

Abstract
Traumatic brain injury (TBI) is a major risk factor for the development of multiple neurodegenerative diseases. With respect to the increasing prevalence of TBI, new therapeutic strategies are urgently needed that will prevent secondary damage to primarily unaffected tissue. Consistently, neuroinflammation has been implicated as a key mediator of secondary damage following the initial mechanical insult. Following injury, there is uncertainty regarding the role that accumulating CCR2+ macrophages play in the injury-induced neuroinflammatory sequelae and cognitive dysfunction. Using CX3CR1GFP/+CCR2RFP/+ reporter mice, we show that TBI initiated a temporally restricted accumulation of peripherally derived CCR2+ macrophages, which were concentrated in the hippocampal formation, a region necessary for learning and memory. Multivariate analysis delineated CCR2+ macrophages' neuroinflammatory response while identifying a novel therapeutic treatment window. As a proof of concept, targeting CCR2+ macrophages with CCX872, a novel Phase I CCR2 selective antagonist, significantly reduced TBI-induced inflammatory macrophage accumulation. Concomitantly, there was a significant reduction in multiple proinflammatory and neurotoxic mediators with this treatment paradigm. Importantly, CCR2 antagonism resulted in a sparing of TBI-induced hippocampal-dependent cognitive dysfunction and reduced proinflammatory activation profile 1 month after injury. Thus, therapeutically targeting the CCR2+ subset of monocytes/macrophages may provide a new avenue of clinical intervention following TBI.
Introduction
Traumatic brain injury (TBI) initiates a robust immune response in the CNS involving resident glial cells, microglia, as well as infiltration of blood leukocytes (Woodcock and Morganti-Kossmann, 2013). Principal to this milieu response is the induction of multiple immunomodulatory cytokine and chemokine signaling factors (Ziebell and Morganti-Kossmann, 2010; Giunta et al., 2012). Notably, within hours following the initial trauma in patients surviving TBI, there are increased CSF levels of CCL2, the cognate ligand for CCR2 (Semple et al., 2010). These findings have been recapitulated in studies modeling TBI in rodents (Israelsson et al., 2008; Semple et al., 2010), further suggesting the involvement of CCL2-CCR2 signaling axis in the acute term following TBI. Importantly, activation of this signaling axis has been shown to recruit CCR2-expressing monocytes into the injured tissue where they become activated macrophages, expressing multiple proinflammatory mediators (Serbina and Pamer, 2006; Auffray et al., 2007; Prinz and Priller, 2010). Microglia, the brain's resident tissue macrophage, phenotypically resemble monocyte-derived macrophages in both appearance and functionality following TBI (Raivich et al., 1999; Town et al., 2005; Cao et al., 2012). This biological phenomenon has prevented the accurate delineation of the contribution of infiltrated macrophages in the propagation of TBI-induced inflammatory response.
Recent work has defined two subpopulations of peripheral blood monocytes as “inflammatory” and “circulating/resident” monocytes (Auffray et al., 2009). These two cell types can be accurately distinguished from each other on the basis of their relative cell-surface expression of two chemokine receptors CCR2 and CX3CR1, with CCR2+ monocytes representing the inflammatory subset. Although brain microglia can resemble activated macrophages following TBI, they do not express CCR2 natively (Saederup et al., 2010; Mizutani et al., 2012) or following CNS injury (Schilling et al., 2009). Therefore, the accumulation of CCR2+ cells into the injured brain is considered to be from peripheral blood-borne monocytes. Interestingly, in the brain, CCL2-CCR2 signaling has been shown to be both neuroprotective (El Khoury et al., 2007; Naert and Rivest, 2011) and neurotoxic (Mildner et al., 2009; Saederup et al., 2010; Belarbi et al., 2013) in various animal models of neurodegenerative disease. However, information regarding the accumulation and inflammatory role of CCR2+ peripheral macrophages in the TBI-induced inflammatory sequelae remains ambiguous.
Herein we used the unique CX3CR1GFP/+CCR2RFP/+ reporter mice (Saederup et al., 2010), referred to here as double-heterozygous (Dbl-Het), to define the temporal kinetics of TBI-induced CCR2 macrophage accumulation in the brain. We found that accumulation of CCR2+ macrophages is temporally restricted to a narrow time frame following injury. Moreover, multivariate analysis revealed distinct relationships between macrophage accumulation and inflammatory gene expression, which then identified a novel therapeutic window for targeting CCR2+ macrophage accumulation. Treatment with CCX872, a novel Phase 1 CCR2 antagonist, reduced the accumulation of peripheral macrophages, disrupted the neurotoxic macrophage polarization, and prevented the increased expression of the superoxide-generating enzyme NOX2, which forms ROS, 1 d after injury. Most importantly, this treatment strategy prevented TBI-induced hippocampal-dependent learning and memory deficits measured 28 d after injury. These data suggest that the accumulation of CCR2+ macrophages significantly contributes to TBI-induced cognitive decline and supports the use of early pharmacological intervention to prevent cognitive decline in patients with TBI.
Materials and Methods
Animals.
All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of California (San Francisco). Adult 6- to 7-month-old male and female Dbl-Het and wild-type (WT; C57BL6/J) male mice were used for all experiments. Dbl-Het mice were generated as previously described (Saederup et al., 2010) and genotyped using a commercial service (Transnetyx), whereas WT mice were purchased from The Jackson Laboratory. Mice were group housed in environmentally controlled conditions with reverse light cycle (12:12 h light/dark cycle at 21 ± 1°C) and provided food and water ad libitum.
Surgical procedure.
