrils in the presence or absence of LPS. After 7 days of fibrillization, the LPS-free aSyn fibrils or LPS-aSyn fibrils were injected into the striatum of mice. Six months after injection, it was observed that LPS-treated fibrils promoted the misfolding of endogenous aSyn into oligomeric forms. These LPS-treated aSyn aggregates had different conformations compared to the wild-type aSyn fibrils, suggesting that LPS-treated aSyn could form distinct configurations based on different environmental factors such as those within the gut and could present different clinical features or lead to different aSyn-related pathological trajectories.
Along with LPS inducing aSyn aggregation, there are certain bacteria such as curli-producing E. coli that can propagate aSyn pathology in the gut of animals [95]. The E. coli genome encodes an amyloidogenic protein, CsgA that is the main component of biofilm that encompasses several bacteria [61]. Like other amyloid proteins, CsgA proteins are unfolded, but can self-induce fibrilization and form protein aggregates [96]. In a study by Sampson et al. [60], oral administration of CsgA was shown to accelerate aSyn aggregation and induce motor deficits in a Thy1-aSyn mouse model that overexpresses human aSyn protein, whereas in wild-type mice these effects were not observed after CsgA administration. While results from this study suggest that CsgA does not have a primary role in the initiation of aSyn aggregation and PD pathology, the summation of small events may cause a significant impact and exacerbate aSyn aggregation once PD pathology reaches a tipping point.
TYPES OF MICROBIAL THERAPY IN PD
Probiotics
Probiotics composed of beneficial bacteria have been shown to be a useful treatment to combat altered gut microbiota composition and alleviate PD-like symptoms. As described above, the local environment that surrounds the gut is heavily influenced by the microbiota. In an in vivo animal study, oral ingestion of probiotic Malaysian LAB-fermented cow’s milk containing various species of Lactobacillus significantly reduced inflammatory cytokines and increased antioxidants in LPS-treated mice [97]. In another study, Magistrelli and colleagues [98] extracted peripheral blood mononuclear cells from 40 PD patients and 40 age-matched controls and cultured the cells against six different strains of probiotics belonging to the Bifidobacterium and Lactobacillus genera. By assessing the total amount of cytokines and reactive oxygen species released from the cells, it was noted that all probiotic strains (L. salivarius, L. plantarum, L. acidophilus, L. rhamnosus, B.animalis subsp. lactis, and B. breve) lowered inflammatory cytokines and oxidative damage in both controls and PD patients. In rats, treatment with probiotics reduced activity of an elevated HPA axis response induced by a partial restraint model. Within 2 weeks of probiotic treatments, levels of plasma adrenocorticotropin (ACTH), corticosterone, and the hypothalamic corticotropin-releasing factor (CRF) diminished [99]. Other animal studies showed that administration of a probiotic strain, Lactobacillus plantarum PS128 (PS128), to germ-free mice increased serotonin and dopamine production in the striatum compared to controls [100]. In humans, a double-blind, placebo-controlled study was conducted on 38 healthy volunteers. Participants were either given a probiotic formulation containing species of Lactobacillus and Bifidobacterium or a placebo for 6 weeks. At the end of the study, participants in the treatment group reported improved mood, reduced depression, and better sleep quality [101]. This effect of probiotic-induced elevation in mood was thought to be due in part to the increased levels of tryptophan and subsequent synthesis of serotonin in the brain [102, 103]. Lastly, probiotic supplementation was also shown to decrease constipation severity in PD individuals. In a double-blind randomized trial, 72 PD patients were either given a multi-strain capsule containing eight bacterial strains (Lactobacillus acidophilus, Lactobacillus reuteri, Lactobacillus gasseri, Lactobacillus rhamnosus, Bifidobacterium bifidum, Bifidobacterium longum, Enterococcus faecalis, Enterococcus faecium) or a placebo maltodextrin capsule for four weeks. PD patients that received the probiotic capsules had more spontanteous bowel movements and improved their quality of life in regard to constipation symptoms compared to those that received the placebo capsule [104]. Given that the striatum is heavily affected in PD with a decrease in dopamine levels, the use of certain probiotic strains could potentially be effective in helping PD patients. This suggests that probiotic treatments may prolong dopaminergic neuronal survival in the brain and alleviate motor and non-motor impairments [100, 105].
