
More Information
Submitted: 11 November 2019 | Approved: 26 November 2019 | Published: 27 November 2019
How to cite this article: Guglielmi V, Correale M, Leandro G. Gaucher’s disease and liver involvement: A review and our experience. Ann Clin Gastroenterol Hepatol. 2019; 3: 031-034.
DOI: 10.29328/journal.acgh.1001012
Copyright License: © 2019 Guglielmi V, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Gaucher disease; Chronic liver disease; Nonalcoholic fatty liver disease
Abbreviations: GD: Gaucher’s Disease; ERT: Enzyme Replacement; NAFLD: Nonalcoholic Fatty Liver Disease

Gaucher’s disease and liver involvement: A review and our experience
Vitantonio Guglielmi*, Mario Correale and Gioacchino Leandro
National Institute of Gastroenterology, “S.de Bellis” Research Hospital, Castellana Grotte, Bari, Italy
*Address for Correspondence: Vitantonio Guglielmi, National Institute of Gastroenterology, “S.de Bellis” Research Hospital, Castellana Grotte, Bari, Italy, Tel: +393389803392; Email: vitantonioguglielmi@tiscali.it
Background: This article reviews current knowledge of Gaucher’s disease (GD) and liver involvement and reports our experience: how many patients with chronic liver disease of unknown origin could be affected by Gaucher’s disease.
Patients and methods: Over 24 months, we tested 75 sine causa chronic liver disease patients (30 women and 45 men, mean age 55 years, range 15 to 77).
Results: None of the 75 patients was affected by Gaucher’s disease.
Conclusion: We believe that the chronic liver disease patient is unlikely to be affected by Gaucher’s disease. Probably this disease is to be found in cases of coexistence of hepatic disease and other symptoms of Gaucher’s disease (bone, neurological, bone marrow involvement).
Gaucher’s disease (GD) is an autosomal recessive disorder resulting from a mutation in the beta-glucocerebrosidase gene. The reduced enzyme activity causes the accumulation of glucocerebroside in cells of the monocyte-macrophage system throughout the body, but mainly in the spleen, liver and bone marrow [1,2]. There are 3 clinical forms. Type 1 is non neuropathic and is the most frequent (90%), the signs are skeletal and visceral. Type 2 is denominated infant: the patient dies before the age of 4 years owing to severe progression of the neurological involvement. Type 3 has been termed juvenile: the neurological signs and symptoms are less severe, and several different degrees of visceral involvement are observed. Liver involvement in GD is common, especially in splenectomized patients [3-6]. In the world population, type I Gaucher’s disease shows a prevalence of 1:40,000, but in the Ashkenazi Jews it is 1:800. In Italy the prevalence rates of GD have been documented as between 1:57.000 and 1:111.000. Currently, specific therapy is available, of 2 types, namely enzyme replacement (ERT) given every 2 weeks intravenously, or eliglustat administered orally. Both have the same effectiveness. Eliglustat is an analogue of glucocerebroside which partially inhibits the enzyme glucocerebroside synthetase, thereby reducing the production of glucocerebroside [7-12].
According to the study by Nagral, [13] (Table 1), liver involvement includes hepatomegaly (63% of GD type 1 patients) and ranges from mildly elevated liver enzymes to cirrhosis. Hepatic fibrosis can occur but cirrhosis and portal hypertension are uncommon except in splenectomized patients. Hepatopulmonary syndrome with hypoxia on standing, clubbing and cyanosis may be seen in patients with advanced cirrhosis, and especially splenectomized patients. In the work by James, [6], 24 of 25 GD patients had hepatomegaly and the majority had serum transaminases and alkaline phosphatase abnormalities. Percutaneous liver biopsies were obtained in 20 of these patients, while in 2, liver tissue was obtained at post-mortem exanimation: 3 had cirrhosis and portal hypertension, 14 had infiltration of the liver with Gaucher cells mainly in the centrilobular area and 5 had scattered foci of Gaucher cells throughout the liver. Linari, et al. [14] indicated significant liver disease (severe hepatomegaly, infarcts, varices, portal hypertension, hepatitis) as one of the symptoms in the highest risk patients that need tailored initial and maintenance ERT doses. In the Rosenbloom classification of clinical characteristics for all GD types [15], splenomegaly (85%), thrombocytopenia (68%), hepatomegaly (63%), osteopenia (55%), growth retardation (36%), anemia (34%), bone pain (33%), bone crises (7%) are listed. In the same work the author reports that liver function abnormalities may occur but are rarely severe. Acute elevation of liver enzymes may sometimes be attributable to cholecystitis as patients with GD type 1 often suffer from gallbladder stones. Cassineiro, et al. [16] confirmed the presence in GD of an enlarged liver which can be associated with ischemia or fibrosis nodules in about 20% of patients, while the development of liver failure or cirrhosis is rare, usually associated with portal hypertension, ascites or oesophageal varices. In the Baris study [17] hepatomegaly was almost universally present in GD type 1 but not usually massive unless massive splenomegaly was also present. In splenectomized patients advanced liver disease with portal hypertension and hepatopulmonary syndrome may occur. In a cohort of 103 pediatric patients, using ultrasonography Patlas, et al. [18] reported 100% prevalence of hepatomegaly. Hill, et al. [19]. Employed Magnetic Resonance, demonstrating some degree of liver enlargement in the entire cohort of 46 adult patients.
