
Abstract
Predicting the dislocation nucleation rate as a function of temperature and stress is crucial for understanding the plastic deformation of nanoscale crystalline materials. However, the limited time scale of molecular dynamics simulations makes it very difficult to predict the dislocation nucleation rate at experimentally relevant conditions. We recently develop an approach to predict the dislocation nucleation rate based on the Becker–Döring theory of nucleation and umbrella sampling simulations. The results reveal very large activation entropies, which originated from the anharmonic effects, that can alter the nucleation rate by many orders of magnitude. Here we discuss the thermodynamics and algorithms underlying these calculations in greater detail. In particular, we prove that the activation Helmholtz free energy equals the activation Gibbs free energy in the thermodynamic limit and explain the large difference in the activation entropies in the constant stress and constant strain ensembles. We also discuss the origin of the large activation entropies for dislocation nucleation, along with previous theoretical estimates of the activation entropy.










Similar content being viewed by others


References
X. Li, Y. Wei, L. Lu, K. Lu, and H. Gao: Dislocation nucleation governed softening and maximum strength in nano-twinned metals. Nature 464, 877 (2010).
J. Li: The mechanics and physics of defect nucleation. MRS Bull. 32, 151 (2007).
T. Zhu, J. Li, S. Ogata, and S. Yip: Mechanics of ultra-strength materials. MRS Bull. 34, 167 (2009).
J. Li, K.J. Van Vliet, T. Zhu, S. Yip, and S. Suresh: Atomistic mechanisms governing elastic limit and incipient plasticity in crystals. Nature 418, 307 (2002).
C.A. Schuh, J.K. Mason, and A.C. Lund: Quantitative insight into dislocation nucleation from high-temperature nanoindentation experiments. Nature Mater. 4, 617 (2005).
P. Schall, I. Cohen, D.A. Weitz, and F. Spaepen: Visualizing dislocation nucleation by indenting colloidal crystals. Nature 440, 319 (2006).
W.D. Nix, J.R. Greer, G. Feng, and E.T. Lilleodden: Deformation at the nanometer and micrometer length scales: Effects of strain gradients and dislocation starvation. Thin Solid Films 515, 315 (2007).
S. Izumi, H. Ohta, C. Takahashi, T. Suzuki, and H. Saka: Shuffle-set dislocation nucleation in semiconductor silicon device. Philos. Mag. Lett. 90, 707 (2010).
G. Xu, A.S. Argon, and M. Ortiz: Critical configurations for dislocation nucleation from crack tips. Philos. Mag. A 75, 341 (1997).
F. Frank: Symposium on the Plastic Deformation of Crystalline Solids; Carnegie Institute of Technology and Office of Naval Research, Pittsburgh, PA, 1950; p. 89.
S. Aubry, K. Kang, S. Ryu, and W. Cai: Energy barrier for homogeneous dislocation nucleation: Comparing atomistic and continuum models. Scripta Mater. 64, 1043 (2011).
M.A. Tschopp, D.E. Spearot, and D.L. McDowell: Atomistic simulations of homogeneous dislocation nucleation in single crystal copper. Model. Simul. Mater. Sci. Eng. 15, 693 (2007).
E.M. Bringa, K. Rosolankova, R.E. Rudd, B.A. Remington, J.S. Wark, M. Duchaineau, D.H. Kalantar, J. Hawreliak, and J. Belak: Shock deformation of face-centered-cubic metals on subnanosecond timescales. Nat. Mater. 5, 805 (2006).
T. Zhu, J. Li, A. Samanta, A. Leach, and K. Gall: Temperature and strain-rate dependence of surface dislocation nucleation. Phys. Rev. Lett. 100, 025502 (2008).
P. Hanggi, P. Talkner, and M. Borkovec: Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 62, 251 (1990).
R. Becker and W. Döring: The kinetic treatment of nuclear formation in supersaturated vapors. Ann. Phys. (Weinheim) 24, 719 (1935).