Prebiotics
Like probiotics, prebiotics can be used to modify the gut bacteria composition within the gut and promote health benefits. In contrast to probiotics, prebiotics are not living organisms and are referred to as “selective fermented ingredients that result in specific changes, both in the composition and/or activity of the gastrointestinal microbiota that confers benefits” [106]. Prebiotics are composed of dietary fiber that cannot be processed in the upper GI tract and must travel down to the lower GI tract where they are fermented in the colon [107]. While prebiotics are considered dietary fibers, not all dietary fibers are prebiotics. Once in the colon, prebiotics can be broken down by Lactobacilli and Bifidobacteria to enhance gut barrier function, prevent pathogenic bacteria from occupying space in the GI tract and enhance SCFA levels such as acetate and butyrate [107]. These effects were demonstrated in humans in a clinical trial (NCT02784145) as Becker and colleagues [108] showed the effectiveness of using prebiotic resistant starch (RS) after 8 weeks in PD patients as butyrate concentrations increased and levels of intestinal inflammatory marker, calprotectin, decreased in their stool samples. In addition, a significant improvement in depressive symptoms were reported whereby Becker et al. [108] suggested this outcome was likely attributable to increased butyrate levels. Despite the therapeutic effects of RS, there is conflicting evidence published regarding the use of RS given that improvement of GI function is not always observed [109]. Whereas Becker and colleagues [108] suggest that differences in outcomes may be owing to RS types that were used, along with the duration of the studies (8 weeks vs. 12 weeks). The effects of prebiotics have also been demonstrated in vitro and in vivo whereby several in vitro studies have shown beneficial outcomes linked with butyrate where it acts by protecting dopaminergic neurons from apoptosis [110] and also reduces cellular inflammatory markers [111]. Meanwhile, in vivo studies have also shown butyrate to improve intestinal and BBB permeability [112, 113], prevent aSyn aggregation [105], and alleviate motor deficits induced in PD animal models [112, 114]. These studies suggest that the consumption of prebiotics may benefit PD pathological outcomes.
Fecal microbial transplantation
Fecal microbial transplantation (FMT) refers to the transfer of healthy gut microbiota from a healthy donor in a fecal suspension into the recipient’s gut to re-establish a healthy gut microbial community [115]. Currently, FMT treatment is generally considered safe with few short adverse effects (e.g., bloating, abdominal pain, nausea, and diarrhea) and long-term adverse effects (e.g., obesity, irritable bowel syndrome, and rheumatoid arthritis) [116]. Over the decade, FMT has gained interest in treating gastrointestinal diseases to which in 2013 the Food and Drug Administration (FDA) had issued a statement requiring physicians to submit an Investigational New Drug (IND) application for the use of FMT in patients. With feedback from the medical community, the FDA eased up regulations with FMT and allowed its use to patients who do not respond to standard therapy for Clostridium difficile infections (CDI) [117]. Nonetheless, the FDA is still imposing tight regulations with rigorous screening and informed consent to patients receiving FMT as cases of pathogenic bacteria were found in stool bank samples and the potential risk of SARS-CoV-2 RNA transmission from infected individuals [118]. Demonstrated in a clinical trial, fecal samples from 25 participants were frozen and stored prior to their stem cell transplantation. After being given antibiotics to prevent bacterial infection, participants either received auto-FMT treatment or standard care and were revaluated for their gut microbiome compositions. From their results, 79% of participants that received auto-FMT restored 75% or more of their original bacteria composition, whereas 27% of participants that received standard care had their gut microbiota composition similar to their original fecal sample [119]. FMT treatment is primarily used to treat Clostridium difficile infections, but FMT treatment has spread to treat various diseases that are also affected by gastrointestinal disorders such as PD [120]. In a preliminary study, fifteen PD patients received FMT treatment, whereby 10 patients received FMT via colonoscopy (colonic FMT group) and the remaining 5 patients received FMT via nasal-jejunal tube (nasointestinal FMT group). Compared to the colonic FMT group, the nasointestinal FMT group did not report improvement in their non-motor symptoms such as anxiety and depression after a 1- and 3-month follow up. Whereas PD patients in the colonic FMT group reported more satisfaction with the treatment and out of the 15 FMT patients, only 5 patients reported adverse effects from FMT that included diarrhea, flatulence, and abdominal pain [115]. Recently, more evidence for this therapy to restore the microbial communities months after treatment has emerged. In a study by Kuai et al. [121], PD patients that received FMT reported an improvement in motor and non-motor symptoms. In addition, the abundance of Prevotella and butyrate-producing bacteria, Faecalibacterium and Blautia, increased after FMT, while the abundance of Bacteriodetes decreased. In PD animal models, the use of FMT treatment re-established a healthy microbiome in the gut and alleviated the intestinal and BBB permeabilities [122, 123]. In a study by Zhao et al. [123], FMT treatment was neuroprotective in a rotenone-induced PD mouse model. More specifically, FMT treatment prevented dopaminergic neuronal loss and inhibited aSyn aggregation in the SN, leading to an improvement in motor deficits that were otherwise induced by rotenone administration. In another in vivo study, MPTP-treated mice that received FMT partly recovered striatal neurotransmitter levels such as dopamine and serotonin and alleviated motor dysfunction in mice [122]. Furthermore, FMT treatment in animals was shown to reduce the number of activated microglia and astrocytes in the SN and decrease the level of neuroinflammation via the inhibition of the TLR4/TANK-binding kinase 1 (TBK1)/TNF-α signaling pathway. Given the FDA’s recommendation that requires rigorous screening protocols prior to FMT use as a way to minimize the transmission of pathogenic bacteria, along with the beneficial effects demonstrated in the studies above (like the restoration of a healthy microbiota), FMT treatment may provide a safe therapeutic alternative to PD patients.