All animals were randomly assigned and divided as equally possible between sexes (Dbl-Het) to their treatment group. Animals were anesthetized and maintained with 2.5% isoflurane with a nonrebreathing nose cone and passive exhaust system connected to a stereotaxic frame (David Kopf). Once animals were secured with nontraumatic ear bars, eye ointment was applied and their heads were cleared of any hair around the scalp. After betadine application, a midline incision was made through the scalp. TBI was reproduced in the parietal lobe using the controlled cortical impact (CCI) model (Smith et al., 1995). Mice received a craniectomy ∼3.5 mm in diameter using an electric microdrill with the center point determined by a digitally calibrated manipulator arm (Leica) to the coordinates: anteroposterior, −2.0 mm; mediolateral, 2.0 mm, with respect to bregma. Explicit attention was paid to prevent damage to the dura during craniectomy; any animal in which the dura was disrupted, as assessed by excessive bleeding, was omitted from the study and replaced by another littermate. After craniectomy, contusion was achieved using a 3.0 mm convex tip attached to an electromagnetic impactor (Leica) mounted to the digitally calibrated manipulator arm. To impact flush with the natural curvature of the head/tissue, the manipulator arm was rotated 20° on the vertical axis. The parameters for impact were for a contusion depth of 0.95 mm (from dura), velocity was constant at 4.0 m/s, and the impact was sustained for of 300 ms. Importantly, these injury parameters penetrated all layers of the cortex stopping short of disrupting the dorsal hippocampal structure (anteroposterior −2.0 mm, mediolateral 2.0 mm, dorsoventral −0.95 mm). After CCI injury, the scalp was sutured and each animal received 0.5 ml of physiologic saline (i.p.) before being placed in a water-heated incubation chamber (37°C) until they fully recovered as exhibited by resumption of movement and grooming. Sham animals were treated to the above parameters, except that the CCI injury was omitted. Surgical procedures were coordinated in batches such that two time points of mice were scheduled to be killed at the same time, this allowed for each cohort of animals to be killed at approximately the same point during the day over the course of the study. All animals fully recovered from surgical procedures and exhibited normal weight gain for the duration.
Tissue collection.
All mice were lethally overdosed using a mixture of ketamine (150 mg/kg)/xylazine (15 mg/kg). Once the animal was completely anesthetized, the chest cavity was opened and blood was removed using 21.5 Ga needle from the atrial cavity. Blood was subsequently expelled into standard plastic 1.5 ml conical tubes for serum collection. After blood removal, each animal was transcardially perfused with ice-cold Hanks balanced salt solution without calcium and magnesium (Invitrogen). Immediately after perfusion, mice were decapitated and the ipsilateral brain hemisphere was placed into ice-cold RPMI-1640 medium without phenol (RPMI; Invitrogen).
Flow cytometry.
Brain hemispheres in RMPI were used for leukocyte isolation following standard procedures (Cardona et al., 2006). Fc receptor blocking was performed before all staining procedures using an anti-CD16/32 antibody (BD Biosciences PharMingen). The following reagents were used for labeling isolated macrophages: 7AAD (Sigma-Aldrich), CD11b AlexaFluor-700 (BD Biosciences PharMingen), F4/80 APC (Invitrogen), and CD45 FITC (WT only; AbD Serotec). Mandibular blood draws from naive CCR2RFP/RFP and CX3CR1GFP/GFP mice were used as positive controls for RFP and GFP expression, respectively. Additionally, naive WT isolated leukocytes served as negative control for RFP and GFP expression. Spectral compensation was achieved using polystyrene microparticles (BD Biosciences PharMingen) in combination with each of the above listed conjugated antibodies following the manufacturer's suggested protocol. Standard staining procedures were conducted as previously described (Cardona et al., 2006) before analysis on FACSAria III cell sorter (BD Biosciences). All samples were run in duplicate.
Serum isolation.
Atrial collected blood was allowed to clot at room temperature for ∼30 min. Upon formation of a solid clot, serum was isolated by centrifugation at 1500 × g for 10 min at 4°C. The resulting serum supernatant was aspirated and stored at −80°C.
qRT-PCR.
Aliquots of each leukocyte isolation or dissected hippocampi were used for gene expression analyses. Briefly, isolated leukocyte samples were washed with cold Hanks balanced salt solution and pelleted two times before final storage at −80°C. For RNA isolation, samples were thawed on ice before being placed in Qiazol reagent (QIAGEN). Isolated leukocytes were homogenized using repeated trituration with a pipette, whereas hippocampal tissues were homogenized using disposable plastic pestles (USA Scientific), both in 1.5 ml microcentrifuge tubes. RNA was isolated using RNEasy mini-columns (QIAGEN) following the manufacturer's suggested protocol. RNA concentration and quality were measured using a NanoDrop Lite (Thermo Scientific). A total of 300 ng of total RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Amplifications of multiple gene transcripts were performed in duplicate using SYBR Green Master Mix (Applied Biosystems) following the manufacturer's suggested protocol. The relative expression of target genes was determined by the 2−ΔΔCt method and normalized against cyclophilin gene expression using a Statagene Mx3005P Real-Time PCR system. Specifically, the multiple genes were analyzed using the following primer sequences (5′ to 3′ sense/antisense): CD68 (GACCTACATCAGAGCCCG/CGCCATGAATGTCCACTG), CD45 (CTTCAGTGGTCCCATTGTGGTG/TCAGACACCTCTGTCGCCTTAG), MHCII (GCTCTCGGAGACCTATGACG/ACAGGCAAACCTCTGGACAC), MARCO (GCACTGCTGCTGATTCAAGTTC/AGTTGCTCCTGGCTGGTATG), NOS2 (GTTCTCAGCCCAACAATACAAGA/GTGGACGGGTCGATGTCAC), TNFα (TGCCTATGTCTCAGCCTCTTC/GAGGCCATTTGGGAACTTCT), CCL2 (GCTGACCCCAAGAAGGAATG/GTGCTTGAGGTGGTTGTGGA), IL-1β (TGTAATGAAAGACGGCACACC/TCTTCTTTGGGTATTGCTTGG), IL-6 (TACCACTTCACAAGTCGGAGGC/CTGCAAGTGCATCATCGTTGTTC), IFN-γ (TCAAGTGGCATAGATGTGGAAGAA/TGGCTCTGCAGGATTTTCATG), Arg1 (GAACACGGCAGTGGCTTTAAC/TGCTTAGCTCTGTCTGCTTTGC), Ym1 (GGCTACACTGGAGAAAATAGTCCCC/CCAACCCACTCATTACCCTGATAG), FIZZ1 (TCCAGCTAACTATCCCTCCACTGT/GGCCCATCTGTTCATAGTCTTGA), CD206 (CCTCTGGTGAACGGAATGAT/ CTTCCTTTGGTCAGCTTTGG), IL-4 (SA Biosciences; #PPM03013F), IL-13 (SA Biosciences; #PPM03021B), CD36 (GGACATTGAGATTCTTTTCCTCTG/ GCAAAGGCATTGGCTGGAAGAAC), CD163 (GCTAGACGAAGTCATCTGCACTGGG/ TCAGCCTCAGAGACATGAACTCGG), TGFβ (TGATACGCCTGAGTGGCTGTCT/ CACAAGAGCAGTGAGCGCTGAA), IL-10 (GCCAAGCCTTATCGGAAATG/ CACCCAGGGAATTCAAATGC), IL-4Rα (ACCAGATGGAACTGTGGGCTGA/ AGCAGCCATTCGTCGGACACAT), IL-1Ra (TGTGCCTGTCTTGTGCCAAGTC/ GCCTTTCTCAGAGCGGATGAAG), gp91phox (ACTCCTTGGGTCAGCACTGG/ GTTCCTGTCCAGTTGTCTTCG), p22phox (GCTCATCTGTCTGCTGGAGTATC/ CGGACGTAGTAATTCCTGGTGAG), p40phox (CAAAGACCTGCTAGCGCTCATG/ CCACATCCTCATCTGACAGCAG), p47phox (GCTGACTACGAGAAGAGTTCGG/ CCTCGCTTTGTCTTCATCTGGC), p67phox (GCAGAAGAGCAGTTGGCATTGG/ CTGCCTCTCATTTGGACGGAAC), and SOD1 (GGTGAACCAGTTGTGTTGTCAGG/ ATGAGGTCCTGCACTGGTACAG). All primer pairs were independently validated using a standard curve of serially diluted mouse cDNA before use in any endpoint. In each PCR analysis, template and RT controls were included to account for contamination. Gene expression data are represented as the fold change relative to sham (time course experiments) or relative to vehicle-sham values.
Tissue sectioning, immunostaining, and imaging.
All brain tissue used for imaging was sectioned on a Microm cryostat. For fluorescent imaging of endogenous RFP (CCR2) and GFP (CX3CR1), 40 μm free-floating sections were mounted onto Superfrost Plus slides (Fisher) and allowed to dry overnight. Slides were rinsed in buffered saline solution before counterstaining with DAPI (Sigma) followed by coverslipping in Vectashield fluorescent mounting medium (Vector). For imaging of CD45+ cells, standard staining procedures using free-floating sections were conducted as previously described (Morganti et al., 2012) using a CD45 primary antibody (AbD Serotec) and biotin-conjugated secondary antibody (Vector). All imaging was achieved using a Zeiss Imager.Z1 Apotome microscope controlled by ZEN software (Zeiss 2012).
ELISA analysis.
Serum CCL2 concentrations were quantified using standard ELISA technique. Serum samples were diluted 1:2 with the supplied diluent and run in duplicate according to the manufacturer's suggested protocol (Quansys Biosciences). All incubations were performed using a MixMate (Eppendorff) sample vortexer at 700 rpm. Raw intensity values for each ELISA were measured on LiCor near infrared scanner, and sample concentrations were calculated based upon the supplied standard curve using Q-view software (Quansys Biosciences 2013).
CCX872 pharmacokinetic analysis.
CCX872 (Chemocentryx), a small molecule antagonist for the human ortholog of CCR2, was dissolved in a solution of 1% hydroxypropyl methylcellulose (vehicle; HPMC + 0.1% Tween 80) at 20 mg/ml. Approximately 6-month-old WT mice were randomly divided into four groups: vehicle, 1, 12, and 24 h (n = 6/group). Animals received daily subcutaneous injections of CCX872 at 100 mg/kg beginning 2 d before sham surgery (day −2) and continued until the day of surgery (day 0). At the prescribed time following the last injection (1, 12, and 24 h groups), animals were killed by lethal overdose of a ketamine (150 mg/kg)/xylazine (15 mg/kg). Vehicle-injected mice were killed in the same manner 24 h after receiving the final injection. Blood was withdrawn as described above and ejected into lithium heparin plasma collection tubes (BD Biosciences) before being placed on ice. Brain tissues comprising either the ipsilateral hemisphere (n = 3/time point) or hippocampus (n = 3/time point) from each animal were rapidly dissected and snap frozen. Analysis of the ipsilateral hemisphere as well as the ipsilateral hippocampus was conducted to assess the penetrance of CCX872 in light of this study's experimental design examining both cell infiltration (ipsilateral hemisphere) as well as cognitive function (hippocampus) to compare with circulating plasma levels of the compound. Plasma was obtained after spinning the collected blood at 1700 × g for 20 min at 18°C. CCX872 concentrations for tissue and plasma were quantified using high pressure liquid chromatography. Briefly, brain tissues were washed in deionized water, dried, and weighed. After homogenization of the tissue in water (0.1 g tissue/ml) on ice, a 50 μl aliquot of the homogenized sample was extracted with 200 μl 90:10 acetonitrile:MeOH/0.1% formic acid containing an internal standard. A 150 μl aliquot of the supernatant was mixed with an equal volume of 0.1% formic acid in water for LC-MS/MS analysis with an Agilent 1100 high pressure liquid chromatography binary pump coupled with an Applied Biosystems API3000 triple quadruple mass spectrometer. The high pressure liquid chromatography column was Sunfire C18 2.0 × 30 mm 3.5 μm, and the mobile phase was a gradient of 0.1% formic acid in acetonitrile (B) over 0.1% formic acid in water (A) (2%-2%-98%-98%-2%-2% B at 0-0.08-1.58-2.58-2.67-3.50 min). The mass spectrometer was operated in the positive electrospray mode with multiple-reaction monitoring (m/z 495 → 223 for CCX872; m/z 520 → 262 for internal standard). A linear calibration curve was constructed through a quadratic regression with CCX872 standard samples prepared with blank brain extract spiked with CCX872, and the calibration range was 1–5000 ng/ml.
