

Abstract
Complex nervous systems achieve proper connectivity during development and must maintain these connections throughout life. The processes of axon and synaptic maintenance and axon degeneration after injury are jointly controlled by a number of proteins within neurons, including ubiquitin ligases and mitogen activated protein kinases. However, our understanding of these molecular cascades is incomplete. Here we describe the phenotype resulting from mutation of TMEM184b, a protein identified in a screen for axon degeneration mediators. TMEM184b is highly expressed in the mouse nervous system and is found in recycling endosomes in neuronal cell bodies and axons. Disruption of TMEM184b expression results in prolonged maintenance of peripheral axons following nerve injury, demonstrating a role for TMEM184b in axon degeneration. In contrast to this protective phenotype in axons, uninjured mutant mice have anatomical and functional impairments in the peripheral nervous system. Loss of TMEM184b causes swellings at neuromuscular junctions that become more numerous with age, demonstrating that TMEM184b is critical for the maintenance of synaptic architecture. These swellings contain abnormal multivesicular structures similar to those seen in patients with neurodegenerative disorders. Mutant animals also show abnormal sensory terminal morphology. TMEM184b mutant animals are deficient on the inverted screen test, illustrating a role for TMEM184b in sensory-motor function. Overall, we have identified an important function for TMEM184b in peripheral nerve terminal structure, function, and the axon degeneration pathway.
SIGNIFICANCE STATEMENT Our work has identified both neuroprotective and neurodegenerative roles for a previously undescribed protein, TMEM184b. TMEM184b mutation causes delayed axon degeneration following peripheral nerve injury, indicating that it participates in the degeneration process. Simultaneously, TMEM184b mutation causes progressive structural abnormalities at neuromuscular synapses and swellings within sensory terminals, and animals with this mutation display profound weakness. Thus, TMEM184b is necessary for normal peripheral nerve terminal morphology and maintenance. Loss of TMEM184b results in accumulation of autophagosomal structures in vivo, fitting with emerging studies that have linked autophagy disruption and neurological disease. Our work recognizes TMEM184b as a new player in the maintenance of the nervous system.
Introduction
Axons relay chemical and electrical information between cell bodies and axon terminals (sensory endings and synapses), traversing lengths up to a meter in the human body. The fidelity of this connection is critical for sensory detection, muscle contraction, and neural network function. When an axon is damaged, it initiates a molecular cascade leading to axon degeneration. Axon damage occurs during traumatic or repetitive injury, following chemotherapy treatment with microtubule-disrupting agents (paclitaxel and vincristine), in diabetic neuropathy, and in many inherited neurodegenerative conditions (Neukomm and Freeman, 2014). Thus, understanding the events leading to axon demise is critical for interfering with the progression of these conditions.
Axon degeneration and death of cell bodies are separable processes. For example, axon degeneration and synaptic terminal loss precede cell body loss in Parkinson's disease patients (Burke and O'Malley, 2013) and in mouse models of CNS ischemia (Zhang et al., 2010). Conversely, suppressing cell death does not necessarily result in axon maintenance of diseased neurons (Gould et al., 2006). In recent years, a number of proteins required for axon degeneration across species have been discovered. These include DLK (wallenda in flies; Miller et al., 2009), Phr1 (highwire; Xiong et al., 2012; Babetto et al., 2013), MORN4 (retinophilin; Bhattacharya et al., 2012), mitochondrial permeability transition pore (Barrientos et al., 2011), and SARM1 (ect4; Osterloh et al., 2012; Gerdts et al., 2013). While recent work has delineated relationships among some of these players (Gerdts et al., 2015; Yang et al., 2015), we do not fully understand how this cascade is initiated and controlled, limiting our ability to interfere in situations of pathological axon damage (Gerdts et al., 2016).
In an RNA interference-based screen in Drosophila for genes that participate in axon degeneration following paclitaxel (taxol) intoxication (Bhattacharya et al., 2012), we identified TMEM184b, a uncharacterized 7-pass transmembrane protein. In mice, we find that TMEM184b is highly expressed in the nervous system, localizes to recycling endosomes within neuronal cell bodies and their axons, and is required for axon degeneration following sciatic nerve injury. Despite this protective effect of TMEM184b loss in axons, TMEM184b mutants display progressive structural abnormalities at the neuromuscular junction (NMJ), swellings in intraepidermal sensory fibers, and functional sensory-motor deficiencies. Our data demonstrate a critical role for TMEM184b in regulating both axon degeneration and peripheral nervous system morphology and function.
Materials and Methods
Sequence similarity and hydrophobicity.
Homologous proteins were identified using the Protein Basic Local Alignment Search Tool (National Center for Biotechnology Information). Sequence identity and similarity were calculated using online tools (http://imed.med.ucm.es/Tools/sias.html). Hydrophobicity analysis was completed using the TMpred software provided by ExPASy (http://www.ch.embnet.org/software/TMPRED_form.html). All graphs and statistical analyses were done in Igor Pro (WaveMetrics).
Mutant mice.