H. Eyring: The activated complex in chemical reactions. J. Chem. Phys. 3, 107 (1935).
G.H. Vineyard: Frequency factors and isotope effects in solid state rate processes. J. Phys. Chem. Solids 3, 121 (1957).
H. Jónsson, G. Mills, and K.W. Jacobsen: Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; B.J. Berne, G. Ciccotti, and D.F. Coker, Eds.; World Scientific: New York; 1998; pp. 385–404.
A.F. Voter: Introduction to the kinetic Monte Carlo Metho; Springer: New York; 2007.
C. Jin, W. Ren, and Y. Xiang: Computing transition rates of thermally activated events in dislocation dynamics. Script. Mater. 62, 206 (2010).
D. Chandler: Introduction to Modern Statistical Mechanics; Oxford University Press: Oxford, UK; 1987.
W. E. Ren and E. Vanden-Eijnden: String method for the study of rare events. Phys. Rev. B 66, 052301 (2002).
W. E. Ren and E. Vanden-Eijnden: Finite temperature string method for the study of rare events. J. Phys. Chem. B 109, 6688 (2005).
S. Ryu, K. Kang, and W. Cai: Entropic effect on the rate of dislocation nucleation. Proc. Natl. Acad. Sci. USA 108, 5174 (2011).
D. Frenkel and B. Smit: Understanding Molecular Simulation: From Algorithms to Applications, Academic Press: San Diego, CA; 2002.
U.F. Kocks, A.S. Argon, and M.F. Ashby: Thermodynamics and kinetics of slip. Prog. Mater. Sci. 19, 1 (1975).
J.F. Nye: physical Properties of Crystals: Their Representation by Tensors and Matrices; Clarendon Press: New York; 1957.
H. Xiao, O.T. Bruhns, and A. Meyers: Logarithmic strain, logarithmic spin and logarithmic rate. Acta Mech. 124, 89105 (1997).
R. Bechmann, A.D. Ballato, and T.J. Lukaszek: Higher-order temperature coefficients of the elastic stiffinesses and complinaces of alpha-quartz. In Proceedings of the Institute of Radio Engineers, Vol. 50; 1962; p. 1812.
E. Whalley: Use of volumes of activation for determining reaction mechanisms. In Advances in Physical Organic Chemistry; V. Gold, Ed.; Academic Press: London; 1964; pp. 93–162.
M.L. Tonnet and E. Whalley: Effect of pressure on the alkaline hydrolysis of ethyl acetate in acetone–water solutions. Parameters of activation at constant volume. Can. J. Chem. 53, 3414 (1975).
W.C. Overton Jr. and J. Gaffney: Temperature variation of the elastic constants of cubic elements. I. Copper. Phys. Rev. 98, 969 (1955).
J.W. Cahn and F.R.N Nabarro: Thermal activation under shear. Philos. Mag. A 81, 1409 (2001).
A.H. Cottrell: Thermally activated plastic glide. Philos. Mag. Lett. 82, 65 (2002).
Y. Mishin, M.R. Sorensen, and A.F. Voter: Calculation of point-defect entropy in metals. Philos. Mag. A 81, 2591 (2001).
D. Gupta, Ed.: Diffusion Processes in Advanced Technological Materials; Springer: New York; 2002; p.140.
C. Kittel: Introduction to Solid State Physics, 8th ed.; Wiley: New York; 2004.
A.S. Argon, R.D. Andrews, J.A. Godrick, and W. Whitney: Plastic deformation bands in glassy polystyrene. J. Appl. Phys. 39, 1899 (1968).
R.J. DiMelfi, W.D. Nix, D.M. Barnett, J.H. Holbrook, and G.M. Pound: An analysis of the entropy of thermally activated dislocation motion based on the theory of thermoelasticity. Phys. Status Solidi B 75, 573 (1976).
R.J. DiMelfi, W.D. Nix, D.M. Barnett, and G.M. Pound: The equivalence of two methods for computing the activation entropy for dislocation motion. Acta Mater. 28, 231 (1980).
