
Abstract
Energy storage is a critical hurdle to the success of many clean energy technologies. Batteries with high energy density, good safety, and low cost can enable more efficient vehicles with electrified drive trains, such as hybrid electric vehicles, electric vehicles, and plug-in hybrid electric vehicles. They can also provide energy storage for intermittent energy sources, such as wind and solar. Today, and for the foreseeable future, rechargeable lithium batteries deliver the highest energy per unit weight or volume at reasonable cost. Many of the important properties of battery materials can be calculated with first-principles methods, making lithium batteries fertile ground for computational materials design. In this article, we review the successes and opportunities in using first-principles computations in the battery field. We also highlight some technical challenges facing the accurate modeling of battery materials.
Similar content being viewed by others

Introduction
Li-ion batteries are now ubiquitous in portable electronics because of their high specific energy, leading to low weight of the battery, and high energy density, leading to low volume of the battery. Materials researchers continually seek to improve the specific energy either by increasing the operating voltage or reducing the weight of the electrode materials. However, as Li-ion batteries are introduced into commercial vehicles, several additional factors such as safety, minimum time to charge and discharge (rate capability) , and cycle life are becoming increasingly important (See the Whittingham article in the April 2008 issue of MRS Bulletin .).
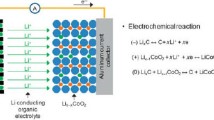
A Li-ion battery is composed of two electrodes (a cathode and an anode) separated by an electrolyte (i.e., a good Li conductor but poor electron conductor), as presented in Figure 1. Upon discharge, lithium ions travel through the electrolyte from the high lithium chemical potential present at the anode to the low lithium chemical potential present at the cathode. The electron traveling in the external circuit can be used to perform external work. During charge, an external electrical potential is applied, and the process is reversed. Typical cathode materials contain at least lithium, a transition metal, and oxygen (e.g., LiFePO4 or LiCoO2). Today, almost all commercial anodes are carbon based, though silicon is also being considered due to its potential for very high volumetric and gravimetric capacity to store Li. The electrolytes are organic solvents containing a Li salt.

Diagram of a Li-ion battery. Li-ion batteries operate by shuttling Li+ ions between the cathode and anode through an electronically insulating, ion-conducting electrolyte. During discharge, lithium ions migrate from the anode, where they are at a higher chemical potential, through the electrolyte, and into the cathode, where they are at a lower chemical potential. At the same time, an electron travels through the external circuit to perform external work. The free energy change of the lithium migration process is the maximum reversible work that can be obtained. During charge, the application of an external potential forces lithium ions to migrate out of the cathode, through the electrolyte, and back into the anode.
In this article, we review how computational methods can elucidate several key properties of Li-ion batteries, encompassing both electrolyte stability and electrode properties such as cell voltage, ionic and electronic conductivities, phase stability, and thermal stability.
Voltage
The energy density and specific energy of a battery are the products of its voltage and its volumetric or gravimetric charge capacity (i.e., the amount of lithium ions that can be reversibly removed and replaced in the materials during charge and discharge). However, one cannot simply increase the voltage without considering the limitations of other battery components. Electrolytes currently used in commercial cells limit the usable voltage to less than about 4.5 V versus Li/Li+, though the accessible stability up to 5 V is good enough to test new materials in laboratories.
The average equilibrium voltage of a cathode can be related to the difference in the Gibbs free energy between its charged state (delithiated phase) and discharged state (lithiated phase), taking into account the reference state of lithium on the anode.1 Because the contributions of entropy and volume effects to the Gibbs free energy difference are small, one may approximate the voltage using energy differences computed at zero temperature and pressure.
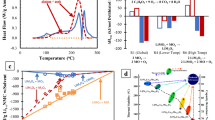
Standard approximations to density functional theory (DFT), such as the local density approximation (LDA) and generalized gradient approximation (GGA), generally lead to large errors in predicted voltages.2 This failure can be attributed to the incomplete cancellation of the non-physical electron self-interaction when an electron is transferred between significantly different environments. This prediction error is particularly significant in redox processes where electron transfer occurs between distinct orbitals. For example, in a typical Li-ion cell, electrons are transferred from the anode material (either Li metal or Li in carbon) to the localized d orbitals of an intercalation oxide. The self-interaction error in voltage predictions can be overcome with methods such as DFT+ U (Reference 3) or hybrid functionals.4–6 Although the U parameter in DFT+ U can be determined by self-consistent linear response theory (i.e., by calculating how the occupation of the transition metal orbitals change with the local potential),2,7 it is often fitted for each transition metal element to experimental oxidation energies.9 In hybrid functionals, some self-interaction is removed without relying on an adjustable parameter by incorporating a fraction of the exact Hartree-Fock exchange (the non-local effect of the Pauli exclusion principle) though at considerably greater computational expense. Figure 2 demonstrates the good agreement of voltages computed using GGA+ U and the Heyd-Scuseria-Ernzerhof (HSE) functional, with experimental voltages for a variety of cathode materials, with differences on the order of only a few hundred mV.