Sperm from the Texas A&M Institute for Genomic Medicine allele Tmem184bGt(IST10294F4) were obtained and used to artificially inseminate C57BL/6 female mice. Mouse husbandry and surgeries were performed in accordance with Washington University Institutional Animal Care and Use Committee guidelines. In some instances, multiple tissues were removed from the same animals to minimize the number of animals used (foot pads, skeletal muscles, and brain tissue for quantitative PCR). Both male and female animals were used in all experiments in approximately equal numbers per genotype or comparison group. For all graphs in all figures, ** indicates p < 0.01, and * indicates p < 0.05.
Genotyping.
Genotyping was done using three primers, two flanking the insertion site (left primer TCTGGGCAGAGATCTTGTGA, right primer TTTTGCCAACACTCGTCACT) and a primer in the long terminal repeat region of the gene-trap construct (Mut LTR2, AAATGGCGTTACTTAAGCTAGCTTGC). Wild-type alleles produced a 461 bp product, while mutant alleles produced a 266 bp product.
Xgal staining.
Tissues were dissected from adult heterozygous mice (∼1 year old), briefly fixed for 5–10 minutes in 5% paraformaldehyde, and then treated with Xgal substrate (adult dorsal root ganglia for 40 min; spinal cords for 1.25 h). Samples were then postfixed, immersed in 30% sucrose overnight, and embedded in Optimal Cutting Temperature (OCT) compound. Sections 10 μm thick were cut, allowed to fix for 10 min, and then rinsed and mounted in 70% glycerol. Images were taken on a Leica TCS SPE confocal microscope. Sections were additionally stained with antibodies to Tuj1 (1:500; Biolegend) or ChAT (1:100; Millipore).
Quantitative RT-PCR.
Mice analyzed included four wild types (three males and one female) and three mutants (two males and one female). They were ∼1 year old. Mice were killed with carbon dioxide using a SmartBox. Tissues were quickly removed and homogenized in Trizol (Invitrogen). Total RNA was prepared, DNase-treated to remove genomic contamination, and quantified (NanoDrop spectrophotometer). cDNA was synthesized from 200 ng of total RNA (cDNA Synthesis Kit, Quanta Biosciences), and 4% of this was used as a template for each qPCR (Perfecta SYBR Green Fastmix, ROX, Quanta Biosciences). Reactions were done in triplicate for each sample on an Applied Biosciences 7900HT quantitative PCR machine. Ct values were computed from amplification plots using a threshold in the linear range of amplification. GAPDH was used to normalize samples and compute relative abundance. Primers used were as follows: GAPDH, forward AGAGACGGCCGCATCTTCTTG, reverse GCCGTTGAATTTGCCGTGAG; TMEM184b forward CTCTCCTTCTGGCAAGGCAT, reverse GTCCAGTCTCTTGTCAGCGT; TMEM184a, forward ATGGACTGCTCTGCTGCTAA, reverse CAATGAACAGGAGGCGGATG; TMEM184c, forward CGCGGTTCCGATATGCATTT, reverse TGACACGGGTATGGTTAGCA. All primers were validated on positive control tissue (testes for TMEM184a; brain for TMEM184b and TMEM184c).
Transfection, infection, and immunostaining.
To create TMEM184b-Myc virus, TMEM184b was amplified by PCR using a Myc-containing primer on the 3′ end, cloned into in an FCIV backbone (CMV promoter, Ires-Venus) and packaged using VSV-G and Δ8.9 transfected into HEK293T cells using Fugene (Promega). For sensory neuron expression and colocalization with markers, the Ires-Venus cassette was removed to allow for visualization of proteins using 488 nm excitation.
Primary embryonic dorsal root ganglion neurons were cultured from CD1 embryos at day 13.5 as described previously (Miller et al., 2009) for 7–8 d. Viruses were add on the first day in vitro. On the day of the experiment, neurons were fixed for 20 min in 4% paraformaldehyde/1×PBS, rinsed in PBST, blocked with 5% goat serum, and incubated with primary antibodies overnight. Antibodies used included rabbit anti-Myc (1:100; Sigma-Aldrich C956), mouse anti-Myc (1:5000; clone 9B11, Cell Signaling Technology), mouse anti-EEA1 (1:500; BD Transduction Labs), rabbit anti-Rab11 (1:200; Cell Signaling Technology), mouse anti-GM130 (1:1000; BD Transduction Labs), Ms anti-Tuj1 (see above). Secondary antibodies from Invitrogen (Alexa 488 anti-Ms or anti-Rb) or Jackson Immunoresearch (Cy3 anti-Rb or Cy3 anti-Ms) were used at a dilution of 1:500. Results were confirmed with four independent neuronal preparations. Images were taken on a Leica TCS SPE confocal microscope or a Nikon Eclipse E600 confocal microscope. TMEM184b colocalization with Rab11 was quantified by examining single confocal slices through neuronal soma (10–20 cells per dissection analyzed). In ImageJ, background fluorescence was subtracted from red and green channels, and the Manders' correlation coefficient was calculated using the Coloc2 plugin with the Costes method for thresholding.