G. Kemeny and B. Rosenberg: Compensation law in thermodynamics and thermal death. Nature 243, 400 (1973).
A. Yelon, M. Movagha, and H.M. Branz: Origin and consequences of the compensation (Meyer–Neldel) law. Phys. Rev. B 46, 12243 (1992).
M. Born: Thermodynamics of crystals and melting. J. Chem. Phys. 7, 591 (1939).
S. Brochard, P. Hirel, L. Pizzagalli, and J. Godet: Elastic limit for surface step dislocation nucleation in face-centered cubic metals: Temperature and step height dependence. Acta Mater. 58, 4182 (2010).
M. Khantha, D.P. Pope and V. Vitek: Dislocation screening and the brittle-to-ductile transition: A Kosterlitz–Thouless type instability. Phys. Rev. Lett. 74, 684 (1994).
Y. Mishin, M.J. Mehl, D.A. Papaconstantopoulos, A.F. Voter, and J.D. Kress: Structural stability and lattice defects in copper: Ab initio, tight-binding, and embedded-atom calculations. Phys. Rev. B 63, 224106 (2001).
J.P. Hirth and J. Lothe: Theory of Dislocations; Krieger: New York; 1992.
M. Parrinello and A. Rahman: Crystal structure and pair potentials: A molecular dynamics study. Phys. Rev. Lett. 45, 1196 (1980).
H.C. Anderson: Molecular dynamics at constant pressure and/or temperature. J. Chem. Phys. 72, 2384 (1980).
W.G. Hoover: Canonical dynamics: Equilibrium phase space distribution. Phys. Rev. A 31, 1695 (1985).
T. Zhu, J. Li, K.J. Van Vliet, S. Ogata, S. Yip, and S. Suresh: Predictive modeling of nanoindentation-induced homogeneous dislocation nucleation in copper. J. Mech. Phys. Sol. 52, 691 (2004).
A.H.W Ngan, L. Zuo, and P.C. Wo: Size dependence and stochastic nature of yield strength of micron-sized crystals: A case study on Ni3Al. Proc. Royal Soc. A 462, 1661 (2006).
S. Ryu and W. Cai: Validity of classical nucleation theory for Ising models. Phys. Rev. E 81, 030601(R) (2010).
T. Zhu, J. Li, A. Samanta, H.G. Kim, and S. Suresh: Interfacial plasticity governs strain rate sensitivity and ductility in nanostructured metals. Proc. Natl. Acad. Sci. USA 104, 3031 (2007).
S.M. Foiles: Evaluation of harmonic methods for calculating the free energy of defects in solids. Phys. Rev. B 49, 14930 (1994).
M. de Koning, C.R. Miranda, and A. Antonelli: Atomistic prediction of equilibrium vacancy concentraions in Ni3Al. Phys. Rev. B 66, 104110 (2002).
S. Ryu and W. Cai: Comparison of thermal properties predicted by interatomic potential models. Model. Simul. Mater. Sci. Eng. 16, 085005 (2008).
A. Yelon, B. Movaghar, and R.S. Crandall: Multi-exitation entropy: Its role in thermodynamics and kinetics. Rep. Prog. Phys. 69, 1145 (2006).
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper has been selected as an Invited Feature Paper.
Appendices
Appendix A: Equality of Critical Sizes \(n_c^{{\sigma }}\) and \(n_c^{{\gamma }}\)
Suppose that the Gibbs free energy G(n,σ,T)is maximized at \(n_c^{{\sigma }}\), then
Note that T is held constant throughout this section. Through the Legendre transform, Eq. (5), we have the following property forthe Helmholtz free energy F(n,γ,T):
By definition, F(n,γ,T) reaches maximum at \(n = n_c^{{\gamma }}\) at constant γ and T,
Therefore, we establish that \(n_c^{{\gamma }} = n_c^{{\sigma }},\) that is, the maxi-mizer \(n_c^{{\sigma }}\) of G(n,σ,T) is also the maximizer \(n_c^{{\gamma }}\) of F(n,γ,T).