| Table 1: Experiences of other authors. | |||||
| Nagral | Hepatomegaly 63% | Elevated liver enzymes | Cirrhosis | ||
| James [6] | Hepatomegaly 96% | The majority had transaminase and alkaline phosphatase abnormalities 12% | Cirrhosis | Gaucher cells in centrilobular area 63% | Scattered foci of Gaucher cells throughout the liver 23% |
| Linari [14] | Hepatomegaly | Liver Infarcts | Varices | Portal hypertension | Hepatitis |
| Rosenbloom [15] | Splenomegaly 85% | Thrombocytopenia 68% | Hepatomegaly 63% | Osteopenia 55% | Growth retardation 36% |
| Cassineiro [16] | Hepatomegaly | Cirrhosis | Liver failure | ||
| Baris [17] | Hepatomegaly | Cirrhosis in splenectomized patients | |||
| Patlas [18] | In pediatric patients | Hepatomegaly 100% using ultrasonography | |||
| Hill [19] | In adult patients | Hepatomegaly 100% using Magnetic resonance | |||
The rationale of the study was to assess the prevalence of GD in a cohort of individuals (75) with chronic liver disease sine causa. In 24 months, we tested 75 sine causa chronic liver disease patients (30 women and 45 men, mean age 55 years, range 15 to 77). All of them presented altered liver function tests (hypertransaminasemia lasting at least 6 months); 30 patients also showed increased cholestasis enzymes. All patients were of Caucasian ethnicity except for one Mexican; none were Jewish. The patients had been consecutively admitted to our hospital (30) or visited in our liver clinic (45) over a period of two year (2017-2018). Two patients were obese and 3 were overweight. All patients underwent blood tests (Table 2), including viral serology for Hepatitis A, Hepatitis B, Hepatitis C, Human immunodeficiency Virus, Epstein-Barr Virus, Herpes Simplex Virus, and Cytomegalovirus and total gamma globulin, fasting lipid profile, glucose level, Homa Test, antitransglutaminases, ceruloplasmin, alpha 1 anti-trypsin, ferritin levels, total iron-binding capacity, antibody anti-smooth muscle, anti-mitochondrial, perinuclear anti-neutrophil cytoplasmic, anti-nuclear, anti-DNA, anti-liver/kidney microsomal, anti-soluble liver antigen, TSH. Lastly, drug and alcohol liver injury were excluded. All were subjected to abdominal ultrasound. No patient was splenectomized. Once all blood tests and abdominal ultrasound had excluded a possible liver disease etiology we looked for GD. After giving written informed consent, they underwent venous sampling, and the samples were sent to the Centogene laboratory in Germany. Here, the enzyme was assayed by tandem mass spectrometry and in cases showing a deficit, genetic-molecular confirmation was performed by Sanger gene sequencing.