For muscle fiber analysis, tibialis anterior muscles (from two females and one male per genotype) were removed and flash frozen, sectioned at 12 μm thickness, and incubated with primary antibodies to LC3 (1:250; Nanotools) and Lamp2 (1:400; Santa Cruz Biotechnology). Images were brightened and contrast was enhanced in the red channel so as to see the borders of myofibers in images.
Sciatic nerve injuries and analysis.
For sciatic nerve studies, mice (∼3 months old) were anesthetized with isoflurane and the right hindleg of each was shaved and cleaned. Skin and muscle dorsal to the sciatic nerve were opened with a small incision. A small chunk (1–2 mm) of sciatic nerve was removed to ensure no regrowth across the injury site. Following surgery, skin was stitched with three stitches, and animals were transferred to recovery cages. Three or 5 d after surgery, animals were killed with carbon dioxide and the distal section of the injured nerve was removed and fixed. For toluidine blue analysis, semithin sections (200 nm thick) were cut and stained with toluidine blue. The uninjured sciatic nerve was used as a control and for normalization purposes.
Quantification of myelinated axon integrity was done by examining four 100× images from nonoverlapping regions of toluidine blue-stained sections of sciatic nerves from each mouse. Analysis was done blinded to genotype. Axons were considered intact if they contained undamaged cytoplasm and also contained clear mitochondria (small pinpoints within cytoplasm). Empty or collapsed axons were not considered intact.
NMJ analysis.
To examine NMJs, a fluorophore labeling all nerves (thy1-YFP line 16; Feng et al., 2000) was bred into control and mutant animals. Extensor hallucis longus (EHL) muscles were removed, fixed, and incubated intact with fluorescently labeled α-bungarotoxin (BTX; Invitrogen) for 30 min. Muscles were then rinsed and mounted for confocal microscopy. Entire muscles were imaged at 10× and a stitched image was created. Using these stitched images, and blinded to genotype, all NMJs were scored for the presence or absence of swellings at nerve terminals (75–125 NMJs scored per muscle per animal). Faint terminals where morphology was not clear were not included in analysis. To analyze apposition, synapses of 6.5-month-old animals were manually identified on a multicolor image, and channels were then separated and thresholded using ImageJ. Green (YFP) and red (BTX) areas were calculated for each synapse, and the red area was divided by the green area to give a fractional apposition. Total acetylcholine receptor expression per synapse was calculated by defining postsynaptic area using thresholding, followed by summation of total pixel area multiplied by average intensity.
Intra-epidermal nerve fiber analysis.
Animals (two females and one male per genotype) were humanely killed, and foot pads of mutant and age-matched wild-type animals were removed, fixed, embedded in OCT freezing media, and sectioned at 30 μm thickness. Sections were stained with rabbit anti-PGP9.5 (1:1000; Millipore) and DAPI to show nuclei (contained within Vectashield mounting media; Vector Laboratories). Quantification of total intraepidermal nerve fibers was done by adapting a method of human skin biopsy analysis (Lauria et al., 2005). Sixty to 90 individual fibers were analyzed per animal. Fibers with swellings measuring >3 fiber diameters were included in swelling counts.
Electron microscopy.
Samples for electron microscopy were fixed in 4% glutaraldehyde and processed according to standard protocols as described previously (Bhattacharya et al., 2012). Imaging was done in the Washington University Molecular Microbiology Electron Microscopy Facility.
Functional and electrophysiological analysis.
Electrophysiology was performed on 6–7-month-old mutant (five males, three females) and wild-type (four males, four females) mice using a Nicolet Viking II electromyography system (Nicolet Biomedical). Throughout nerve conduction studies, mice received 2.5% isoflurane anesthesia. Motor nerve velocity and amplitude of the left sciatic nerve were determined by inserting the recording electrode subcutaneously into the plantar surface of the foot between the first and second digit and the reference electrode 2–3 mm lateral to it. For distal stimulation, the stimulating electrode was inserted above the ankle; for proximal stimulation it was placed at the sciatic notch. The nerve was stimulated with single square-wave pulses of 0.1 ms duration of increasing intensity until supramaximal responses were generated. For sensory nerve recordings, the active recording electrode was inserted 1 cm distal to the proximal tip of the tail and the stimulating electrode 30 mm distally to the active recording electrode. Supramaximal responses were generated using 0.1 ms square-wave pulses and a train of 15 recordings was averaged. Motor and sensory conduction velocities were calculated using the recording program. Slow (2 Hz) and fast (50 Hz) repetitive nerve stimulation was performed by stimulating at the sciatic notch and recording from the gastrocnemius muscle. Each mouse received three trains of 10 stimuli at 2 and 50 Hz with a pause of 2 min between each train of stimuli.
Tests with the inverted screen were performed on 6–7-month-old mice. The time mice held on to an inverted screen was recorded. The experiment was stopped after 120 s or when mice fell off the screen, whatever came first. Each mouse was tested three times and the average time holding on to the screen was calculated.