Comparison between experimental and computed (using generalized gradient approximation [GGA]+U or Heyd-Scuseria-Ernzerhof [HSE]) voltages. As titanium does not require a U value to reproduce accurate energies, no U value was used for Li2Ti2O4 and LiTiS2. Data from Chevrier et al.6
Screening materials through voltage prediction before any experimental work is conducted is now quite common. For instance, Zhou et al. predicted LiNiPO4 to have a voltage of 5.1 V,10 which was subsequently measured experimentally to lie between 5.1 V and 5.3 V.11 Likewise, the Co2+/3+ redox couple in Li2CoSiO4 was predicted to occur at a voltage of 4.4 V12 and confirmed at 4.3 V experimentally.13
While the previously described approach gives the average voltage as Li is exchanged, for most materials, the voltage changes as Li is inserted into a crystal structure. The actual voltage profile of a material can be obtained in great detail by predicting the Li configurations in the structure and their associated energy as a function of Li content by subdividing the lithiation interval and predicting the local average voltage within each interval,14–17 or through more advanced methods such as the cluster expansion.18,19 The cluster expansion method consists of fitting a model Hamiltonian to a large number of ab initio computations. This model Hamiltonian can subsequently be used to evaluate the energy of any lithium-vacancy arrangement at a much lower computational cost. By combining a cluster expansion with Monte Carlo simulation, configurational entropy can be accounted for to generate an accurate voltage profile.
Ionic and electronic conductivities
Charge and discharge of a lithium-ion battery consist of a series of events: during discharge, Li+ ions diffuse out of the anode material, solvate in the electrolyte, transfer through the electrolyte in the porous separator, and the process reverses at the cathode where Li+ desolvates and diffuses into the cathode. Electrons follow a different path, migrating from the anode material to the current collector, through the external circuit where they perform work, and through the carbon black in the cathode until they reach the intercalation cathode material, where they must reside on a transition metal ion close to the Li+ ion that was inserted from the electrolyte. Although all of these events can, in principle, be rate limiting, researchers have focused mostly on understanding and improving the electron and ion transport limitations in the cathode active materials.
Ionic conductivity
Li+ diffusion in electrode materials has been described in great detail by Van der Ven.18,20,21 Diffusion requires a low enough migration barrier for the ion as well as the presence of vacancies. The latter is usually not a problem, as the process of delithiation (battery charging) creates the necessary vacancies for Li motion. While dilute diffusion theory can be a reasonable guide in estimating the rate capability of a material, it needs to be realized that upon delithiation of the cathode, the concentration of vacancies and lithium are similar, and complex ordering phenomena or other phase transformations can occur.18 Diffusion in such non-dilute limits has been studied with first-principles methods by combining kinetic Monte Carlo with cluster expansions and activation energies obtained from DFT.21 Results on LiCoO2 showed that Li diffusion for layered materials actually occurs through a di-vacancy mechanism, underscoring the danger of relying too much on simplified diffusion models.
To obtain reasonable rate performance at room temperature, the barrier for Li migration in a crystal has to be below 500–600 meV. The Li migration barrier can be computed using the nudged elastic band method.22 This method approximates the transition state by relaxing a set of initial guesses; the relaxations are constrained by a spring force to prevent transition states from settling into equilibrium and to keep them well-spaced from one another. Systematic studies of the Li activation barrier through computations have given considerable insight into mechanisms to improve Li mobility.
Kang et al. found that Li mobility in layered cathodes such as LiCoO2 and Li(Ni,Mn,Co)O2 is controlled by layer spacing, thereby explaining the very large variation of diffusion constant with Li concentration in LiCoO2.25–27 This insight led to ways of improving the rate capability of layered Li(Ni0.5Mn0.5)O2 through crystal structure engineering.24 The diffusion mechanisms and defect energies in LiCoO2 were also comprehensively probed using atomistic methods by Fisher et al.28 For graphite (the dominant commercial anode material), Toyoura et al. were able to calculate the diffusive prefactor,29 and Persson et al. subsequently showed that diffusion within the two-dimensional graphite layers was intrinsically high-rate30 while significantly more sluggish in other directions. This finding elucidated the wide range of experimental diffusion coefficients reported in the literature.