Appendix B: Equality of Activation Gibbs and Helmholtz Free Energies
The activation Gibbs free energy is the free energy difference between state 0, a perfect crystal, and state 1, a crystal containing a critical dislocation loop under a same shear stress σ. Because of the plastic shear deformation caused by the dislocation loop, state 1 has a higher strain (γ) than state 0 (γ0). It has been shown that the maximizer \(n_c^{{\sigma }}\) of G(n,σ,T) equals the maximizer of \(n_c^{{\gamma }}\) of F(n,γ,T) when σ equals σ(nc,γ,T), as defined in Eq. (6). Note that we keep σ to be the stress at ncγ, and T. Then at the same σ, but for n = 0, the strain becomes γ0. Hence, the activation Gibbs free energy barrier can be written as
Note that F(nc,γ,T) and F(0,γ0,T) do not correspond to the same strain state, so that their difference is not the activation Helmholtz free energy. To construct the activation Helmholtz free energy, we subtract and add the F(0,γ,T) term in the right hand side,
Note that \({{\gamma }}V\,\, = \,\,\, - \partial G\left( {{n_{\text{c}}},{{\sigma,}}T} \right)/\partial {{\sigma }}\) and \({{{\gamma }}_0}V\,\, = \,\,\, - \partial G\left( {{n_{\text{c}}},{{\sigma,}}T} \right)/\partial {{\sigma }}\). Then \(\left( {{{\gamma }}V\,\, - \,\,{{{\gamma }}_0}V} \right)\) is equivalent to \( - \left( {{\partial \mathord{\left/ {\vphantom {\partial {\partial {{\sigma }}}}} \right. } {\partial {{\sigma }}}}} \right)\left( {G\left( {{n_{\text{c}}},{{\sigma,}}T} \right) - G\left( {0,{{\sigma,}}T} \right)} \right) = - \left( {\partial {G_{\text{c}}}/\partial {{\sigma }}} \right) \equiv {\Omega _c}\),that is, the activation volume. By plugging \(\left( {{{\gamma }} - {{{\gamma }}_0}} \right){\Omega _c}/V\) into the equation, we have
In the thermodynamics limit \(\left( {V \to \infty } \right)\), we have Gc = Fc. Hence, the nucleation rate does not depend on whether the crystal is subjected to constant stress or constant strain loading. The equality allows us to compute the activation Gibbs free energy\({G_c}\left( {{{\sigma,}}T} \right)\) by combining the activation Helmholtz free energy Fc(γ,T) and the stress–strain relations of the perfect crystal shown in Fig. 2(b) and (d).
Appendix C: Physical Interpretation of Activation Entropy Difference ΔSc
It is well known that entropy is a thermodynamic state variable that is independent of the ensemble of choice, that is, \(S\left( {n,{{\gamma }},T} \right) \equiv \partial F{\left. {{{\left( {n,{{\gamma }},T} \right)} \mathord{\left/ {\vphantom {{\left( {n,{{\gamma }},T} \right)} {\partial T}}} \right. } {\partial T}}} \right|_{n{\text{,}\gamma }}}\) and \(S\left( {n,{{\sigma }},T} \right) \equiv \partial G{\left. {{{\left( {n,{{\sigma }},T} \right)} \mathord{\left/ {\vphantom {{\left( {n,{{\sigma }},T} \right)} {\partial T}}} \right. } {\partial T}}} \right|_{n{\text{,}\sigma }}}\) equal to each other as long as \({{\sigma }} = {V^{ - 1}}{\left. {{{\partial F} \mathord{\left/ {\vphantom {{\partial F} {\partial {{\gamma }}}}} \right. } {\partial {{\gamma }}}}} \right|_{n,T}}\). At the same time, the activation entropy is just the entropy difference between the activated state and the metastable state, that is, \({S_c}\left( {{{\gamma,}}T} \right) = S\left( {{n_c},{{\gamma,}}T} \right) - S\left( {0,{{\gamma }},T} \right)\) and \({S_c}\left( {{{\sigma,}}T} \right) = S\left( {{n_c},{{\sigma,}}T} \right) - S\left( {0,{{\sigma }},T} \right)\). If the entropies in two ensembles can equal each other, it may seem puzzling how the activation entropies can be different.