Results
TMEM184b is a multipass transmembrane protein. Hydrophobicity analysis suggests that this protein contains seven transmembrane domains (Fig. 1A). The amino acid sequence of TMEM184b has been highly conserved from flies to mice (57% identical and 66% similar). To determine where TMEM184b is expressed, we performed quantitative PCR on wild-type mouse tissues. Our results show that TMEM184b is expressed in the nervous system, including both central (spinal cord, brain) and peripheral (dorsal root ganglia) nervous tissues (Fig. 1B). In contrast, TMEM184b is expressed at minimal levels in muscle tissue. This expression profile is consistent with RNA-seq (European Molecular Biology Laboratory–European Bioinformatics Institute Expression Atlas) and in situ hybridization data (Allen Brain Atlas) showing abundant brain, spinal cord, and dorsal root ganglion expression and scarce muscle expression of TMEM184b in mouse. These studies show that TMEM184b is enriched in the nervous system.

TMEM184b is expressed in central and peripheral neurons. A, Hydrophobicity plot of fly, mouse, and human TMEM184b showing seven transmembrane domains. B, Relative expression of TMEM184b mRNA in wild-type tissues by quantitative PCR (n = 3–4 animals per tissue). S.C., Spinal cord; aDRG, adult dorsal root ganglia. Error bars are SEM. C, Quantitative PCR of TMEM184 family members from brain tissue of wild-type and mutant animals. Data are normalized to wild-type TMEM184b expression. n.s., Not significant. Student's t test values for wild-type versus mutant: TMEM184a, p = 0.62; TMEM184b, p = 2.0 × 10−6; TMEM184c, p = 0.26. D, E, Sections of adult dorsal root ganglion (D) and spinal cord (E) from a TMEM184b heterozygous mouse containing one copy of the gene-trapped allele. Xgal stain is shown in blue. In D, an entire ganglion is shown to illustrate the extent of expression within sensory neurons. Red staining is β-III tubulin (tuj1). In E, red staining is ChAT, a marker of acetylcholine-producing motor neurons. Scale bars: D, 100 μm; inset, 20 μm; E, 10 μm.
To undertake a functional analysis of TMEM184b, we obtained mice in which the TMEM184b gene is disrupted by a gene-trap cassette between the first and second exons (Texas A&M Institute for Genomic Medicine allele Tmem184bGt(IST10294F4)). Mice homozygous for the TMEM184b gene-trap allele are homozygous viable and fertile. Using adult brain tissue, we found that in homozygous TMEM184b gene-trap (GT/GT) animals, RNA expression of TMEM184b was severely reduced [5.5 ± 4.5% (SE) relative to wild-type; Fig. 1C, middle bars]. This represents an ∼20-fold reduction in TMEM184b RNA in GT/GT mice. We will subsequently refer to these animals as TMEM184b mutants. No change in the expression of other TMEM184 family members was observed in mutant animals (Fig. 1C), demonstrating that they are not transcriptionally upregulated in response to loss of TMEM184b.
To characterize the expression of TMEM184b in the nervous system, we attempted to make a TMEM184b-specific antibody, but our antibody was not able to recognize endogenous TMEM184b protein (data not shown). Instead, we used the β-galactosidase expression derived from the gene-trap cassette to visualize the cellular expression pattern of TMEM184b. In dorsal root ganglia, TMEM184b is expressed in virtually all sensory neuron cell bodies (Fig. 1D). In the ventral spinal cord, TMEM184b expression is found in motor neurons expressing ChAT (Fig. 1E). In summary, our data demonstrate expression of TMEM184b in both motor and sensory neurons.
To study the intracellular localization of TMEM184b, we generated a Myc-tagged version of the protein and introduced this into neurons with a lentivirus. In primary cultures of dorsal root ganglion neurons, TMEM184b is found in a vesicular pattern. We see extensive colocalization of TMEM184b and Rab11, a marker of recycling endosomes, in the soma (Fig. 2A). TMEM184b protein is trafficked into axons, where it continues to occupy Rab11-positive compartments (Fig. 2B,C). Therefore, TMEM184b is found in recycling endosomes within distal axons, where it could function in axon degeneration. In contrast to the clear colocalization with Rab11, we find very little TMEM184b in early endosomes (labeled by EEA1; Fig. 2D; Mu et al., 1995). TMEM184b is found in areas adjacent to the Golgi apparatus (labeled by GM130; Fig. 2E; Nakamura et al., 1995), but does not colocalize with this compartment. The presence of TMEM184b in recycling endosomes but not in Golgi or early endosomes suggests that its localization is not an artifact of TMEM184b overexpression. Additionally, because recycling endosomes carry cargo between the plasma membrane and internal compartments (van Ijzendoorn, 2006), it suggests that TMEM184b protein is present on the plasma membrane of neurons.