The dimensionality of diffusion is important for the macroscopic transport behavior. Some of the highest rate cathode materials have two- and three-dimensional diffusion. In layered materials, such as LiCoO2, diffusion is two-dimensional, and Li+ moves rapidly within a single layer. Removal of too much lithium from the layer can lead to a contraction of the layer spacing, thereby increasing the activation barrier for motion. In three-dimensional diffusers such as spinel oxides, the Li diffusion constant varies less with Li content, as the framework of the structure remains intact in all dimensions as Li is removed. Spinels are therefore some of the highest rate cathode materials available.31,32
One-dimensional diffusion is somewhat pathological; even when high lithium mobility can be achieved in a channel, blocking of channels by defects limits the distance over which such high transport rate can be achieved, and may explain why a material such as LiFePO4 (which has one-dimensional diffusion channels) only functions well as a nanomaterial.33 Indeed, while first-principles computations20 predict that the migration barrier for Li in the channels of LiFePO4 is only about 200 meV, indicating ultrafast one-dimensional diffusion, measurements on large single crystals34,35 indicate somewhat lower three-dimensional diffusion. The paradox of this size-dependent diffusion was recently resolved by incorporating channel blocking defects into the diffusion model33 and confirmed the prediction that nanoscale LiFePO4 would have extremely high charge and discharge capability.
Based on the first-principles predictions, a high-rate LiFePO4 has been designed with the help of first-principles phase diagrams.36 Using off-stoichiometry that creates Li-conducting surface phases,37,38 materials could be made that, when embedded in optimized electrode configurations, achieved charge and discharge times approaching only a few minutes (Figure 3) or even seconds.37

(a) Calculated phase diagram and (b) experimental electrochemical performance of an off-stoichiometric LiFePO4. Off-stoichiometric experiments are typically performed with Li-excess or Li-deficiencies (directions B and C), but these directions do not improve electrochemical performance in LiFePO4. The calculated phase diagram shows that off-stoichiometry in an unconventional direction (mixed Li/P excess, direction A) leads to equilibrium with the highly conductive glass Li4 P2O7. Li4P2O7 forms a surface coating that facilitates Li+ transfer into the cathode, thus allowing for very high rates (up to 44 C, or full discharge in less than 1.5 minutes) to be achieved in laboratory experiments (b). Adapted from Reference 37.
Electronic conductivity
High rates of charge and discharge require not only fast ionic conductivity, but also fast electron transport within the electrode material. Electronic conduction in electrode materials has not been studied in as much detail as Li-ion transport, though it is likely to become more important as highly insulating materials are being considered as cathode compounds.39,40
Most intercalation compounds are probably polaronic, though some such as LiCoO2 undergo an insulator to metal transition upon delithiation due to the overlap of the hole states created by Li+ removal.41,42 Experimental papers have often attributed low electronic conductivity to a large bandgap, even though this may not always be the relevant quantity to determine electronic conductivity. In polaronic materials, conductivity depends on both the availability of carriers and their mobility. The charge compensation on the transition metal ions that accompanies Li+ insertion or removal creates mixed valence compounds with a large amount of potential carriers. Hence, focus should be more on the migration energy of polarons and their binding to Li+ or its vacancies. The application of DFT+ U to LiFePO4 has revealed43 small polaron migration barriers of ~200 meV, but relatively high binding energies between polarons and Li+ ions/vacancies (~370–500 meV). Methods that remove self-interaction, such as DFT+U, are crucial for studying polarons, as standard LDA and GGA approximations to DFT tend to incorrectly delocalize excess carriers in most of these materials. As many promising new cathode material classes such as silicates, borates, and sulfates are electrically insulating, predictive theories for polaron conduction would greatly clarify the rate capability of these materials.
Phase stability
Several important properties of electrode materials are highly dependent on structure: diffusion, voltage, and stability can vary significantly from one crystal structure to another. DFT has been used early on with a high level of success to predict structures and to provide insights into the structural stability of cathode materials: for instance, LiMnO2 forms an orthorhombic structure instead of the more common layered α-NaFeO2 structure because of magnetic effects.44 More recently, DFT has been used to predict stable new electrode materials. Arroyo et al. predicted a new high-pressure polymorph for Li2MnSiO4, confirmed subsequently by experiments.45 Recent developments in crystal structure prediction46–49 are likely to lead to more new cathode materials guided by computations.