CONCLUSIONS
Although the presence of motor symptoms is necessary for the diagnosis of PD, it may be equally important to investigate non-motor symptoms as early indicators of this disease. Accordingly, emerging evidence has described that within the gut, dysbiosis of the microbiota can potentially increase the risk of inflammation, gut permeability, and induce aSyn aggregation. However, an important caveat to consider in human studies is that the beneficial outcomes that were described are self-reported. Hence, more research is needed to provide direct evidence that bacterial mechanisms can contribute to PD symptomatology. This leads to future research aimed at identifying specific bacterial strains that initiate PD pathology may be a viable therapeutic approach for the treatment of this disease. Taken together, by focusing on restoring a healthy microbiome composition, in addition to combating the microbial metabolites that interact with the environment that promote a pro-inflammatory state or induce aSyn misfolding, some of the symptoms like irritable bowel syndrome and constipation would likely be improved if not alleviated. This would mark the start to the improvement of QoL for the patients which has been a long-standing goal of the healthcare industry.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | GBD ((2016) ) Parkinson’s Disease Collaborators (2018) Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the global burden of disease Study 2016. Lancet Neurol 17: , 939–953. |
[2] | Elbaz A , Carcaillon L , Kab S , Moisan , F ((2016) ) Epidemiology of Parkinson’s disease. Rev Neurol (Paris) 172: , 14–26. |
[3] | Klein C , Westenberger A ((2012) ) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2: , a008888. |
[4] | Gazewood JD , Richards DR , Clebak K ((2013) ) Parkinson disease: an update. Am Fam Physician 87: , 267–273. |
[5] | Dauer W , Przedborski S ((2003) ) Mechanisms and models. Neuron 39: , 889–909. |
[6] | Cheng HC , Ulane CM , Burke RE ((2010) ) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67: , 715–725. |
[7] | Surmeier DJ ((2018) ) Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J 285: , 3657–3668. |
[8] | Goedert M , Jakes R , Spillantini MG ((2017) ) The synucleinopathies: twenty years on. J Parkinsons Dis 7: , S51–S69. |
[9] | Baltic S , Perovic M , Mladenovic A , Raicevic N , Ruzdijic S , Rakic L , Kanazir S ((2004) ) α-Synuclein is expressed in different tissues during human fetal development. J Mol Neurosci 22: , 199–203. |
[10] | Sprenger FS , Stefanova N , Gelpi E , Seppi K , Navarro-Otano J , Offner F , Vilas D , Valldeoriola F , Pont-Sunyer C , Aldecoa I , Gaig C , Gines A , Cuatrescasas M , Högl B , Frauscher B , Iranzo A , Wenning GK , Vogel W , Tolosa E , Poewe W ((2015) ) Enteric nervous system α-synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 85: , 1761–1768. |
[11] | Butler B , Saha K , Rana T , Becker JP , Sambo D , Davari P , Goodwin JS , Khoshbouei H ((2015) ) Dopamine transporter activity is modulated by α-synuclein. J Biol Chem 290: , 29542–29554. |
[12] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[13] | Spillantini MG , Schmidt ML , Lee VM-Y , Trojanowski JQ , Jakes R , Goedert M ((1997) ) alpha synuclein in Lewy bodies. Nature 388: , 839–840. |
[14] | Menezes R , Tenreiro S , Macedo D , Santos CN , Outeiro TF ((2015) ) From the baker to the bedside: yeast models of Parkinson’s disease. Microb Cell 2: , 262–279. |
[15] | Braak , H , Del Tredici K ((2017) ) Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis 7: (Suppl 1), S71–S85. |
[16] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag AE , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Primers 3: , 1–53. |
[17] | Poirier AA , Aubé B , Côté M , Morin N , Di Paolo T , Soulet D ((2016) ) Gastrointestinal dysfunctions in Parkinson’s disease: symptoms and treatments. Parkinsons Dis 2016: , 6762528. |
[18] | Ueki A , Otsuka M ((2004) ) Life style risks of Parkinson’s disease: association between decreased water intake and constipation. J Neurol 251: (Suppl 7), vII18–vII23. |
[19] | Nair AT , Ramachandran V , Joghee NM , Antony S , Ramalingam G ((2018) ) Gut microbiota dysfunction as reliable non-invasive early diagnostic biomarkers in the pathophysiology of Parkinson’s disease: a critical review. J Neurogastroenterol Motil 24: , 30–42. |