TMEM184b localizes to recycling endosomes within neuronal axons and soma. In all panels, TMEM184b-Myc has been virally expressed and identified with antibody to Myc (red). A, Cell bodies of embryonic dorsal root ganglia neurons expressing TMEM184b-Myc. Box in the upper portion of the merged image shows region from which inset (bottom right corner of each image) is taken. Yellow in the merged image indicates colocalization of Rab11 (green), a marker of recycling endosomes, and TMEM184b-Myc (red); 90 ± 7% (SD) of TMEM184b-Myc signal overlaps with Rab11 signal; 92 ± 5% (SD) of Rab11 signal overlaps with TMEM184b-Myc signal (Manders' correlation coefficient calculation, n = 53). Arrows within insets show clusters of recycling endosomes that contain TMEM184b. B, Axons of embryonic DRG neurons (7 d in vitro) stained for Tuj1 (green) and Myc (red). C, Co-occurrence of TMEM184b (red) and Rab11 (green) within axons. D, E, Double immunostaining of TMEM184b-Myc (red) with (D) early endosomes (EEA1, green) or with (E) Golgi (GM130, green). Little to no overlap is seen for either compartment. Representative images from four independent experiments are shown. Scale bars: A, D, E, 5 μm; C, 10 μm.
To ascertain whether TMEM184b is required for axon degeneration in mammals, we transected the sciatic nerves of mutant and wild-type mice and examined axon integrity using toluidine blue staining of distal nerve cross sections. Transected axons in TMEM184b mutant animals show remarkable preservation 3 d following nerve injury, with ∼6 times the number of intact myelinated axons remaining at this time point when compared with wild-type controls (Fig. 3A–C). This protection is not prolonged, as mutant axons are lost by 5 d postinjury (Fig. 3B). These data show that TMEM184b loss affords a short but significant delay in axon degeneration following a severe nerve injury. Remak bundles containing small-diameter C-fiber nociceptive axons did not appear preserved in TMEM184b mutant animals following nerve injury (Fig. 3D), suggesting that these unmyelinated fibers may be more difficult to protect than myelinated ones. Electron microscopic analysis of mutant and wild-type axons revealed normal morphology of uninjured mutant axons (Fig. 3C,D). Thus, TMEM184b promotes the degeneration of myelinated peripheral axons in vivo.

TMEM184b participates in the axon degeneration cascade. A, Toluidine blue-stained sections through sciatic nerves (SN) of spared and injured (distal) sciatic nerves in wild-type and mutant animals. B, Quantification of the percentage of intact myelinated axons (showing cytoplasm and mitochondria) on the injured versus spared side at 3 and 5 d following sciatic nerve cut. N = 4 animals per genotype and treatment. **p = 0.0004 (two-tailed t test). C, Electron micrographs of representative myelinated axons in the sciatic nerves of wild-type and mutant animals. D, Representative sensory (Remak) axon bundles in each genotype and treatment. Scale bars: A, 20 μm; C, 1 μm; D, 500 nm.
To further characterize the nervous system phenotype of the TMEM184b mutant mouse, we examined NMJs formed at the EHL muscle. We noticed dramatic morphological changes in the NMJs of mutant animals (Fig. 4A–D). In mutant animals, large dystrophic swellings are present in presynaptic terminals. These swellings are also found in synapses onto the tibialis anterior and diaphragm muscles (data not shown), suggesting a widespread problem with NMJ architecture. The frequency of these swellings increases with age in mutant animals (Fig. 4G), a finding suggesting that TMEM184b loss causes progressive neurodegenerative disease in these animals. In areas of presynaptic swelling, postsynaptic receptor expression is virtually absent (Fig. 4C,D). Overall, we find that apposition of presynaptic terminals declines dramatically in TMEM184b mutant animals, with ∼50% of mutant synapses having less than half of their presynaptic area opposed by acetylcholine receptors (Fig. 4H). We did not observe an overall decrease in the amounts of receptor per synapse (mutants had slightly increased expression, 1.6-fold of wild-type, n = 200 synapses; data not shown), arguing that terminal swellings may have asymmetrically expanded the presynaptic side. Thus, TMEM184b is required for proper NMJ morphology, and its disruption results in defects in acetylcholine receptor apposition.

TMEM184b loss causes morphological defects at motor and sensory terminals. A, B, NMJs at the EHL muscle in heterozygous (A) or mutant (B) 8–10-week-old mice. All nerves are green (YFP); acetylcholine receptors in muscle are red (labeled with BTX). Arrows point to synaptic swellings at mutant synapses; yellow asterisk indicates a morphologically normal synapse. C, D, TMEM184b mutant NMJs showing individual nerves (YFP, green) and acetylcholine receptors (BTX, red) in 6.5-month-old mice. Dashed lines in D indicate locations of presynaptic swellings where little receptor apposition is found. E, F, Intraepidermal nerve fibers of wild-type and mutant animals, stained with PGP9.5 (red) and DAPI (blue). Dashed line shows the border between dermis (left) and epidermis (right). Mutants show abnormal swellings within epidermal fibers (arrows). G, Quantification of synaptic swellings in control (filled boxes) and mutant (open boxes) animals over time. p = 2.5 × 10−4 at 18 d; 1.6 × 10−4 at 8–10 weeks; 2.1 × 10−5 at 6.5 months. N = 3–4 animals per genotype (**p < 0.01). H, Quantification of the percentage of presynaptic terminal area opposed by acetylcholine receptors (BTX). Black bars, Wild type; gray bars, mutant. Graph shows the percentage of analyzed synapses (n = 40–60 synapses per mouse, 3 mice per genotype) that have the indicated levels of apposition. P values calculated using one-way ANOVA (p = 0.025, 0.16, and 0.0033 for lowest, middle, and highest levels of apposition, respectively). Scale bars: (in A) A, B, 20 μm; (in C) C, D, 10 μm; (in E) E, F, 20 μm.