While the as-synthesized structures of electrode materials can often be accurately determined with diffraction techniques, it is important to understand the changes in structure that occur upon lithium removal. For instance, a new phase of Lix CoO2 present at high states of charge was detected through first principles computations18 and later confirmed in experiments.42 In most cases, partially delithiated states present a unique electronic structure problem, as Li removal leads to mixed valence on the transition metal ions. A material such as Lix FePO4 should therefore be written as (Lix, VLi(1–x ))(Fe3+(1–x )Fe2+x) PO4, where VLi represents a Li vacancy. The coupling between the Fe2+/Fe3+ charge ordering and the Li-vacancy arrangement has been found to be crucial in reproducing the Lix FePO4 phase diagram. Zhou et al. found the DFT+U scheme could localize electrons and holes on the different Fe sites and explain the unusual shape of the experimental phase diagram by modeling the electronic entropy of the system (see Figure 4).50 Recently, the approach used in Zhou’s work has been extended to study the more complex Li(Mn,Fe)PO4 system.51 The use of standard LDA or GGA creates a single Fe type of intermediate valence by delocalizing Fe2+ and Fe3+, leading to a qualitative failure in predicting the phase diagram of this material;50 in LDA and GGA, the system is predicted to form a solid solution with intermediate Li-vacancy ordering rather than a two-phase miscibility gap. Given the fundamental difference in how such systems would lithiate and delithiate (two-phase reaction versus solid solution), a proper treatment of electron correlation is essential for making relevant predictions on battery materials. Only in Lix CoO2, which becomes metallic upon delithiation, is the delocalized description of GGA and LDA appropriate.

Comparison between the Lix FePO4 experimental phase diagram from Delacourt et al.52 and Dodd et al.53 and computed phase diagrams obtained with electronic entropic contribution (middle) and without (bottom).50 The triphylite LiFePO4 is referred to by T; the fully delithiated FePO4 phase, heterosite, by H; and the solid solution region is indicated by SS. Only the introduction of the electronic entropy contribution reproduces the experimentally observed solid solution region. © 2006, American Physical Society.
While most of the work on lithium battery electrodes has concerned intercalation, electrodes operating without preserving a host framework are currently of interest due to their much larger theoretical capacities.55 For instance, FeF3 has been demonstrated experimentally as a potential electrode following the reaction: FeF3 + Li → 3LiF + Fe.56 In this so-called conversion mechanism, the charged and discharged products are not structurally related. First-principles computations also have been used to investigate conversion cathodes such as FeF3 and BiF3.57,58
Studying conversion batteries by first-principles computations presents unique challenges. DFT errors in reaction energies are typically smaller when the reaction involves phases similar in chemical bonding. As conversion reactions may involve very different chemical phases (e.g., metals and fluorides), their associated computed voltage can show larger discrepancies with experiments than insertion voltages. In addition, the kinetics of ionic rearrangements are typically more difficult to model than diffusion in intercalation compounds, as reaction paths are largely unknown. The understanding and design of conversion electrodes will require significant advances in the currently available modeling techniques.
Thermal stability
Safety is a major concern of lithium batteries, and developing computational approaches toward predicting safety has been a major focus of several federally supported energy programs. At elevated temperatures, cathode materials may potentially release oxygen, which can combust the electrolyte and lead to runaway reactions and fire. While the discharged cathode is usually relatively stable with respect to oxygen release, cathodes based on layered LiCoO2, LiNiO2, and spinel LiMn2O4 decompose with oxygen evolution59,60 when heated in the highly oxidized charged state. Olivine LiFePO4,61 on the other hand, offers much better thermal stability, and it has been tacitly assumed that all materials based on the phosphate polyanion would share this advantage.62,63 With the increasing application of lithium rechargeable batteries to large scale applications such as hybrid electric vehicles and fully electric vehicles, the thermal stability of the battery system is becoming ever more important.
Recent computational thermodynamics approaches can approximate the intrinsic thermal stability of an electrode material. Wang et al.64 first studied the thermal stability of layered LiCoO2 and LiNiO2 and spinel LiMn2O4 by computing their first-principles phase diagrams and calculating the heats for structural transformation and decomposition reactions of the delithiated states. Subsequently, Ong et al. developed an oxygen grand potential phase diagram approach to determine the oxygen chemical potential at which an electrode decomposes to evolve oxygen.65 Using this approach, it has been shown that delithiated MnPO4 has significantly worse thermal stability than delithiated FePO4 (see Figure 5), both in terms of the critical oxygen evolution temperature as well as the amount of oxygen evolved. The calculations agree with previous experimental results59,66 and have cast doubt on the assumption that all phosphate polyanion compounds share the excellent thermal stability of LiFePO4.