| Table 2: Blood tests to rule out other causes of liver disease. | |||
| Viruses | Metabolic diseases | Autoimmune diseases | Others |
| HBV | Lipid profile | Antibody | Antitransglutaminases |
| HCV | Glucose level | Anti smooth muscle | TSH |
| HAV | Homa test | Anti mithocondrial | Drugs |
| HIV | Ceruloplasmin | Perinuclear anti neutrophil cytoplasmic | Alcohol |
| EBV | Alpha 1 anti trypsin | Anti nuclear | |
| Herpes | Ferritin level | Anti DNA | |
| CMV | Total iron binding capacity | Anti liver-kidney microsomal | |
| Anti soluble liver antigen | |||
Of the 75 patients tested (40 men and 35 women, mean age 55, range 15-77) with elevated transaminase levels lasting at least 6 months and unknown etiology, the test was doubtful in 4 patients, and repeated with negative results in 3, while 1 patient failed to return for further tests. Negative results were obtained in all the other patients. In 30 patients the diagnosis was hypertransaminasemia and in 16 of these abdominal sonography demonstrated diffuse steatosis: nonalcoholic fatty liver disease (NAFLD) [20,21]. In 19 patients the diagnosis was liver cirrhosis (in 5 diagnosed histologically and in 14 clinically; four had esophageal varices). Ten patients had hepatomegaly defined as a liver mass > 1.25 times the estimated normal volume [22], 7 splenomegaly (1500-3000 cc in size, compared to 50-200 cc in the average adult), 3 hepatosplenomegaly, 2 short stature, 2 thrombocytopenia, 1 chorea minor and 1 patient was diagnosed with chronic hepatitis of unknown etiology (Figure 1). Even the daughter of a Mexican patient suffering from Gaucher’s disease was negative. In six patients liver biopsy was performed: in 5 the diagnosis was liver cirrhosis and in 1 liver fibrosis (Metavir 2). In no case did the histology suggest an etiology. No patient was diagnosed with hepatocellular carcinoma [23]. The following associated diseases were found: diabetes mellitus (8 patients), arterial hypertension (7 patients), obesity (2 patients), one case of Parkinson’s disease, one of celiac disease with a gluten-free diet, one of oligophrenia, one of idiopathic portal thrombosis, one of multiple sclerosis, one of chronic renal failure.
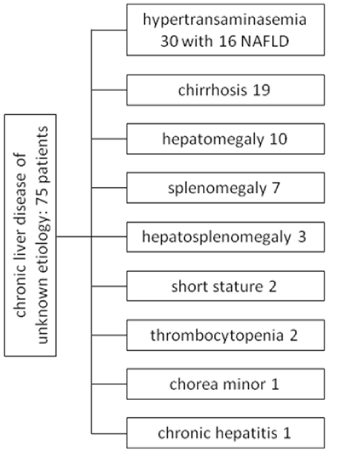
Figure 1: Chronic liver disease of unknown etiology: 75 patients.
This work, albeit within the limits of the small number of cases, demonstrates how rare GD is in patients with chronic liver disease of unknown etiology: of 75 patients we had only 1 doubtful case, who failed to return for further tests. Most probably, GD is more likely to be found in those patients who, apart from chronic liver disease, show signs and symptoms of other organs or systems involvement: Nervous System, Bone Marrow and Bone Tissue. We think the only important warning is that a priori the pretest probability is low even in this selected cohort, as GD is a rare disease. So it is not surprising that there were null results. For this reason we are still continuing our research to see whether as the numbers of patients increase the results change. In any case, we believe that GD must be included in the differential diagnosis of patients with chronic liver disease of an undetermined nature because the disease is, in fact, treatable [24-28], specific therapy is available, namely enzyme replacement (ERT) given every 2 weeks intravenously, or eliglustat administered orally. Both have the same effectiveness. We must also consider that many patients with chronic liver disease still lack a clear etiology. It is probable that other hepatotropic viruses, other metabolic diseases or other genetic diseases that can cause chronic liver disease have still to be identified.
We believe that the patient with chronic liver disease of unknown etiology is unlikely to be affected by Gaucher’s disease, although larger numbers of patients are needed to draw more precise conclusions. Probably this disease is to be found in cases with coexistent hepatic disease and other symptoms of Gaucher’s disease (bone, neurological, bone marrow involvement) [29-31]. In any case, faced with patients with liver markers alterations, this rare disease that includes liver involvement must be borne in mind, particularly since resolution therapy is available.
- Zirman A, Elstein D. Lipid storage diseases in: Williams Hematology, 8th ad McGraw-Hill, New York. 2010; 1065-1071.
- Grabowski GA, Horowitz M. Gaucher’s disease: molecular, genetic and enzymological aspects. Baillieres Clin. Haematol. 1997; 10: 635-656. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9497856
- Zirman A. How I treat Gaucher disease. Blood, 2011; 118: 1463-1471. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21670466
- Lachmann RH, Wight DG, Lomas DJ, Fisher NC, Schofield JP, et al. Massive hepatic fibrosis in Gaucher’s disease: clinic-pathological and radiological features. QJM. 2000; 93: 237-244.
- James SP, Stroymeyer FW, Stowens DW. Gaucher disease: hepatic abnormalities in 25 patients, in: DRJ, S Gatt (Eds), Gaucher disease: A Century of delineation and Research. New York. 1982; 131-142.