Given the high expression of TMEM184b in sensory as well as motor neurons, we tested whether abnormalities occur in sensory terminals of mutant animals. Foot pad intraepidermal nerve fiber morphology was examined by staining for PGP9.5 (for review, see Mellgren et al., 2013). We found dystrophic sensory fibers in the epidermis of TMEM184b mutant animals, a phenotype strikingly similar to that in motor terminals (Fig. 4E,F). Mutant animals had significantly more swollen fibers than wild-type controls [13.2 ± 4.4% (SD) in mutant; 0.8 ± 0.7% (SD) in wild type; Student's t test p = 0.008]. This experiment shows that TMEM184b is required for proper sensory terminal morphology.
When we examined TMEM184b mutant NMJs by electron microscopy, we found examples of swollen presynaptic terminals with membranous aggregates (Fig. 5A–D). In the muscle, we found aggregates of vesicles, some of which are clearly surrounded by a double membrane characteristic of autophagosomes (Fig. 5E,F). Furthermore, mutant muscle shows occasional myofibers with abnormal accumulations of autophagosomes and/or lysosomes in their center (Fig. 5G,H). In summary, the abnormalities observed by both light and electron microscopy in TMEM184b mutant mice suggest impairments in autophagy.
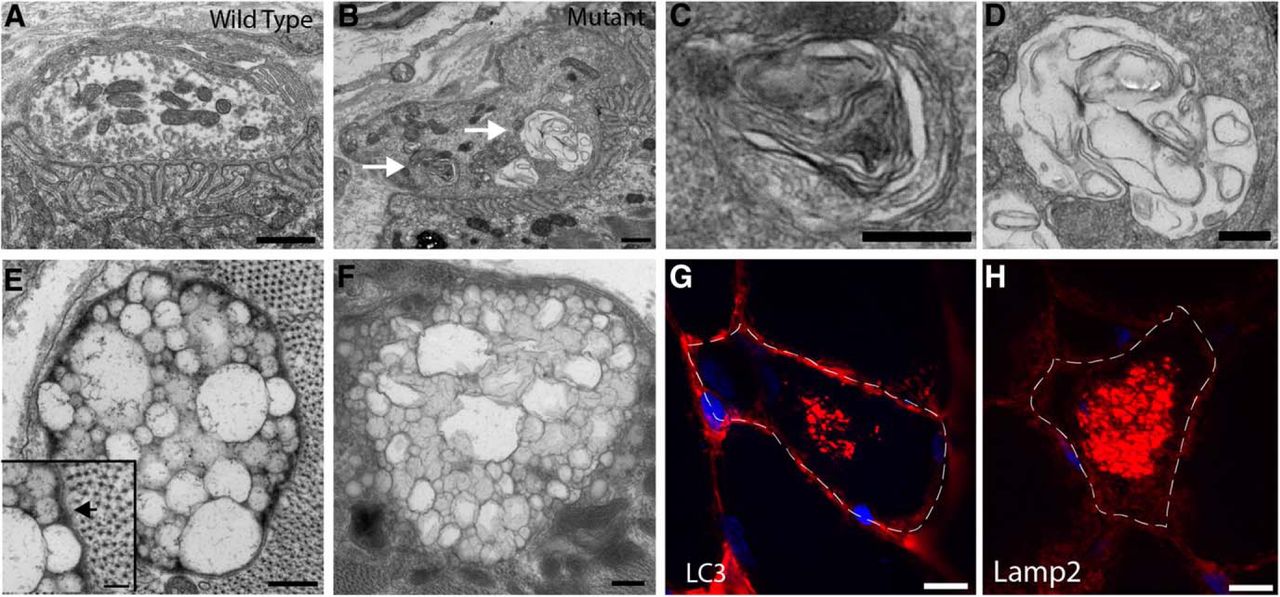
Electron and light microscopy reveal abnormal autophagosomal structures in TMEM184b mutant mice. A–F, Electron micrographs of TMEM184b mutant and control EHL muscles. A, B, NMJs on the EHL in wild-type (A) or mutant (B) animals. Mutant synaptic terminals are abnormally shaped (protrusion in upper right corner) and contain membranous inclusions (arrows). C, D, Enlargements of regions in B (mutant) shown by the white arrows. E, F, Examples of abnormal vesicular structures found near the muscle fiber surface in TMEM184b mutant mice. The inset in E shows a double membrane surrounding this structure (arrow). G, H, Sections of skeletal muscle (tibialis anterior) stained with the autophagosomal marker LC3 (G) or the lysosomal marker Lamp2 (H). In mutant animals, occasional accumulations of LC3 or Lamp2-positive structures are found within muscle fibers. Dotted lines show borders of a single myofiber. Scale bars: A, B, 1 μm; C, D, 500 nm; E, F, 250 nm; inset in E, 100 nm.