Calculated thermal stabilities of delithiated MnPO4 and FePO4 (represented by MPO4 on the y -axis). The figure shows the number of moles of O2 evolved per mole of cathode material as a function of temperature. It can be seen that the onset of O2 evolution is at a much higher temperature (~700°C) for FePO4 compared to MnPO4 (~370°C), and the initial amount of O2 evolved is also much less. This difference in thermal behaviors can be explained by the fact that Fe3+ and Mn2+ both have the exchange-stabilized 3d 5 electron configuration, which suggest that the decomposition of FePO4 (requiring the reduction of Fe3+ to Fe2+) would be relatively more difficult than the decomposition of MnPO4 (requiring the reduction of Mn3+ to Mn2+). Adapted from Reference 65.
Electrolytes
The electrolyte functions as an ionic conductor and comprises a solvent, typically a mixture of a cyclic carbonate, such as ethylene carbonate (EC), and a linear carbonate, such as dimethyl carbonate (DMC), and a lithium salt, typically LiPF6. An ideal electrolyte should have a wide enough electrochemical window to accommodate the operating voltage of the battery, chemical stability against the electrodes and other components, a high ionic conductivity with extremely low electronic conductivity, low flammability, and low cost. Current EC/DMC + LiPF6 electrolytes are an imperfect compromise of these objectives. The electrochemical windows of EC/DMC + LiPF6 electrolytes of up to approximately 5 V versus Li/Li+ 67 are sufficient for current electrode systems but would be severely tested by higher voltage cathodes (e.g., LiNiPO4, which has a predicted voltage of 5.1 V).10 Current electrolytes are also unstable against the graphitic anode and function by forming a passivating solid-electrolyte interphase whose composition, structure, and transport mechanisms are still not well understood.67
A sizable proportion of the work on electrolytes has been focused on understanding the reductive and oxidative stabilities and decomposition mechanisms of EC, DMC, and other solvent additives, with and without coordination with Li+ or PF6–.68–71 I n some cases, a dielectric continuum solvation model,72 where the complex system of the salt and solvent is modeled as a “solute” embedded in a dielectric continuum, was used. Another body of work involves using molecular dynamics simulations with only a few repulsion-dispersion parameter types to obtain excellent descriptions of the heats of vaporizations, densities, self-diffusion coefficients, structural factors, and conformational dynamics of solvents.73,74 A few computational studies have also been devoted to the study of the lithium salt, especially with regard to the oxidative stability of the anions, ion conductivities, and dissociation energies.75,76
In recent years, room-temperature ionic liquids (ILs), which are comprised entirely of ions, have emerged as promising replacement electrolytes due to their wide electrochemical windows, low flammability, and low vapor pressure.77,78 The properties of ILs can be tuned by the suitable modification of the functional groups attached to the cation and anion, and therefore, ILs present an extremely large chemical design space that is amenable to computational design approaches. Ong et al. recently showed that qualitative trends in cation electron affinities and anion ionization energies are in good agreement with experimental data and found that electron-donating groups stabilize cations (leading to better reductive stability), and electron-withdrawing groups stabilize the anions (leading to better oxidation stability).79 However, it should be pointed out that the complex nature of the interactions in ILs presents a serious challenge for standard computational methods. For instance, Izgorodina et al. showed that the errors in the ion binding energies in ILs were predicted by many standard DFT functionals to be fairly large (above 12 kJ mol‒1).80 Nonetheless, computational approaches have been applied successfully to the investigation of the dielectric constants,81 decomposition mechanisms,82 and formation energies and stabilities of ILs.83,84 The development of force fields designed especially for ILs also has resulted in burgeoning research in the transport properties of these complex systems.85–89
In addition to the electrolyte solvent and salt, computational studies have shown that the oxidation potentials of redox shuttle additives, which provide overcharge and overdischarge protection for lithium batteries, can be estimated to a high degree of accuracy using first-principles calculations coupled with a dielectric continuum model.90,91
Computational resources
First-principles methods are more accessible than ever due to more efficient codes and reductions in the cost of computing over the last two decades, and most individual computations presented in this article can be run with 8–32 computing cores. A typical DFT calculation, including structural relaxations on a cathode material, may take 100–200 CPU hours on such resources. A voltage prediction encompasses two such calculations, and the accurate evaluation of voltage as a function of lithium content requires about 50–200 DFT calculations (7500–30000 CPU hours) in order to evaluate many possible Li vacancy arrangements at multiple intermediate compositions. Such a study requires at least a small computing cluster. In terms of computing activation barriers for Li diffusion, a single nudged elastic band calculation gives the barrier for one hop at one lithiation level and typically consumes about 5000 CPU hours. Because diffusivity may vary with the amount of lithium in the electrode, or “state of charge,” a comprehensive study of diffusion in a material may calculate activation barriers for several paths at several lithiation levels, thus multiplying this number by about a factor of 10, or 50000 CPU hours. Evaluating phase stability and thermal stability is typically less computationally expensive; a quaternary phase diagram may contain about 50–100 phases, which each require a DFT computation, and such a phase diagram can often be recycled to evaluate other materials in that chemical space.