CCX872 administration.
CCX872 was administered for two separate studies examining acute and chronic effects of CCR2 antagonism. For both studies, CCX872 and vehicle were injected subcutaneously as described above. Briefly, the acute studies began administration on day −2 and continued through day 0, whereas chronic studies received injections through day 5. Controls received injection volumes of vehicle analogous to that of treatment animals with respect to the animal's weight.
Behavioral analysis.
At 28 d after surgery, animals were tested for hippocampal-dependent cognitive function using the radial arm water maze (RAWM) as previously described (Alamed et al., 2006). The RAWM was constructed as a modification to the design previously described by Alamed et al. (2006). Specifically, the maze consisted of a circular pool (diameter 118.5 cm) with eight swim lanes (arms) radiating from an open central area (diameter 36.5 cm) with a platform located at the end of one goal arm. The pool's eight arms were formed from sheet metal inserts (41 cm long × 30.5 cm high) each bent at a 45° angle. Procedurally, spatial learning and memory were assessed as previously described (Alamed et al., 2006). Briefly, in this forced-swim behavioral model, animals must locate a hidden platform in one of eight arms using navigational cues set within the testing space. The first day of testing is the acquisition phase wherein the animal is given multiple attempts, through 60 s trials (3 trials per block), to acquire the test's parameters using an alternating scheme of either an above water or hidden escape platform (blocks 1–4) that is located in a semirandomly determined goal arm for each animal. At the conclusion of the first day (block 5), all trials are conducted using the hidden escape platform. The second day of testing is the retention phase wherein the animal must swim to the hidden platform located in the previous day's arm. If, at any point, during either the acquisition or retention phases, the animal enters an incorrect goal arm or remains static for 15 s, an error penalty is tallied for that trial. All trials were completed by either finding the escape platform or by default if the animal had not reached the platform within 60 s. Each trial was recorded using an overhead camera connected to a video tracking and analysis system (Ethovsion XT version 8.5, Noldus Information Technology). All mice were tested until the vehicle sham animals had, on average, reached the criterion of less than one error in a block of three trials. Following the conclusion of the 2 d paradigm, mice were assessed for motor impairments in the pool with the eight arms removed, but this time with an elevated and flagged escape platform (Alamed et al., 2006).
Statistical analyses.
All analyses were performed in Prism version 6.0 (GraphPad) or SPSS version 19 (IBM). Prism was used for cell counts from flow cytometry and gene expression analyses using one-way ANOVA with Dunnett's correction for multiple comparisons (time course experiments) or two-way ANOVA with Tukey's HSD correction for multiple comparisons (CCX872 experiments). SPSS was used for principal component analysis (PCA) using base, regression, advanced models, and missing value add-ons. PCA was performed using eigenvalue decomposition of the cross-correlation matrix of all genes over time. PCs were retained using the eigenvalue >1 and Scree plot criteria. PC scores were calculated using the regression method. PC loadings are represented using arrows where gauge indicates loading magnitude and heat reflects directionality (red represents positive; blue represents inverse). Loading values >|0.3| were retained for PC interpretation. Significant differences were assessed using GLMs. Multivariate ANOVA (MANOVA) was used to assess multivariate changes along PC1–3 axes, followed by post hoc univariate ANOVA and Tukey's post hoc testing. Assessment of learning and memory on the RAWM was assessed using three-way mixed repeated-measures ANOVA. Significance for all measures was assessed at p < 0.05.
Results
TBI initiates a temporally restricted accumulation of CCR2+ macrophages
Recent work has delineated two subsets of monocytes based upon their relative surface expression of the CCR2 chemokine receptor, allowing for the distinction between resident versus peripheral CNS macrophages (Auffray et al., 2009; Prinz and Priller, 2010; Saederup et al., 2010). To determine when CCR2+ macrophages accumulate in the ipsilateral brain parenchyma following TBI, we used flow cytometry to examine cell populations in Dbl-Het reporter mice over acute (3–6 h), subacute (12 h-2 d), and chronic (7–28 d) time points (Fig. 1a). Interestingly, the accumulation of activated peripheral macrophages, characterized by the following markers, CD11b+F4/80hiCCR2+CX3CR1−, into the ipsilateral hemisphere, was restricted to a distinct temporal window beginning at 12 h and peaking at 24 h following injury (Fig. 1b). At 24 h following injury, the accumulation of CCR2+ macrophages dissipated. However, a subset of the CD11b+F4/80hiCCR2+ macrophages subsequently differentiated into CD11b+F4/80hiCCR2+CX3CR1+ resident-like macrophages. The increase in the number of differentiated CCR2+ resident-like macrophages was elevated by 12 h and peaked by 48 h but persisted at significant levels through 7 d following injury (Fig. 1c). TBI induced a dramatic increase in the number of CD11b+F4/80hiCCR2−CX3CR1+ macrophages at all time points, with a clear peak 12 h following injury (Fig. 1d). This finding was similar to recent observations in a skull compression model of brain injury (Roth et al., 2014). Similar to the temporal pattern of accumulation of CCR2+ macrophages, there was an increase in the serum levels of CCL2, with a peak 12–24 h following injury (Fig. 1e); these data are corroborated by similar findings observed in humans following TBI (Semple et al., 2010). At the approximate site of the CCI impact, the accumulation of CCR2+ and CCR2+CX3CR1+ cells was concentrated within hilar and CA3/4 regions of the dorsal hippocampus, and noticeably not as numerous around the cavitation (Fig. 2a). Moreover, there were numerous CCR2+ cells lining the third ventricle as well as within the choroid plexus, which has been previously reported to occur following CNS injury (Shechter et al., 2013). Visually, 1 d following injury, CCR2+CX3CR1− and CCR2+CX3CR1+ cells had a similar histologic appearance, which was distinct compared with CCR2−CX3CR1+ cells (Fig. 2b–iii).