[20] | Braak H , Ghebremedhin E , Rüb , U , Bratzke H , Del Tredici K ((2004) ) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318: , 121–134. |
[21] | Chen H , Wang K , Scheperjans F , Killinger B ((2022) ) Environmental triggers of Parkinson’s disease –Implications of the Braak and dual-hit hypotheses. Neurobiol Dis 163: , 105601. |
[22] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: a hypothesis. J Parkinsons Dis 9: (Suppl 2), S281–S295. |
[23] | Borghammer P , Horsager J , Andersen K , Van Den Berge N , Raunio A , Murayama S , Parkkinen L , Myllykangas L ((2021) ) Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 161: , 105557. |
[24] | Wakabayashi K , Takahashi H , Takeda S , Ohama E , Ikuta F ((1988) ) Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol 76: , 217–221. |
[25] | Cersosimo MG ((2015) ) Gastrointestinal biopsies for the diagnosis of alpha-synuclein pathology in Parkinson’s disease. Gastroenterol Res Pract 2015: , 476041. |
[26] | Uemura N , Yagi H , Uemura MT , Hatanaka Y , Yamakado H , Takahashi R ((2018) ) Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 13: , 21. |
[27] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103: , 627–641. |
[28] | Schemann M ((2005) ) Control of gastrointestinal motility by the “gut brain”–the enteric nervous system. J Pediatr Gastroenterol Nutr 41: (Suppl 1), S4–S6. |
[29] | Breit S , Kupferberg A , Rogler G , Hasler G ((2018) ) Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front Psychiatry 9: , 44. |
[30] | Harsanyiova J , Buday T , Kralova Trancikova A ((2020) ) Parkinson’s disease and the gut: future perspectives for early diagnosis. Front Neurosci 14: , 626. |
[31] | Wang HX , Wang YP ((2016) ) Gut microbiota-brain axis. Chin Med J (Engl) 129: , 2373–2380. |
[32] | Carabotti M , Scirocco A , Maselli MA , Severi C ((2015) ) The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 28: , 203–209. |
[33] | Bonaz B , Bazin T , Pellissier S ((2018) ) The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci 12: , 49. |
[34] | Yano JM , Yu K , Donaldson GP , Shastri GG , Ann P , Ma L , Nagler CR , Ismagilov RF , Mazmanian SK , Hsiao EY ((2015) ) Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161: , 264–276. |
[35] | Matijašić M , Meštrović T , Paljetak HČ , Perić M , Barešić A , Verbanac D ((2020) ) Gut microbiota beyond bacteria-mycobiome, virome, archaeome, and eukaryotic parasites in IBD. Int J Mol Sci 21: , 2668. |
[36] | Furness JB ((2016) ) Integrated neural and endocrine control of gastrointestinal function. Adv Exp Med Biol 891: , 159–173. |
[37] | Hasan N , Yang H ((2019) ) Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 7: , e7502. |
[38] | Carding S , Verbeke K , Vipond DT , Corfe BM , Owen LJ ((2015) ) Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 26: , 26191. |
[39] | Fukui H ((2016) ) Increased intestinal permeability and decreased barrier function: does it really influence the risk of inflammation? Inflamm Intest Dis 1: , 135–145. |
[40] | Lobionda S , Sittipo P , Kwon HY , Lee YK ((2019) ) The role of gut microbiota in intestinal inflammation with respect to diet and extrinsic stressors. Microorganisms 7: , 271. |
[41] | Ranjan R , Rani A , Metwally A , McGee HS , Perkins DL ((2016) ) Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun 469: , 967–977. |
[42] | Peterson D , Bonham KS , Rowland S , Pattanayak CW ((2021) ) Comparative analysis of 16S rRNA gene and metagenome sequencing in pediatric gut microbiomes. Front Microbiol 12: , 670336. |
[43] | Romano S , Savva GM , Bedarf JR , Charles IG , Hildebrand F , Narbad A ((2021) ) Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis 7: , 27. |
[44] | Keshavarzian A , Green SJ , Engen PA , Voigt RM , Naqib A , Forsyth CB , Mutlu E , Shannon KM ((2015) ) Colonic bacterial composition in Parkinson’s disease. Move Disord 30: , 1351–1360. |
[45] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Move Disord 30: , 350–358. |
[46] | Petrov VA , Saltykova IV , Zhukova IA , Alifirova VM , Zhukova NG , Dorofeeva YB , Tyakht AV , Kovarsky BA , Alekseev DG , Kostryukova ES , Mironova YS , Izhboldina OP , Nikitina MA , Perevozchikova TV , Fait EA , Babenko VV , Vakhitova MT , Govorun VM , Sazonov AE ((2017) ) Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 162: , 734–737. |