Conclusions
Energy storage is an important societal problem; one where computational models can greatly assist in developing better materials. Density functional theory computations have been used to predict the voltage of several materials prior to synthesis10,12 and are now a viable pre-screening tool for new materials. In addition, computations have provided insights into the conductivity of existing Li-ion battery materials, which have led to methods for improving rate capability. Examples include the high-rate LiNi0.5Mn0.5O2 by minimizing interlayer disorder24 and extremely fast LiFePO437 by the use of surface coatings.
Improved techniques to model the complex properties of liquid electrolytes would further advance the field, as would a better understanding of intercalation dynamics in systems with mixed redox couples (e.g., LiFex Mn1‒x PO4). In addition, modeling conversion reactions that involve large structural changes remains problematic, despite some progress in this area.57,58 Finally, even when promising new materials are designed computationally, the discovery of appropriate synthesis and optimization routes still presents a difficult and often time-intensive challenge, which computations still only marginally address. We are optimistic that further work in these areas will yield new insights and electrode design possibilities.
The development of new electrode materials combines aspects of materials science, physics, and chemistry and can function as a test bed for new predictive computational approaches across a variety of disciplines.
References
M.K. Aydinol, A.F. Kohan, G. Ceder, K. Cho, J. Joannopoulos, Phys. Rev. B 56, 1354 (1997).
F. Zhou, M. Cococcioni, C.A. Marianetti, D. Morgan, G. Ceder, Phys. Rev. B 70, 235121 (2004).
S.L. Dudarev, S.Y. Savrasov, C.J. Humphreys, A.P. Sutton, Phys. Rev. B 57, 1505 (1998).
J. Heyd, G.E. Scuseria, M. Ernzerhof, J. Chem. Phys. 118, 8207 (2003).
A.V. Krukau, O.A. Vydrov, A.F. Izmaylov, G.E. Scuseria, J. Chem. Phys. 125, 224106 (2006).
V.L. Chevrier, S.P. Ong, R. Armiento, M.K.Y. Chan, G. Ceder, Phys. Rev. B 82, 075122 (2010).
H. Kulik, M. Cococcioni, D. Scherlis, N. Marzari, Phys. Rev. Lett. 97, 1 (2006).
F. Zhou, M. Cococcioni, C.A. Marianetti, D. Morgan, G. Ceder, Phys. Rev. B 70, 235121 (2004).
L. Wang, T. Maxisch, G. Ceder, Phys. Rev. B 73, 1 (2006).
F. Zhou, M. Cococcioni, K. Kang, G. Ceder, Electrochem. Commun. 6, 1144 (2004).
J. Wolfenstine, J. Allen, J. Power Sources 142, 389 (2005).
M. Arroyo-de Dompablo, M. Armand, J. Tarascon, U. Amador, Electrochem. Commun. 8, 1292 (2006).
C. Lyness, B. Delobel, A.R. Armstrong, P.G. Bruce, Chem. Commun. 4890 (2007).
J. Bréger, Y.S. Meng, Y. Hinuma, S. Kumar, K. Kang, Y. Shao-Horn, G. Ceder, C.P. Grey, Chem. Mater. 18, 4768 (2006).
D. Morgan, G. Ceder, M.Y. Saïdi, J. Barker, J. Swoyer, H. Huang, G. Adamson, Chem. Mater. 14, 4684 (2002).
A. Yamada, N. Iwane, Y. Harada, S. Nishimura, Y. Koyama, I. Tanaka, Adv. Mater. 8501 (2010).
B.J. Hwang, Y.W. Tsai, D. Carlier, G. Ceder, Chem. Mater. 15, 3676 (2003).