Peripheral CCR2+ macrophage accumulation in the brain parenchyma is temporally restricted following TBI. Flow cytometric data were obtained from CX3CR1+/GFPCCR2+/RFP adult mice ∼6 months of age at the time of injury. a, Representative pseudocolored scatterplots (n = 5 mice per time point, except 48 h, which had n = 4) of ipsilateral CD11b+F4/80hi macrophages over a time course covering acute through chronic time points. CD11b+F4/80hi macrophages were delineated by the expression of CCR2 (RFP+) along the x-axis and CX3CR1 (GFP+) along the y-axis, with the upper-right quadrant representing CX3CR1+CCR2+ double-positive macrophages. b, Accumulation of CD11b+F4/80hiCX3CR1−CCR2+ macrophages was confined to 12–24 h following injury before returning to sham levels by 2 d after injury. c, Increased numbers of differentiated CD11b+F4/80hiCX3CR1+CCR2+ macrophages began to accumulate at 12 h following injury and persisted at significant levels through 7 d before returning to sham levels. d, Accumulation of CD11b+F4/80hiCX3CR1+CCR2− macrophages began at 3 h before peaking at 12 h following injury, however, an elevated trend that persisted through 28 d. e, Serum levels of CCL2 (n = 4/group) increased acutely and peaked by 12–24 h after injury, although there was an elevated trend compared with sham for the remaining time points following injury. Data were analyzed using one-way ANOVA with Dunnett's correction for multiple comparisons wherein the means for each time point were compared with mean of the sham group. Data are mean ± SEM. Dumbell-style bars represent the same level of significance among the those data points relative to sham. *p < 0.05. **p < 0.01. ***p < 0.001.

Controlled cortical impact model of TBI induces the accumulation of CCR2+ cells in the hippocampus. a, One day following controlled cortical impact, CCR2+ macrophages (black) are distributed throughout the dorsal hippocampus but primarily within the CA3/4 and hilar subregions of the dentate gyrus. However, concentrations of CCR2+ macrophages can be seen in the third ventricle yet more diffuse in number around the cavitation (Cav.). b–d, Inset magnification (black box) of dentate/CA3 macrophages in their respective pseudocolor images and merged with DAPI nuclei. b, Yellow ovals highlight CX3CR1−CCR2+ cells. c, Magenta arrowheads indicate CX3CR1+CCR2− cells. d, White arrowhead indicates CX3CR1+CCR2+ cells. i–iii, Single-cell magnification with DAPI. i, CX3CR1−CCR2+ cell. ii, CX3CR1+CCR2− cell. iii, CX3CR1+CCR2+ cell. Representative image is a stitched mosaic of multiple individual high-magnification images using mCherry (RFP), FITC (GFP), and ultraviolet (DAPI) epifluorescent filter sets. Pseudocolored image was converted to black and white.
Macrophage polarization spans the M1-M2 continuum following TBI
Following injury, the polarization of tissue macrophages into M1 or M2 subtypes is a heterogeneous response dependent upon various stimuli present in their microenvironment (Gordon, 2003; Mantovani et al., 2004; Mosser and Edwards, 2008; Lawrence and Natoli, 2011). We examined mRNA expression for multiple genes associated with macrophage polarization from the ipsilateral purified leukocytes obtained at several time points after injury. Gene expression analysis was conducted to quantify classically activated or proinflammatory M1, alternatively activated M2a, and acquired deactivation M2c mediators. In general, TBI induced a broad response, spanning the M1-M2 continuum across all time points, wherein TNFα, CCL2, IL-1β, MARCO, and Arg1, Ym1 showed the highest expression for proinflammatory/M1 and M2a associated responses, respectively (Fig. 3a). Moreover, we observed a delayed, but significant, increase in gene expression occurring at later time points (Fig. 3b) for the cell surface antigens CD68, CD45, and MHC II, which is similar to findings in other neurodegenerative diseases (Prinz et al., 2011) as well as postmortem tissue samples from long-term survivors of TBI (Ramlackhansingh et al., 2011; Johnson et al., 2013). Additionally, significant induction of the M2a-associated cytokines IL-4 and IL-13 was only found in the late subacute phase following injury (Fig. 3a,b). Restoration of CNS homeostasis following injury may require shifting of macrophages from a proinflammatory state to one supporting repair through expression of genes that are associated with an anti-inflammatory phenotype (Colton, 2009). Compared with the M1 and M2a response, gene expression of markers associated with M2c “acquired deactivation” polarization state varied depending upon the time point examined after injury (Fig. 3b). Specifically, our data show that IL-1Ra is markedly increased in the acute through subacute time points, whereas IL-4Rα, IL-10, TGF-β, and CD36 were significantly increased in the subacute time span (Fig. 3a,b).

Isolated leukocytes from the ipsilateral hemisphere (n = 4 or 5 per time point) exhibit a broad-spectrum of M1–M2 polarization mRNA gene expression following TBI. a, Log2-converted fold change from sham of M1 (magenta), M2a (turquoise), and M2c (orange) mRNA gene expression. Heat map visualization reveals that the soluble proinflammatory ligands of TNFα, CCL2, and IL-1β along with the cellular marker MARCO represented the greatest induction following TBI relative to all other M1 measures. Similarly, we found the greatest induction of M2a mRNA in Ym1 and Arg1 relative to all other M2a measures and IL-1Ra for M2c. b, Average of Log2-converted values for each fold change with their respective significance relative to sham indicated. Bold type indicates statistically significant fold change values. Data were analyzed using one-way ANOVA with Dunnett's correction for multiple comparisons wherein the means for each time point were compared with mean of the sham group. Data are mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
Multivariate analysis of inflammatory signaling modules
Given the multidimensional response of macrophage accumulation and polarization over time after injury, we examined the relationship of these variables using multivariate PC analysis (Ferguson et al., 2013). This analysis yielded four orthogonal PCs, which accounted for 83% of the total variance in inflammatory signaling (Fig. 4). PC1, reflecting both CX3CR1+ and CCR2+ macrophages and their associated genes (Fig. 4a), was characterized by an immediate induction during the acute phase and persisted through the subacute time points (Fig. 4b). The PC1 module represented the systems-biological inflammatory response reflecting induction of all polarization subtypes: M1, M2a, and M2c. PC1 peaked by 3 h after injury resolving to low-grade sustained activation through 28 d after injury. The PC2 module was not significantly correlated with changes in the accumulation of macrophages (Fig. 4c); however, the genes associated within this module were predominantly M2a and M2c mediators. Temporally, the PC2 module was significantly induced in the late subacute phase 2 d after injury (Fig. 4d). The PC3 module associated with increased numbers of each of the three macrophage subsets: CX3CR1+, CCR2+, and CCR2+CX3CR1+, with accumulation of CCR2+ macrophages as the greatest contributors (Fig. 4e). PC3 gene expression displayed a biphasic response over time, repressed in the acute phase, and induced during the early subacute phase (Fig. 4f). Similar to what we observed in the PC1 module, PC3 expressed both M1 and M2 mediators, with concomitant expression of both Arg1 and NOS2 predominating. Last, PC4-reflected CCR2+CX3CR1+-differentiated macrophages and their associated genes (Fig. 4g) followed a distinctive temporal pattern characterized by early repression during the acute phases and significant upregulation during the early chronic phase following injury (Fig. 4h).