[47] | Lin A , Zheng W , He Y , Tang W , Wei X , He R , Huang W , Su Y , Huang Y , Zhou H , Xie H ((2018) ) Gut microbiota in patients with Parkinson’s disease in southern China. Parkinsonism Relat Disord 53: , 82–88. |
[48] | Toh TS , Chong CW , Lim SY , Bowman J , Cirstea M , Lin CH , Chen CC , Appel-Cresswell S , Finlay BB , Tan AH ((2022) ) Gut microbiome in Parkinson’s disease: New insights from meta-analysis. Parkinsonism Relat Disord 94: , 1–9. |
[49] | Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Bürmann J , Faßbender K , Schwiertz A , Schäfer KH ((2016) ) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32: , 66–72. |
[50] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480. |
[51] | Aho VTE , Houser MC , Pereira PAB , Chang J , Rudi K , Paulin L , Hertzberg V , Auvinen P , Tansey MG , Scheperjans F ((2021) ) Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol Neurodegener 16: , 6. |
[52] | Braniste V , Al-Asmakh M , Kowal C , Anuar F , Abbaspour A , Tóth M , Korecka A , Bakocevic N , Ng LG , Kundu P , Gulyàs B , Halldin C , Hultenby K , Nilsson H , Hebert H , Volpe BT , Diamond B , Pettersson S ((2014) ) The gut microbiota influences blood-brain barrier permeability in mice. , 263ra. Sci Transl Med 6: , 158. |
[53] | Chen QQ , Haikal C , Li W , Li JY ((2019) ) Gut inflammation in association with pathogenesis of Parkinson’s disease. Front Mol Neurosci 12: , 218. |
[54] | Ferrari CC , Tarelli R ((2011) ) Parkinson’s disease and systemic inflammation. Parkinsons Dis 2011: , 436813. |
[55] | Nishiwaki H , Ito M , Ishida T , Hamaguchi T , Maeda T , Kashihara K , Tsuboi Y , Ueyama J , Shimamura T , Mori H , Kurokawa K , Katsuno M , Hirayama M , Ohno K ((2020) ) Meta-analysis of gut dysbiosis in Parkinson’s disease. Move Disord 35: , 1626–1635. |
[56] | Miquel S , Martín R , Rossi O , Bermúdez-Humarán LG , Chatel JM , Sokol H , Thomas M , Wells JM , Langella P ((2013) ) Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol 16: , 255–261. |
[57] | Sokol H , Pigneur B , Watterlot L , Lakhdari O , Bermúdez-Humarán LG , Gratadoux JJ , Blugeon S , Bridonneau C , Furet JP , Corthier G , Grangette C , Vasquez N , Pochart P , Trugnan G , Thomas G , Blottière HM , Doré J , Marteau P , Seksik P , Langella P ((2008) ) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 105: , 16731–16736. |
[58] | Forsyth CB , Shannon KM , Kordower JH , Voigt RM , Shaikh M , Jaglin JA , Estes JD , Dodiya HB , Keshavarzian A ((2011) ) Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PloS One 6: , e28032. |
[59] | Mirsepasi-Lauridsen HC , Vallance BA , Krogfelt KA , Petersen AM ((2019) ) pathobionts associated with inflammatory bowel disease. Clin Microbiol Rev 32: , e00060–18. |
[60] | Sampson TR , Challis C , Jain N , Moiseyenko A , Ladinsky MS , Shastri GG , Thron T , Needham BD , Horvath I , Debelius JW , Janssen S , Knight R , Wittung-Stafshede P , Gradinaru V , Chapman M , Mazmanian SK ((2020) ) A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. ELife 9: , e53111. |
[61] | Chen SG , Stribinskis V , Rane MJ , Demuth DR , Gozal E , Roberts AM , Jagadapillai R , Liu R , Choe K , Shivakumar B , Son F , Jin S , Kerber R , Adame A , Masliah E , Friedland RP ((2016) ) Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans.. Sci Rep 6: , 34477. |
[62] | Shen X , Yang H , Wu Y , Zhang D , Jiang H ((2017) ) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22: doi:10.1111/hel.12398. |
[63] | Mridula KR , Borgohain R , Chandrasekhar Reddy V , Bandaru VCh , Suryaprabha T ((2017) ) Association of Helicobacter pylori with Parkinson’s disease. J Clin Neurol 13: , 181–186. |
[64] | Akazawa Y , Isomoto H , Matsushima K , Kanda T , Minami H , Yamaghchi N , Taura N , Shiozawa K , Ohnita K , Takeshima F , Nakano M , Moss J , Hirayama T , Nakao K ((2013) ) Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PloS One 8: , e82322. |
[65] | Liao WC , Huang MZ , Wang ML , Lin CJ , Lu TL , Lo HR , Pan YJ , Sun YC , Kao MC , Lim HJ , Lai CH ((2017) ) Statin decreases burden in macrophages by promoting autophagy. Front Cell Infect Microbiol 6: , 203. |
[66] | Kao CY , Sheu BS , Wu JJ ((2016) ) Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed J 39: , 14–23. |