The resolution of this apparent paradox is that under the constant applied stress, the nucleation of a dislocation loop causes a strain increase. Let γ be the strain at the state defined by n = nc, σ, and T, and γ0 be the strain at the state defined by n = 0, σ, and T, then γ > γ0. Hence, we have \(S\left( {{n_c},{{\sigma,}}T} \right) = S\left( {{n_c},{{\gamma,}}T} \right)\) and \(S\left( {{n_c},{{\sigma,}}T} \right) = S\left( {{n_c},{{\gamma,}}T} \right)\), but \(S\left( {0,{{\gamma,}}T} \right) \ne S\left( {0,{{{\gamma }}_0}{\text{,}}T} \right)\).
This shows that the activation entropy difference \(\Delta {S_c} \equiv {S_c}\left( {{\sigma }} \right) - {S_c}\left( {{\gamma }} \right)\) equals to \(S\left( {0,{{\gamma,}}T} \right) - S\left( {0,{{{\gamma }}_0}{\text{,}}T} \right)\), which is the entropy difference of the perfect crystal at two slightly different strains.
In the limit of \(V \to \infty \), because we expect \(\left( {{{\gamma }} - {{{\gamma }}_0}} \right) \to 0\), we might reach a false conclusion that \(\Delta {S_c} = \left( {S\left( {{\text{0,}\gamma },T} \right) - S\left( {{\text{0,}}{{{\gamma }}_0},T} \right)} \right) \to 0\). Instead, the correct behavior in the thermodynamic limit can be obtained by expanding ΔSc in a Taylor series,
where the Maxwell relationship \({\left. {{{\partial S} \mathord{\left/ {\vphantom {{\partial S} {\partial {{\gamma }}}}} \right. } {\partial {{\gamma }}}}} \right|_T} = - V{\left. {{{\partial {{\sigma }}} \mathord{\left/ {\vphantom {{\partial {{\sigma }}} {\partial T}}} \right. } {\partial T}}} \right|_{{{\gamma }},V}}\) is used. The term (γV–γ0V) equals the activation volume Ωc and can be interpreted as plastic strain γpl due to the formation of a dislocation loop times the volume of the crystal, that is,
where b is the magnitude of the Burgers vector and Ac is the area of the critical dislocation loop. Using the relation \(\left( {{{\gamma }} - {{{\gamma }}_0}} \right) = {{{\Omega _c}} \mathord{\left/ {\vphantom {{{\Omega _c}} V}} \right. } V}\), we have
which is exactly the same as Eq. (24).
A similar expression has been obtained for the difference between point defect formation entropies under constant pressure (Sp) and under constant volume (Sv),39 with
where \({{\beta }} \equiv {V^{ - 1}}{\left. {\left( {{{\partial V} \mathord{\left/ {\vphantom {{\partial V} {\partial T}}} \right. } {\partial T}}} \right)} \right|_{p = 0}}\) is the thermal expansion factor at zero hydrostatic pressure p, B is the isothermal bulk modulus, and Vrel is the relaxation volume of the defect. In Cu, the value of Sp–Sv is estimated to be–1.7 kB for a vacancy and 13.7 kB for an interstitial.36 Comparing Eq. (C5) with Eq. (C4), we note that relaxation volume Vrel for point defects corresponds to the activation volume X for dislocation nucleation and that ßß corresponds to the \({\left( {{{\partial {{\sigma }}} \mathord{\left/ {\vphantom {{\partial {{\sigma }}} {\partial T}}} \right. } {\partial T}}} \right)}\) term. The similarity between these two equations is because they both express the entropy difference between two states, and the choice of the two states depends on whether the stress or the strain is kept constant when the defect is introduced. In contrast, there are also some differences between the physics expressed by these two equations. First, thermal expansion plays a prominent role in Eq. (C5) because it focuses on hydrostatic stress and strain effects. In comparison, thermal expansion does not play a role in Eq. (C4) because it focuses on shear stress and strain effects. Second, the formation entropy of a point defect is the entropy difference between two metastable states and governs equilibrium properties, for example, the density of vacancies at thermal equilibrium. In comparison, the activation entropy is the entropy difference between a saddle (i.e., unstable) state and a metastable state and governs kinetics, such as the dislocation nucleation rate. In addition, the saddle state (i.e., the size of the critical nucleus) depends on stress and temperature, whereas such complexity does not arise in the formation entropy of point defects.