A. Van Der Ven, M.K. Aydinol, G. Ceder, J. Electrochem. Soc. 145, 2149 (1998).
J. Bhattacharya, A. Van Der Ven, Phys. Rev. B 81 (2010).
D. Morgan, A. Van Der Ven, G. Ceder, Electrochem. Solid-State Lett. 7, A30 (2004).
A. Van Der Ven, G. Ceder, Mater. Sci. 3, 5 (2000).
H. Jonsson, G. Mills, K.W. Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, B.J. Berne, G. Ciccotti, D.F. Coker, Eds. (World Scientific , River Edge, NJ , 1998 ), pp. 385 .
K. Kang, G. Ceder, Phys. Rev. B 74, 1 (2006).
K. Kang, Y.S. Meng, J. Bréger, C.P. Grey, G. Ceder, Science 311, 977 (2006).
M.D. Levi, G. Salitra, B. Markovsky, H. Teller, D. Aurbach, U. Heider, L. Heider, J. Electrochem. Soc. 146, 1279 (1999).
J. Barker, R. Pynenburg, R. Koksbang, M.Y. Saidi, Electrochim. Acta 41, 2481 (1996).
J. McGraw, C.S. Bahn, P.A. Parilla, J.D. Perkins, D.W. Readey, D.S. Ginley, Electrochim. Acta 45, 187 (1999).
C.A.J. Fisher, M.S. Islam, H. Moriwake, J. Phys. Soc. Jpn. 79, 59 (2010).
K. Toyoura, Y. Koyama, A. Kuwabara, F. Oba, I. Tanaka, Phys. Rev. B 78, 1 (2008).
K. Persson, V.A. Sethuraman, L.J. Hardwick, J. Phys. Chem. Lett. 1, 1176 (2010).
X. Ma, B. Kang, G. Ceder, J. Electrochem. Soc. 157, A925 (2010).
K. Nakahara, R. Nakajima, T. Matsushima, H. Majima, J. Power Sources 117, 131 (2003).
R. Malik, D. Burch, M. Bazant, G. Ceder, Nano Lett. 10, 4123 (2010).
R. Amin, J. Maier, P. Balaya, D.P. Chen, C.T. Lin, Solid State Ionics 179, 1683 (2008).
R. Amin, P. Balaya, J. Maier, Electrochem. Solid-State Lett. 10, A13 (2007).
S.P. Ong, L. Wang, B. Kang, G. Ceder, Chem. Mater. 20, 1798 (2008).
B. Kang, G. Ceder, Nature 458, 190 (2009).
A. Kayyar, H. Qian, J. Luo, Appl. Phys. Lett. 95, 221905 (2009).
N. Recham, J.-N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand J.-M. Tarascon, Nat. Mater. 9, 68 (2010).
N. Pereira, F. Badway, M. Wartelsky, S. Gunn, G.G. Amatucci, J. Electrochem. Soc. 156, A407 (2009).
C.A. Marianetti, G. Kotliar, G. Ceder, Nat. Mater. 3, 627 (2004).
M. Ménétrier, I. Saadoune, S. Levasseur, C. Delmas, J. Mater. Chem. 9, 1135 (1999).
T. Maxisch, F. Zhou, G. Ceder, Phys. Rev. B 73, 1 (2006).
S.K. Mishra, G. Ceder, Phys. Rev. B 59, 22 (1999).
M.E. Arroyo-de Dompablo, R. Dominko, J.M. Gallardo-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik, E. Morán, Chem. Mater. 20, 5574 (2008).
S.M. Woodley, R. Catlow, Nat. Mater. 7, 937 (2008).
G. Hautier, C.C. Fischer, A. Jain, T. Mueller, G. Ceder, Chem. Mater. 22, 3762 (2010).
C.C. Fischer, K.J. Tibbetts, D. Morgan, G. Ceder, Nat. Mater. 5, 641 (2006).
A.R. Oganov, C.W. Glass, J. Chem. Phys. 124, 244704 (2006).
F. Zhou, T. Maxisch, G. Ceder, Phys. Rev. Lett. 97, 1 (2006).
R. Malik, F. Zhou, G. Ceder, Phys. Rev. B 79, 1 (2009).
C. Delacourt, P. Poizot, J.-M. Tarascon, C. Masquelier, Nat. Mater. 4, 254 (2005).
J.L. Dodd, R. Yazami, B. Fultz, Electrochem. Solid-State Lett. 9, A151 (2006).
F. Zhou, T. Maxisch, G. Ceder, Phys. Rev. Lett. 97, 1 (2006).
J. Cabana, L. Monconduit, D. Larcher, M.R. Palacín, Adv. Mater. (2010).
F. Badway, N. Pereira, F. Cosandey, G.G. Amatucci, J. Electrochem. Soc. 150, A1209 (2003).
R.E. Doe, K.A. Persson, G. Hautier, G. Ceder, Electrochem. Solid-State Lett. 12, A125 (2009).