[67] | Kim C , Lv G , Lee JS , Jung BC , Masuda-Suzukake M , Hong CS , Valera E , Lee HJ , Paik SR , Hasegawa M , Masliah E , Eliezer D , Lee SJ ((2016) ) Exposure to bacterial endotoxin generates a distinct strain of α-synuclein fibril. Sci Rep 6: , 30891. |
[68] | Kawahara T , Teshima S , Kuwano Y , Oka A , Kishi K , Rokutan K ((2001) ) Helicobacter pylori lipopolysaccharide induces apoptosis of cultured guinea pig gastric mucosal cells. Am J Physiol Gastrointest Liver Physiol 281: , G726–G734. |
[69] | Upneja A , Paul BS , Jain D , Choudhary R , Paul G ((2021) ) Anxiety in Parkinson’s disease: correlation with depression and quality of life. J Neurosci Rural Pract 12: , 323–328. |
[70] | Tolosa E , Compta Y , Gaig ((2007) ) The premotor phase of Parkinson’s disease. Parkinsonism Relat Disord 13: , S2–S7. |
[71] | Felger JC ((2018) ) Imaging the role of inflammation in mood and anxiety-related disorders. Curr Neuropharmacol 16: , 533–558. |
[72] | Liu Y , Zhao J , Guo W ((2018) ) Emotional roles of mono-aminergic neurotransmitters in major depressive disorder and anxiety disorders. Front Psychol 9: , 2201. |
[73] | Vanuytsel T , van Wanrooy S , Vanheel H , Vanormelingen C , Verschueren S , Houben E , Salim Rasoel S , Tóth J , Holvoet L , Farré R , Van Oudenhove L , Boeckxstaens G , Verbeke K , Tack J ((2014) ) Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut 63: , 1293–1299. |
[74] | Farzi A , Fröhlich EE , Holzer P ((2018) ) Gut microbiota and the neuroendocrine system. Neurotherapeutics 15: , 5–22. |
[75] | Zacharowski K , Zacharowski PA , Koch A , Baban A , Tran N , Berkels R , Papewalis C , Schulze-Osthoff K , Knuefermann P , Zähringer U , Schumann RR , Rettori V , McCann SM , Bornstein SR ((2006) ) Toll-like receptor 4 plays a crucial role in the immune-adrenal response to systemic inflammatory response syndrome. Proc Natl Acad Sci U S A 103: , 6392–6397. |
[76] | Vedder H , Schreiber W , Yassouridis A , Gudewill S , Galanos C , Pollmächer T ((1999) ) Dose-dependence of bacterial lipopolysaccharide (LPS) effects on peak response and time course of the immune-endocrine host response in humans. Inflamm Res 48: , 67–74. |
[77] | Goebel M , Stengel A , Wang L , Reeve J Jr , Taché Y ((2011) ) Lipopolysaccharide increases plasma levels of corticotropin-releasing hormone in rats. Neuroendocrinology 93: , 165–173. |
[78] | Vakharia K , Hinson JP ((2005) ) Lipopolysaccharide directly stimulates cortisol secretion by human adrenal cells by a cyclooxygenase-dependent mechanism. Endocrinology 146: , 1398–1402. |
[79] | Sulakhiya K , Keshavlal GP , Bezbaruah BB , Dwivedi S , Gurjar SS , Munde N , Jangra A , Lahkar M , Gogoi R ((2016) ) Lipopolysaccharide induced anxiety- and depressive-like behaviour in mice are prevented by chronic pre-treatment of esculetin. Neurosci Lett 611: , 106–111. |
[80] | Gershon MD ((2013) ) 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes 20: , 14–21. |
[81] | Hata T , Asano Y , Yoshihara K , Kimura-Todani T , Miyata N , Zhang XT , Takakura S , Aiba Y , Koga Y , Sudo N ((2017) ) Regulation of gut luminal serotonin by commensal microbiota in mice. PloS One 12: , e0180745. |
[82] | Strandwitz P , Kim KH , Terekhova D , Liu JK , Sharma A , Levering J , McDonald D , Dietrich D , Ramadhar TR , Lekbua A , Mroue N , Liston C , Stewart EJ , Dubin MJ , Zengler K , Knight R , Gilbert JA , Clardy J , Lewis K ((2019) ) GABA-modulating bacteria of the human gut microbiota. Nat Microbiol 4: , 396–403. |
[83] | Kaur H , Bose C , Mande SS ((2019) ) Tryptophan metabolism by gut microbiome and gut-brain-axis: an analysis. Front Neurosci 13: , 1365. |
[84] | Jaglin M , Rhimi M , Philippe C , Pons N , Bruneau A , Goustard B , Daugé V , Maguin E , Naudon L , Rabot S ((2018) ) Indole, a signaling molecule produced by the gut microbiota, negatively impacts emotional behaviors in rats. Front Neurosci 12: , 216. |
[85] | Tavassoly O , Sade D , Bera S , Shaham-Niv S , Vocadlo DJ , Gazit ((2018) ) Quinolinic acid amyloid-like fibrillar assemblies seed α-synuclein aggregation. J Mol Biol 430: , 3847–3862. |
[86] | Santos SF , de Oliveira HL , Yamada ES , Neves BC , Pereira A Jr ((2019) ) The gut and Parkinson’s disease-a bidirectional pathway. Front Neurol 10: , 574. |