- James SP, Stroymeyer FW, Chang C, Barranger JA. Liver abnormalities in patients with Gaucher’s disease. Gastroenterology. 1981; 80: 126-133. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7450398
- Perel Y, Bioulac-Sage P, Chateil JF, Trillaud H, Carles J, et al. Gaucher’s disease and fatal hepatic fibrosis despite prolonged enzyme replacement therapy. Pediatrics. 2002; 109: 1170-1173. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12042560
- Mistry PK, Lukina E, Ben Turkia H, Amato D, Baris H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trila, JAMA. 2015; 313: 695-706. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25688781
- Ficicioglu C. Review of Miglustat for clinical management in Gaucher disease type 1. Ther Clin Risk Manag. 2008; 4: 425-431. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18728838
- Beutler E, Kay A, Saven A, Garver P, Thurston D, et al. Enzyme replacement therapy for Gaucher disease. Blood. 1991; 78: 1183-1189.
- Elstein D, Altarescu G, Maayaan H, Philips M, Abrahamov A, et al. Booster-effect with velaglucerase alfa in patients with Gaucher disease switched from long-term imiglucerase therapy: early access program results from Jerusalem. Blood Cells Mod Dis. 2012; 48: 45-50. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22047948
- Pastores GM, Barnett NL, Kolodny EH. An open-label non comparative study of miglustat in type 1 Gaucher disease: efficacy and tolerability over 24 months of treatment. Cli Ther. 2005; 27: 1215-1227. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22047948
- Aabha N. Gaucher disease. J Clin Exp Hepatol. 2014; 4: 37-50. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25755533
- Linari S, Castaman G. Clinical manifestations and management of Gaucher disease. Clin Cases Miner Bone Metab. 2015; 12: 157-164. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26604942
- Rosenbloom BE, Weinrteb NJ. Gaucher disease: a comprehensive review. Critical reviews in oncogenesis. 2013; 18: 163-175.
- Cassineiro E, Graziadei G, Poggiali E. Gaucher disease: a diagnostic challenge for internists. European Journal of Internal Medicine. 2014; 25: 117-124. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24090739
- Baris HN, Cohen IJ, Mistry PK. Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr Endocrinol Rev. 2014; 12: 72-81. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25345088
- Patlas M, Hadas-halpern I, Abrahamov A, Elstein D, Zimran A. Spectrum of abdominal sonographic findings in 103 pediatric patients with gaucher disease. Eur Radiol. 2002; 12: 397-400. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11870441
- Hill SC, Damaska BM, Ling A, Patterson K, Di Bisceglie AM, et al. Gaucher disease: abdominal MR imaging findings in 46 patients. Radiology. 1992; 184: 561-566. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1620865
- Zeqi G, Miaomiao L, Bing H, Xingshun Q. Association of non-alcoholic fatty liver disease with thyroid function: A systemic review and meta-analysis. Digestive and Liver Disease 2018; 50: 1153-1162.
- Chalasani N, Younossi Z, Lavine JE. The diagnosis and management of non-alcoholic fatty liver disease: practice guidance from the American Association for the study of Liver Diseases. Hepatology. 2018; 67: 328-357. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28714183
- Pastores GM, Weinereb NJ, Aerts H, Andria G, Cox TM, et al. Therapeutic goals in the treatment of Gaucher disease. Semi Hematol. 2004; 41: 4-14. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15468045
- De Fost M, Vom Dahl S, Weverling GJ, Brill N, Brett S, et al. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol Dis. 2006; 36: 53-58. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16246599
- Mistry PK. Consequences of diagnostic delays in type 1 Gaucher disease the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007; 82: 697-701. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17492645
- Harmanci O. Gaucher disease: new developments in treatment and etiology. World J Gastroenterol. 2008; 14: 3968-3973. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18609679
- Giraldo P. Neurological manifestations in patients with Gaucher disease and their relatives, it is just a coincidence? J Ineherit Metab Dis. 2011; 34: 781-787. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21384230
- Vom Dahl S. Loss of vision in Gaucher’s disease and its reversal by Enzyme-Replacement Therapy. N Eng J Med. 1998; 338: 1471-1472. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9583981
- Thomas AS. Diagnosing Gaucher disease: an ongoing need for increased awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. A J Hematol. 2007; 82: 697-701.
- Hasan. Huge splenomegaly with pancytopenia due to Gaucher’s desease in 22 years old woman. Mymensingh Med J. 2019; 284: 949-951. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31599267
- Capalbo L. Clinical characteristics of the neurological forms of Gauchers’s disease. Med Clin (Barc). 2011; 137: 6-11.
- Baldini M. Skeletal involvement in type 1 Gaucher disease: Not just bone mineral density. Blood cells Mol Dis. 2018; 68: 148-152. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28693786