To determine whether the structural abnormalities cause deficits in motor unit function in TMEM814b mutant mice, we tested functional and electrophysiological parameters of motor and sensory function. Mutant mice were not able to hold on to an inverted screen, suggesting a deficit in sensory-motor function (Fig. 6A). Action potential amplitudes (both sensory and compound motor) and conduction velocities in both sensory and motor nerves were indistinguishable between mutant and wild-type animals (Fig. 6B–E), indicating that there is no motor or sensory neuropathy. Therefore, while functional deficits exist in TMEM184b mutant animals, they cannot be ascribed to conduction failure in nerves. To ask whether mice had a synaptic transmission defect, we performed repetitive nerve stimulation (2 and 50 Hz) but saw no decrement in the amplitude or area of the compound motor action potential in mutant animals, suggesting the absence of a severe postsynaptic defect at the NMJ (data not shown). However, we cannot rule out a more subtle defect in synaptic transmission. In summary, TMEM184b mutant mice have a sensory-motor functional defect that is consistent with structural abnormalities found at their sensory and motor terminals.

TMEM184 loss causes sensory-motor impairment. Analysis of electrophysiological and functional parameters in TMEM184b mutant mice. A, Mutant mice hold on to an inverted screen for significantly less time than their littermate controls. Two-tailed t test, p = 0.0002. B–E, The compound motor action potential (CMAP) recorded from the plantar muscles of the foot (B), motor nerve conduction velocity of the sciatic nerve (C), mixed orthodromic compound nerve action potential amplitude (D), and conduction velocity of the tail nerve (E) were unchanged between littermate controls and TMEM184b mutants. For B–F, two-tailed t test, p > 0.05. F, The weight of TMEM184b mutant mice is the same as littermate controls. N = 8 mice/genotype/trial.
Discussion
We have described an important role for TMEM184b in the nervous system in both nerve terminal maintenance and responses to nerve injury. This represents the first description of a function for mammalian TMEM184b in vivo.
The mammalian TMEM184 family contains three members, TMEM184a, TMEM184b, and TMEM184c. TMEM184a [also called TMEM34 (human) and lazarus1 (Arabidopsis)] and TMEM184c (also called Sdmg1, or Osta-1 in Caenorhabditis elegans) share a domain structure with TMEM184b that is similar to organic solute transporters. Roles ascribed to TMEM184a and TMEM184c include control of cell migration and tumorigenesis (Akaishi et al., 2007; Fukumoto et al., 2015), cilia architecture (Olivier-Mason et al., 2013), and spermatogenesis (Best and Adams, 2009), although the molecular mechanisms are unknown. Additionally, P-element insertions near Drosophila CG12004 cause defects in innate immunity (Jin et al., 2008) and in synaptic target selection (Kurusu et al., 2008). However, there are no data as to whether these insertions disrupt CG12004 expression or that these phenotypes can be rescued by restoration of CG12004 protein.
We find that TMEM184b localizes to recycling endosomes in mammalian cell bodies and axons (Fig. 2). Furthermore, our light and electron microscopy analyses reveal abnormal accumulations of autophagosomal structures, suggesting possible defects with autophagic clearance of cellular debris (Fig. 5). These two compartments are functionally linked; Rab11-positive recycling endosomes contain the autophagy proteins ULK1 and Atg9, and they contribute membranes to nascent autophagosomes (Longatti et al., 2012). Thus, one possible explanation for the defects seen in TMEM184b mutant mice is a disruption in autophagosomal structure or clearance, leading to the buildup of recycled cargo in nerve terminals and at synapses.
Together, our data implicate TMEM184b in the regulation of autophagy. Disorders involving autophagosomes can cause both nerve and muscle disease. In neurons, autophagy is a constitutive process (Maday et al., 2012). It is thought to function in the degradation of proteins that, if accumulated, cause neurodegenerative diseases like Huntington's disease (Ravikumar et al., 2002; for review, see Wong and Holzbaur, 2015). Correspondingly, defective autophagy is a causative event in some familial forms of amyotrophic lateral sclerosis (ALS; Fecto et al., 2011), and some loci linked to ALS encode proteins regulating autophagosomes (Alegre-Abarrategui et al., 2009; Ju et al., 2009; Hadano et al., 2010).
A number of mouse models of neurodegenerative disease show similar phenotypes to that of TMEM184b. For example, both the light and electron microscopic phenotypes in TMEM184b mutant muscle bear significant similarity to rimmed vacuoles and inclusions caused by dominant mutations in an autophagy regulator, valosin-containing protein (VCP; Ju et al., 2009). VCP mutations can cause not only ALS but also frontotemporal dementia and inclusion body myopathy, illustrating how the dysfunction of protein degradative processes can affect nerves and muscles simultaneously (Weihl et al., 2015). The dystrophic presynaptic swellings we find in TMEM184b mutants have also been reported in AxJ mice, which contain a spontaneous mutation in the deubiquitinating protease Usp14. AxJ mice accumulate proteins in their terminals that were meant for degradation by the proteasome (Chen et al., 2009). Finally, mouse models of infantile neuroaxonal dystrophy caused by mutations in the phospholipase PLA2G6 deletion show ultrastructural terminals similar to those of TMEM184b mutants (Malik et al., 2008; Sumi-Akamaru et al., 2015). Thus, the membranous aggregates and swollen compartments in TMEM184b mutant nerves and muscles could reflect abnormalities in degradation of cellular proteins similar to those known to cause neurodegenerative disease.