[87] | Andrews ZB , Erion D , Beiler R , Liu ZW , Abizaid A , Zigman J , Elsworth JD , Savitt JM , DiMarchi R , Tschoep M , Roth RH , Gao XB , Horvath TL ((2009) ) Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci 29: , 14057–14065. |
[88] | Wang H , Dou S , Zhu J , Shao Z , Wang C , Cheng B ((2020) ) Ghrelin protects dopaminergic neurons against MPTP neurotoxicity through promoting autophagy and inhibiting endoplasmic reticulum mediated apoptosis. Brain Res 1746: , 147023. |
[89] | Caspani G , Kennedy S , Foster JA , Swann J ((2019) ) Gut microbial metabolites in depression: understanding the biochemical mechanisms. Microb Cell 6: , 454–481. |
[90] | Mirzaei R , Bouzari B , Hosseini-Fard SR , Mazaheri M , Ahmadyousefi Y , Abdi M , Jalalifar S , Karimitabar Z , Teimoori A , Keyvani H , Zamani F , Yousefimashouf R , Karampoor S ((2021) ) Role of microbiota-derived short-chain fatty acids in nervous system disorders. Biomed Pharmacother 139: , 111661. |
[91] | Van de Wouw M , Boehme M , Lyte JM , Wiley N , Strain C , O’Sullivan O , Clarke G , Stanton C , Dinan TG , Cryan JF ((2018) ) Short-chain fatty acids: microbial metabolites that alleviate stress-induced brain-gut axis alterations. J Physiol 596: , 4923–4944. |
[92] | Sharma S , Taliyan R , Singh S ((2015) ) Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: Modulation of histone deacetylase activity. Behav Brain Res 291: , 306–314. |
[93] | Getachew B , Csoka AB , Bhatti A , Copeland RL , Tizabi Y ((2020) ) Butyrate protects against salsolinol-induced toxicity in SH-SY5Y cells: implication for Parkinson’s disease. Neuroto Res 38: , 596–602. |
[94] | Lubomski M , Tan AH , Lim SY , Holmes AJ , Davis RL , Sue CM ((2020) ) Parkinson’s disease and the gastrointestinal microbiome. J Neurol 267: , 2507–2523. |
[95] | Chorell E , Andersson E , Evans ML , Jain N , Götheson A , Åden J , Chapman MR , Almqvist F , Wittung-Stafshede P ((2015) ) Bacterial chaperones CsgE and CsgC differentially modulate human α-synuclein amyloid formation via transient contacts. PloS One 10: , e0140194. |
[96] | Evans ML , Chorell E , Taylor JD , Åden J , Götheson A , Li F , Koch M , Sefer L , Matthews SJ , Wittung-Stafshede P , Almqvist F , Chapman MR ((2015) ) The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell 57: , 445–455. |
[97] | Musa NH , Mani V , Lim SM , Vidyadaran S , Abdul Majeed AB , Ramasamy ((2017) ) Lactobacilli-fermented cow’s milk attenuated lipopolysaccharide-induced neuroinflammation and memory impairment in vitro and in vivo. J Dairy Res 84: , 488–495. |
[98] | Magistrelli L , Amoruso A , Mogna L , Graziano T , Cantello R , Pane M , Comi C ((2019) ) Probiotics may have beneficial effects in Parkinson’s disease: in vitro evidence. Front Immunol 10: , 969. |
[99] | Ait-Belgnaoui A , Durand H , Cartier C , Chaumaz G , Eutamene H , Ferrier L , Houdeau E , Fioramonti J , Bueno L , Theodorou V ((2012) ) Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 37: , 1885–1895. |
[100] | Liu WH , Chuang HL , Huang YT , Wu CC , Chou GT , Wang S , Tsai ((2016) ) Alteration of behavior and monoamine levels attributable to Lactobacillus plantarum PS128 in germ-free mice. Behav Brain Res 298: (Pt B), 202–209. |
[101] | Marotta A , Sarno E , Del Casale A , Pane M , Mogna L , Amoruso A , Felis GE , Fiorio ((2019) ) Effects of probiotics on cognitive reactivity, mood, and sleep quality. Front Psychiatry 10: , 164. |
[102] | Wallace CJK , Milev ((2017) ) The effects of probiotics on depressive symptoms in humans: a systematic review. Ann gen Psychiatry 16: , 14. |
[103] | Banskota S , Ghia JE , Khan WI ((2019) ) Serotonin in the gut: Blessing or a curse. Biochimie 161: , 56–64. |
[104] | Tan AH , Lim SY , Chong KK , A Manap MAA , Hor JW , Lim JL , Low SC , Chong CW , Mahadeva S , Lang AE ((2021) ) Probiotics for constipation in Parkinson disease: a randomized placebo-controlled study. Neurology 96: , e772–e782. |
[105] | Hsieh TH , Kuo CW , Hsieh KH , Shieh MJ , Peng CW , Chen YC , Chang YL , Huang YZ , Chen CC , Chang PK , Chen KY , Chen HY ((2020) ) Probiotics alleviate the progressive deterioration of motor functions in a mouse model of Parkinson’s disease. Brain Sci 10: , 206. |
[106] | Gibson GR , Scott KP , Rastall RA , Tuohy KM , Hotchkiss A , Dubert-Ferrandon A , Gareau M , Murphy EF , Saulnier D , Loh G , Macfarlane S , Delzenne N , Ringel Y , Kozianowski G , Dickmann R , Lenoir-Wijnkoop I , Walker C , Buddington R ((2010) ) Dietary prebiotics: Current status and new definition. Food Sci Technol Funct Foods 7: , 1–19. |