Appendix D: Approximation of \({S_c}\left( {{\sigma }} \right)\)
We now introduce a series of simplifying approximations to estimate the magnitude of \({S_c}\left( {{\sigma }} \right)\) in the low temperature, low stress limit. In the temperature range of 0–300 K, the activation entropy is found to be insensitive to temperature. Starting from Eqs. (24) and (38), we have
If we assume the crystal is linear elastic, that is, σ = μγ, then
Similar expressions can be obtained for normal (com-pressive) loading by replacing γ with e and replacing μ by the Young’s modulus.
To gain more intuition, we note that in the limit of σ →0, the line tension model estimates that \({H_c}\left( {{\sigma }} \right) \propto {{{\sigma }}^{ - 1}}\). In addition, in the limit of \(T \to 0,{\Omega _c}\left( {{\sigma }} \right) \approx {{\partial {H_c}} \mathord{\left/ {\vphantom {{\partial {H_c}} {\partial {{\sigma }}}}} \right. } {\partial {{\sigma }}}}\). Under these conditions, \({\Omega _c}\left( {{\sigma }} \right) \times {{\sigma }} \approx {H_c}\left( {{\sigma }} \right)\), so that
Comparing Eq. (D3) with Eq. (38), we have
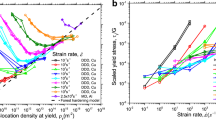
This trend is qualitatively observed in heterogeneous nucleation, when comparing Fig. 8(b) and (d), and is less clear in homogeneous nucleation, when comparing Fig. 8(a) and (c). This is probably because the stress-strain relationship is more nonlinearin the case ofhomogeneous nucleation.
Appendix E: Activation Free Energy Data
Table E.I and Table E.II provide the activation free energy data at all temperature and strain conditions in this study so that interested readers can use them as a benchmark. For homogeneous nucleation, the strain γxy is defined as \({{\Delta x} \mathord{\left/ {\vphantom {{\Delta x} {h_y^0}}} \right. } {h_y^0}}\), where Δx is the displacement of the repeat vector initially in the y direction along the x axis at each pure shear stress condition and \({h_y^0}\) is the height of the cell along the y axis at zero temperature without external loading. Shear stress σxy is determined from the x–y component of the average Virial stress. For heterogeneous nucleation, we take only the elastic strain into account. The elastic strain ϵzz at T is defined as \({{\left[ {{L_z}\left( {{{\sigma }},T} \right) - {L_z}\left( {{{\sigma = 0,}}T} \right)} \right]} \mathord{\left/ {\vphantom {{\left[ {{L_z}\left( {{{\sigma }},T} \right) - {L_z}\left( {{{\sigma = 0,}}T} \right)} \right]} {L_z^0}}} \right. } {L_z^0}}\), where \({{L_z}\left( {{{\sigma }},T} \right)}\) is the length of the repeat vector along the z axis;\({{L_z}\left( {{{\sigma = 0,}}T} \right)}\) is the equilibrium length at temperature T under zero stress; and \(L_z^0 = 20{a_0} = 72.3{\text{{\AA}}}\) is the reference length (before relaxation), where a0 is the lattice constant of copper. The compressional stress σzz is defined by \({{{\sigma }}_{zz}} = < {\text{F}} >/{d^2}\), where F> is the axial force computed from the z–z component of the average Virial stress and d = 15a0 = 54.225 Å is the reference side length of the nanorod.