We find that mutation of TMEM184b in mice results in severe and progressive changes in synaptic and epidermal nerve terminal architecture, and that TMEM184b loss affects sensory-motor function (Figs. 4⇑–6). It is very likely that the morphological changes we observe contribute to the functional deficits; however, other explanations are possible. Given the widespread expression of TMEM184b in the CNS and in particular the cerebellum (Allen Brain Atlas; M.R.C. Bhattacharya, unpublished observation), we cannot rule out the possibility that other cell types participate in the generation of the behavioral phenotypes described here (due to the ubiquitous nature of the gene-trap allele). Interestingly, abnormalities are seen in skeletal muscle (Fig. 5), even though virtually no mRNA for TMEM184b is expressed in this tissue (Fig. 1), indicating that a noncell-autonomous mechanism of action may be responsible for the muscle phenotypes in this mouse.
Loss of TMEM184b has a protective phenotype within axons following nerve injury in vivo (Fig. 3). It is thus one of a growing list of proteins that play roles in axon degeneration following injury or chemotherapy-induced toxicity, including Sarm1 (Osterloh et al., 2012; Gerdts et al., 2013, 2015), Phr1/highwire (Xiong et al., 2012; Babetto et al., 2013), the dual leucine zipper kinase/c-Jun N-terminal kinase pathway (Miller et al., 2009), the mitochondrial permeability transition pore (Barrientos et al., 2011), and Morn4 (Bhattacharya et al., 2012). Overexpression of NMNAT1 (Mack et al., 2001; Araki et al., 2004) or maintenance of NMNAT2 levels (Gilley and Coleman, 2010) also protects axons after injury. Links between Sarm1, DLK, and nicotinamide adenine dinucleotide levels have recently emerged (Gerdts et al., 2015; Yang et al., 2015), but the complete network of required proteins is unknown.
While seemingly paradoxical, other examples exist of proteins that alter synaptic architecture while also protecting axons from degeneration, including Phr1/highwire, which functions as an E3 ubiquitin ligase (Wan et al., 2000; Xiong et al., 2012; Babetto et al., 2013). One possible explanation for our data is that TMEM184b loss and the resulting disruption of membrane trafficking could directly affect the activity of one or more of these known pathways, leading to a protective phenotype in axons. Another possibility is that disrupting synaptic structure by loss of TMEM184b (or Phr1) could cause a low-level cellular stress, triggering neurons to upregulate compensatory responses that protect them from further insults. The relationship between TMEM184b and Phr1, or other axon degeneration pathway components, requires further study. Interestingly, Phr1 mutant mice show similar axon swellings at NMJs (E. Babetto, personal communication), suggesting commonalities in their roles in neurons.
The molecular function of the TMEM184b protein remains to be determined. Though its membrane topology might suggest that it is a G-protein-coupled receptor (GPCR), sequence comparisons suggest that TMEM184b has a protein structure most similar to bile acid transporters, rather than a similarity to GPCRs. Its localization in recycling endosomes and at the plasma membrane puts it in a position to have signaling roles within neurons, and potentially between neurons and muscle. Further work is necessary to identify the biological activity of TMEM184b.
In summary, we have described the first roles for TMEM184b in the mammalian nervous system in vivo. TMEM184b is critical both for axon degeneration and for sensory and motor terminal structure and function. Given that it is highly expressed in both central and peripheral nervous system tissues, and given the importance of proteostasis in neurodegenerative disease, future work should investigate links between mutation in TMEM184b and the development of neurodegenerative disease in humans.
Footnotes
This work was supported by the Children's Discovery Institute at St. Louis Children's Hospital and Washington University in St. Louis (M.B.), a development grant from the Muscular Dystrophy Association (M.B.), a clinical research training fellowship from the American Brain Foundation (S.G.), and the National Institutes of Health [NS065053 (National Institute of Neurological Disorders and Stroke) and DA020812 (National Institute on Drug Abuse) to A.D., NS091448 (National Institute of Neurological Disorders and Stroke) to S.G.]. We thank members of the DiAntonio and Milbrandt laboratories for helpful discussions throughout the development of this manuscript. Technical assistance was kindly provided by Xiaolu Sun, Sylvia Johnson, and Amy Strickland. Antibodies were kindly provided by Andrew Yoo, Phyllis Hansen, and Chris Weihl. We thank Wandy Beatty for assistance with electron microscopy and Pakou Vang for analysis assistance. We appreciate the expert advice of Robert Schmidt and Erica Holzbaur during the preparation of this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Martha R.C. Bhattacharya, St. Louis College of Pharmacy, 4588 Parkview Place, St. Louis, MO 63130. Martha.bhattacharya{at}stlcop.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}