Inflammation patterns in the brain after TBI revealed by PCA. PCA uncovered four orthogonal PC groups that together accounted for 82.7% of the total variance. Data are an analysis of both cell infiltration data (black boxes) obtained from flow cytometry in conjunction with gene expression. a, PC1 (28.8% variance) reflects increased accumulation of both CX3CR1+ and CCR2+ macrophages and their associated genes in the brain following injury. As both macrophage subsets increased in number, a corresponding increase occurred in gene expression for IL-1β, IL-1Ra, TNFα, IL-4Rα, and MARCO, with YM1, Arg1, and CD36 to a lesser degree. b, PC1 peaked dramatically early after injury, gradually waning over time (F = 130.67, p < 0.00001). c, PC2 (19.5% variance) was not linked to changes in the numbers of any accumulating macrophage subsets; however, there the PC2 group did predominantly cluster with M2a and M2c genes. Interestingly, within this cluster, IFNγ was the most strongly correlated, followed by IL-13, IL-10, IL-4, and FIZZ1, with TGFβ and IL-4Rα to lesser degrees. d, PC2 displayed a mild biphasic response, such that there was a minor repression followed by a delayed induction peaking at 2 d following injury (F = 3.909, p = 0.002). e, PC3 (15.1% variance) reflects increased accumulation of all three macrophage subsets; CCR2+, CX3CR1+, and CCR2+CX3CR1+, with CCR2+ macrophages representing the greatest association, and their gene expression spanned the M1–M2 continuum, with NOS2 and Arg1 predominantly associated. f, Temporally, PC3 exhibited a significant biphasic response, such that in the acute phases it was repressed followed by a significant induction in the subacute time frame before waning at later phases (F = 30.230, p < 0.00001). g, PC4 represents increased accumulation of CCR2+CX3CR1+ differentiated macrophages and their respective genes. As CCR2+CX3CR1+ differentiated macrophages increased in number, there was a parallel increase in gene expression of both M2a and M2c, primarily with CD206, CD163, TGFβ, and CD36. h, PC4 had a delayed response increasing by 2 d and peaking on 7 d following injury before returning to basal levels (F = 10.691, p < 0.00001). Arrows indicate PC loading magnitude (equivalent to Pearson correlations between individual variables and PCs). Arrow gauge indicates loading magnitude. Heat reflects loading directionality: red represents positive; blue represents inverse.
Cumulatively, these data represent a sequential activation of the inflammatory response to brain injury such that temporally arranged, inflammatory response peaks in the order of PC1, PC3, PC2, to PC4 orthogonal modules. Importantly, the inflammatory response we observed as a function of time and macrophage accumulation was heterogeneic in nature such that there was not a clear delineation of an exclusive M1, M2a, or M2c response. These analyses are consistent with previous reports of heterogeneity in CNS macrophage populations identified in the injured brain (Wang et al., 2013) and spinal cord (Shechter et al., 2013).
Administration of CCX872 reduces peripheral macrophage accumulation following TBI and alters inflammatory gene expression
After receiving a total of 3 injections of CCX872, one injection per day (Fig. 5a), pharmacokinetic profiling of CCX872 revealed that circulating levels of the compound remained significantly higher in the plasma compared with brain tissue at all time points examined (Fig. 5b). Noticeably, plasma concentrations of CCX872 were relatively flat over the course of 24 h. Although there was some variability in the tissue concentrations of CCX872 between dissected hemibrain and hippocampi, these values were dramatically lower compared with plasma concentrations at their respective time points. We next examined the use of CCX872 to prevent the accumulation of peripheral macrophages following TBI. We hypothesized that the accumulation of CCR2+ macrophages augmented the TBI-induced proinflammatory response due to their correlation within the PC1 and PC3 orthogonal spaces. To test this hypothesis, we administered CCX872 and examined the response 24 h after injury in WT mice (Fig. 5a), reflecting the time point of highest activation within the PC3 group (Fig. 4e). Importantly, WT mice were used in lieu of the Dbl-Het genotype to ensure complete gene expression of CCR2. Previous work has defined CCR2-expressing monocytes/macrophages with a CD45hi phenotype in WT mice (Auffray et al., 2009; Prinz and Priller, 2010; Saederup et al., 2010). Using this treatment paradigm, serial administration of CCX872 significantly decreased the accumulation of CD11b+F4/80hiCD45hi macrophages by 50% (Fig. 5c) compared with vehicle-treated TBI animals. Notably, there was a visible decrease in the number of CD45+ cells in the hilar region of the hippocampus due to CCX872 treatment compared with vehicle (Fig. 5d). Gene expression analyses using the same targets as the time course study above (Fig. 3a,b) showed that CCR2 antagonism significantly decreased multiple genes associated with both M1 and M2 polarization phenotypes. Specifically, there were marked reductions in gene expression for macrophage markers of CD68 and CD45, whereas concomitant reductions were found in CCL2, IL-6, and IL-1β (Fig. 6a). Interestingly, we observed no change in response in both NOS2 and TNFα at this time point (Fig. 6a). These data suggest that CCR2+ cells are not the principle constituents in these response mechanisms following TBI. Corroborating these findings in part, a recent report has shown that CX3CR1+ macrophages are the principle mediators of NOS2 following CNS injury (Donnelly et al., 2011). Internally, these findings are in agreement with our flow cytometry data showing the increased expression of TNFα before significant accumulation of CCR2+ macrophages. We also observed an impact on both M2a and M2c gene expression due to CCR2 antagonism. CCX872 treatment significantly reduced the expression of both Arg1 and Fizz1 (Fig. 6b) as well as TGF-β and IL-10 (Fig. 6c). Moreover, when we analyzed all of these effects simultaneously, multivariate PCA revealed that CCR2 antagonism resulted in a significant blunting of the inflammatory response compared with vehicle-treated animals along the PC1, PC2, and PC3 orthogonals (Fig. 6d).