[107] | Slavin J ((2013) ) Fiber and prebiotics: mechanisms and health benefits. Nutrients 5: , 1417–1435. |
[108] | Becker A , Pierre Schmartz G , Gröger L , Grammes N , Galata V , Philippeit H , Weiland J , Ludwig N , Meese E , Tierling S , Walter J , Schwiertz A , Spiegel J , Wagenpfeil G , Faßbender K , Keller A , Unger MM (2021) Effects of resistant starch on symptoms, fecal markers and gut microbiota in Parkinson’s disease –the RESISTA-PD Trial. Genomics Proteomics Bioinformatics. doi:10.1016/j.gpb.2021.08.009. |
[109] | Alfa MJ , Strang D , Tappia PS , Olson N , DeGagne P , Bray D , Murray BL , Hiebert B ((2017) ) A randomized placebo controlled clinical trial to determine the impact of digestion resistant starch® on glucose, insulin, and insulin resistance in elderly and mid-age adults. Front Med (Lausanne) 4: , 260. |
[110] | Kidd SK , Schneider JS ((2010) ) Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res 1354: , 172–178. |
[111] | Park JS , Lee EJ , Lee JC , Kim WK , Kim HS ((2007) ) Anti-inflammatory effects of short chain fatty acids in IFN-gamma-stimulated RAW 264.7 murine macrophage cells: involvement of NF-kappaB and ERK signaling pathways. Int Immunopharmacol 7: , 70–77. |
[112] | Liu J , Wang F , Liu S , Du J , Hu X , Xiong J , Fang R , Chen W , Sun J ((2017) ) Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J Neurol Sci 381: , 176–181. |
[113] | Feng W , Wu Y , Chen G , Fu S , Li B , Huang B , Wang D , Wang W , Liu J ((2018) ) Sodium butyrate attenuates diarrhea in weaned piglets and promotes tight junction protein expression in colon in a GPR109A-dependent manner. Cell Physiol Biochem 47: , 1617–1629. |
[114] | Zhou W , Bercury K , Cummiskey J , Luong N , Lebin J , Freed CR ((2011) ) Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem 286: , 14941–14951. |
[115] | Xue LJ , Yang XZ , Tong Q , Shen P , Ma SJ , Wu SN , Zheng JL , Wang HG ((2020) ) Fecal microbiota transplantation therapy for Parkinson’s disease: A preliminary study. Medicine (Baltimore) 99: , e22035. |
[116] | Park SY , Seo GS ((2021) ) Fecal microbiota transplantation: is it safe? Clin Endosc 54: , 157–160. |
[117] | Food and Drug Administration. Guidance for Industry: Enforcement Policy Regarding Investigational New Drug Requirements for Use of Fecal Microbiota for Transplantation to Treat Clostridium difficile Infection Not Responsive to Standard Therapies, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/enforcement-policy-regarding-investigational-new-drug-requirements-use-fecal-microbiota, Accessed August 2022. |
[118] | Food and Drug Administration. Safety alert regarding use of fecal microbiota for transplantation and additional safety protections pertaining to SARS-CoV-2 and COVID-19, https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/safety-alert-regarding-use-fecal-microbiota-transplantation-and-additional-safety-protections, Accessed August, 2022. |
[119] | Taur Y , Coyte K , Schluter J , Robilotti E , Figueroa C , Gjonbalaj M , Littmann ER , Ling L , Miller L , Gyaltshen Y , Fontana E , Morjaria S , Gyurkocza B , Perales MA , Castro-Malaspina H , Tamari R , Ponce D , Koehne G , Barker J , Jakubowski A , Papadopoulos E , Dahi P , Sauter C , Shaffer B , Young JW , Peled J , Meagher RC , Jenq RR , van den Brink MRM , Giralt SA , Pamer EG , Xavier JB ((2018) ) Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci Transl Med 10: , eaap9489. |
[120] | Wang JW , Kuo CH , Kuo FC , Wang YK , Hsu WH , Yu FJ , Hu HM , Hsu PI , Wang JY , Wu DC ((2019) ) Fecal microbiota transplantation: Review and update. . J Formos Med Assoc 118: (Suppl 1), S23–S31. |
[121] | Kuai XY , Yao XH , Xu LJ , Zhou YQ , Zhang LP , Liu Y , Pei SF , Zhou CL ((2021) ) Evaluation of fecal microbiota transplantation in Parkinson’s disease patients with constipation. Microb Cell Fact 20: , 98. |
[122] | Sun MF , Zhu YL , Zhou ZL , Jia XB , Xu YD , Yang Q , Cui C , Shen YQ ((2018) ) Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun 70: , 48–60. |
[123] | Zhao Z , Ning J , Bao XQ , Shang M , Ma J , Li G , Zhang D ((2021) ) Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome 9: , 226. |