R.E. Doe, K.A. Persson, Y.S. Meng, G. Ceder, Chem. Mater. 20, 5274 (2008).
G. Chen, T.J. Richardson, J. Power Sources 195, 1221 (2010).
J. Dahn, E. Fuller, M. Obrovac, U. Vonsacken, Solid State Ionics 69, 265 (1994).
A.K. Padhi, K.S. Nanjundaswamya, J.B. Goodenough, J. Electrochem. Soc. 144, 1188 (1997).
A. Yamada, S. Chung, J. Electrochem. Soc. 148, A960 (2001).
S.K. Martha, B. Markovsky, J. Grinblat, Y. Gofer, O. Haik, E. Zinigrad, D. Aurbach, T. Drezen, D. Wang, G. Deghenghi, I. Exnar, J. Electrochem. Soc. 156, A541 (2009).
L. Wang, T. Maxisch, G. Ceder, Chem. Mater. 19, 543 (2007).
S.P. Ong, A. Jain, G. Hautier, B. Kang, G. Ceder, Electrochem. Commun. 12, 427 (2010).
S.-W. Kim, J. Kim, H. Gwon, K. Kang, J. Electrochem. Soc. 156, A635 (2009).
K. Xu, Chem. Rev. 104, 4303 (2004).
L. Xing, W. Li, C. Wang, F. Gu, M. Xu, C. Tan, J. Yi, J. Phys. Chem. B 113, 16596 (2009).
K. Tasaki, K. Kanda, T. Kobayashi, S. Nakamura, M. Ue, J. Electrochem. Soc. 153, 2192 (2006).
J.M. Vollmer, L.A. Curtiss, D.R. Vissers, K. Amine, J. Electrochem. Soc. 151, A178 (2004).
K. Tasaki, J. Phys. Chem. B 109, 2920 (2005).
J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 105, 2999 (2005).
O. Borodin, G.D. Smith, J. Phys. Chem. B 110, 4971 (2006).
O. Borodin, G.D. Smith, J. Phys. Chem. B 110, 6279 (2006).
P. Johansson, Phys. Chem. Chem. Phys. 9, 1493 (2007).
M. Ue, A. Murakami, S. Nakamura, J. Electrochem. Soc. 149, 1572 (2002).
M. Galinski, A. Lewandowski, I. Stepniak, Electrochim. Acta 51, 5567 (2006).
B. Garcia, S. Lavallee, G. Perron, C. Michot, M. Armand, Electrochim. Acta 49, 4583 (2004).
S.P. Ong, G. Ceder, Electrochim. Acta 55, 3804 (2010).
E.I. Izgorodina, U.L. Bernard, D.R. MacFarlane, J. Phys. Chem. A 113, 7064 (2009).
E.I. Izgorodina, M. Forsythb, D.R. MacFarlane, Phys. Chem. Chem. Phys. 11, 2452 (2009).
P.C. Howlett, E.I. Izgorodina, M. Forsyth, D.R. Macfarlane, Z. Phys. Chem. 220, 1483 (2006).
O. Borodin, J. Phys. Chem. B 113, 12353 (2009).
K.E. Gutowski, J.D. Holbrey, R.D. Rogers, D.A. Dixon, J. Phys. Chem. B 109, 23196 (2005).
O. Borodin, G.D. Smith, J. Phys. Chem. B 110, 11481 (2006).
O. Borodin, G.D. Smith, O. Geiculescu, S.E. Creager, B. Hallac, D. DesMarteau J. Phys. Chem. B 110, 24266 (2006).
O. Borodin, G.D. Smith, W. Henderson, J. Phys. Chem. B 110, 16879 (2006).
J. de Andrade, E.S. Boes, H. Stassen, J. Phys. Chem. B 106, 13344 (2002).
M.J. Monteiro, F.F.C. Bazito, L.J.A. Siqueira, M.C.C. Ribeiro, R.M. Torresi, J. Phys. Chem. B 112, 2102 (2008).
R.L. Wang, J.R. Dahn, J. Electrochem. Soc. 153, A1922 (2006).
R.L. Wang, C. Buhrmester, J.R. Dahn, J. Electrochem. Soc. 153, A445 (2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ceder, G., Hautier, G., Jain, A. et al. Recharging lithium battery research with first-principles methods. MRS Bulletin 36, 185–191 (2011). https://doi.org/10.1557/mrs.2011.31
Published:
Issue Date:
DOI: https://doi.org/10.1557/mrs.2011.31