CCR2 antagonism ameliorates TBI-induced peripheral macrophage response 24 h after injury. a, CCX872 and vehicle subcutaneous injection schedule. Each animal received a single injection of CCX872 or vehicle once per day. CCX872-treated mice received three total injections at 100 mg/kg volume by weight; vehicle-treated mice received injection volumes analogous to that of CCX872 animals with respect to their weight. b, Pharmacokinetic profile of CCX872 following three subcutaneous injections shows that CCX872 concentration remains significantly higher in the plasma (n = 6/time point) at all time points relative to both the ipsilateral hemibrain (n = 3/time point) and hippocampus (n = 3/time point). c, CCX872 significantly decreased the accumulation of CD11b+F4/80hiCD45hi macrophages 24 h following injury. This resulted in an ∼50% decrease in the average number of ipsilateral F4/80hiCD45hi macrophages compared with vehicle-treated TBI mice. d, Representative images of CD45+ cells (dark gray) within the hilar subregions of the dentate gyrus 1 d after injury for both vehicle- and CCX872-treated animals. Mirroring flow cytometric data, CCX872 treatment visibly decreased the number of CD45+ cells in the hilar region of the hippocampus compared with vehicle-treated TBI animals. Flow cytometric data were analyzed using two-way ANOVA with Tukey's HSD correction for multiple comparisons. Data are mean ± SEM. # #p < 0.01, pairwise comparison for vehicle-TBI versus CCX872-TBI. Hemi, Ipsilateral hemisphere; HPC, ipsilateral hippocampus; V, vehicle; 1, 1 h; 12, 12 h; 24, 24 h; C, CCX872.

Acute treatment with CCX872 reduced neuroinflammatory response following TBI. Data were obtained from leukocytes (n = 6/group) isolated from the ipsilateral hemisphere. a, CCX872 treatment ameliorated TBI-induced proinflammatory/M1 response 24 h after injury. Specifically, CCX872 significantly reduced CD68, CD45, CCL2, IL-1β, and IL-6 gene expression compared with vehicle-TBI animals. b, CCX872 treatment also reduced the expression of the M2a mediators Arg1 and FIZZ1. c, Further, this treatment paradigm also decreased TBI-induced response in two anti-inflammatory M2c cytokines (TGFβ and IL-10) at this time point. d, Effect of CCR2 antagonism on multidimensional inflammation within the PC1–3 space. MANOVA revealed a significant cumulative multivariate effect on PC1–3 (Wilks λ = 0.087, F = 6.57, p < 0.001). Post hoc univariate ANOVAs revealed a significant effect on PC1 (F = 19.0, p < 0.001), PC2 (F = 4.74, p < 0.05), and PC3 (F = 3.92, p < 0.05). The effect on PC4 did not reach significance (p > 0.05). Gene expression for all groups is relative to vehicle-sham values. Data were analyzed using two-way ANOVA with Tukey's HSD correction for multiple comparisons. Data are mean ± SEM. #p < 0.05,(pairwise comparison for vehicle-TBI versus CCX872-TBI. ##p < 0.01, pairwise comparison for vehicle-TBI versus CCX872-TBI. ###p < 0.001, pairwise comparison for vehicle-TBI versus CCX872-TBI. Black bars represent sham. Red bars represent TBI. V, Vehicle; C, CCX872.