Data for homogeneous nucleation: σxy, (Gpa); Ec \({\tilde E_c}\), and Fc (eV); and \(f_c^ + \left( {{{10}^{14}}{{\text{s}}^{ - 1}}} \right)\).

Data for heterogeneous nucleation: σzz (Gpa), Ec and Fc (eV), and \(f_c^ + \left( {{{10}^{14}}{{\text{s}}^{ - 1}}} \right)\).
Appendix F: Activation Volume and Critical Loop Size
The activation volume Ωc is defined as the derivative of activation free energy with stress, that is, \({\Omega _c}\left( {{{\sigma }},T} \right) = - {\left. {{{\partial {G_c}} \mathord{\left/ {\vphantom {{\partial {G_c}} {\partial {{\sigma }}}}} \right. } {\partial {{\sigma }}}}} \right|_T}\), and measures the sensitivity of the nucleation rate to the stress. Physically, it is interpreted as plastic strain associated with the dislocation loop times the volume of the crystal, that is, Ωc = bAc,where Ac is theareaof the critical dislocation loop (see Appendix C). Here, we use our numerical data to test the validity of the latter interpretation.
Because the activation volume measures the sensitivity of Gc(σ,T) to applied stress [see Eq. (10)], it must be proportional to the Schmid factor S in uniaxial loading. Hence, the hypothesis we wish to test is
where b is the magnitude of the Burgers vector and Aa is the average area each atom occupies on the {111} slip plane. Given that the lattice constant of Cu is a0 = 3.615 Å, we have b = a0/√6 = 1.48 Å and \({A_{\text{a}}} = \sqrt 6 {{a_0^2} \mathord{\left/ {\vphantom {{a_0^2} 4}} \right. } 4} = 5.66{{\text{{\AA}}}^2}\), so that bAa = 8.35 Å3. For the pure shear loading in our homogeneous nucleation case, the Schmid factor S = 1. For the uniaxial loading in our heterogeneous nucleation case, S = 0.471.
Figure E1 plots nc versus Ωc for both homogeneous and heterogeneous dislocation nucleations. In both cases, Ωc appears to be roughly linear with nc as expected from Eq. (F1). For homogeneous nucleations under pure shear [Fig. E.I(a)], the slope of the curves is roughly \({\Omega _c}/{n_{\text{c}}} \approx 10{{\text{{\AA}}}^3}\), close to the expected value of 8.35 Å3. For heterogeneous nucleation under compression, [Fig. E1(a)], the slopes of the curves after correction for the Schmid factor is roug hly \({\Omega _c}/\left( {{n_{\text{c}}}S} \right) \approx 8{{\text{{\AA}}}^3}\), which is similar to the case of homogeneous nucleation. Therefore, our data confirm that the activation volume is proportional to the size of the critical dislocation loop. That \({\Omega _c}/\left( {{n_{\text{c}}}S} \right)\) is somewhat smaller than bAa supports the notion that the Burgers vector of a critical dislocation nucleus is smaller than that of a fully formed dislocation.9,11

The relation between critical dislocation size nc and the activation volume Ω ≡ −(∂Gc/∂σ). (a) Homogeneous nucleation and (b) heterogeneous nucleation. Circles represent the activation volume obtained from the derivative of Gc with respect to σ. Squares represent the activation volume data multiplied by 1/S, where S is the Schmid factor. Dashed lines are linear fits to the data.
Rights and permissions
About this article
Cite this article
Ryu, S., Kang, K. & Cai, W. Predicting the dislocation nucleation rate as a function of temperature and stress. Journal of Materials Research 26, 2335–2354 (2011). https://doi.org/10.1557/jmr.2011.275
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2011.275