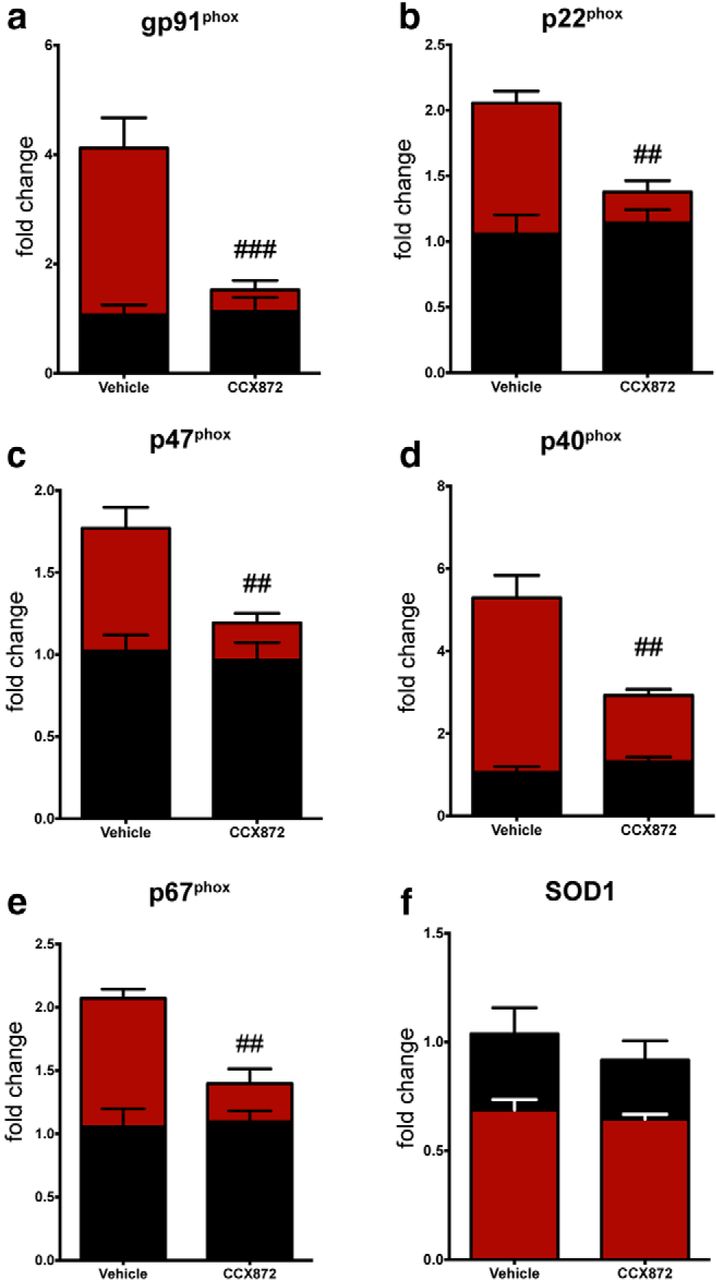
TBI-induced NADPH oxidase overexpression is blocked by CCR2 antagonism
Regulation of the intermediates associated with ROS generation is vital to neuronal health. ROS have been implicated in the propagation of neuronal dysfunction in multiple neurodegenerative diseases (Wu et al., 2003; Boillée and Cleveland, 2008; Hernandes and Britto, 2012). Leukocyte production of ROS is primarily associated with the formation of the NADPH oxidase (NOX2), which is comprised of multiple subunits (Block et al., 2007). In addition to ROS production, NOX2 activation has been shown to maintain redox proinflammatory signaling cascades within microglia/macrophages (Block et al., 2007) as well as regulate macrophage polarization mechanisms (Choi et al., 2012). Importantly, recent data indicate that inhibition of NOX2 decreased neuronal damage and improved functional recovery following TBI (Zhang et al., 2012; Loane et al., 2013). Given that TBI induced a spectrum of proinflammatory/neurotoxic mediators following injury in the acute to subacute time frames and that this response was blunted with administration of CCX872, we next tested its effects on NOX2 expression. Using this treatment paradigm, CCX872 significantly reduced the induction of multiple NOX2 subunits following TBI compared with vehicle-treated animals. Specifically, gene expression analysis revealed that CCR2 antagonism reduced the expression of gp91phox, p22phox, p47phox, p40phox, and p67phox; however, there was no treatment effect on the TBI-induced downregulation of SOD1 (Fig. 7a–f, respectively). Acutely, our data suggest that the bulk of NOX2 activation following TBI is mediated through the influx of CCR2+ macrophages, as treatment with CCX872 significantly blunted this response. Importantly, our findings are corroborated by data showing that CCR2−/− mice are have reduced ROS production (Seki et al., 2009).

CCR2 antagonism mitigates TBI-induced upregulation of the NOX2 subunits. All data were obtained from leukocytes (n = 6/group) isolated from the ipsilateral hemisphere. a–e, mRNA gene expression for the multiple subunits associated with NOX2 activation, which results in increased ROS production, were downregulated in CCX872-treated TBI animals compared with vehicle-treated TBI. f, However, CCX872 treatment was unable to rescue TBI-induced downregulation of SOD1. Data were analyzed using two-way ANOVA with Tukey's HSD correction for multiple comparisons. Data are mean ± SEM. ##p < 0.01, pairwise comparison for vehicle-TBI versus CCX872-TBI. ###p < 0.001, pairwise comparison for vehicle-TBI versus CCX872-TBI. Black bars represent sham. Red bars represent TBI.
CCR2 antagonism prevents TBI-induced cognitive dysfunction
Based upon the marked reduction of accumulated macrophages, the reduced proinflammatory response, and the reduced expression of the NOX2 complex, we extended our hypothesis to examine whether CCR2 antagonism would reduce TBI-induced cognitive dysfunction. Again using WT mice, we delivered CCX872 or vehicle for 5 d following surgery (Fig. 8a). Hippocampal-dependent cognitive function was assessed 28 d following surgery using the 2 d RAWM (Fig. 8b), which tests spatial learning and memory (Alamed et al., 2006). TBI induced a significant increase in the average number of errors during the acquisition phase of RAWM for vehicle-treated TBI animals, but not in CCX872-treated mice (Fig. 8c). Notably, during the retention phase of the RAWM test, we observed a significant decrease in the average number of errors for CCX872-treated TBI animals compared with vehicle (Fig. 8d). Next, we quantified the gene expression of markers associated with macrophage polarization response from the isolated ipsilateral hippocampus. Similar to the response we observed at 24 h following injury (Fig. 6), CCX872 treatment significantly blunted the chronic elevation of multiple proinflammatory markers. Specifically, we observed markedly decreased expression of CD68, CD45, TNFα, CCL2, and IL-1β (Fig. 8e,f). Conversely, there was an inverse M2 response such that Arg1 and FIZZ1 expression was significantly increased, whereas YM1 and IL-10 had visible trends for increased expression (Fig. 8g,h).

CCR2 antagonism abrogates TBI-induced hippocampal-dependent cognitive dysfunction 28 d following injury. a, Experimental timeline. b, Over the course of the 2 d paradigm, all four groups were able to learn the RAWM paradigm as exhibited by a progressive decrease in the average number of errors in relation to time spent in the maze. c, TBI induced a significant increase in the average number of errors during the first day (experimental day 28) of RAWM in the vehicle-treated group compared with sham. TBI did not increase the average errors for CCX872-treated mice compared with sham CCX872 animals; however, this trend was not significantly different from vehicle-treated TBI mice. d, By day 2 (experimental day 29), vehicle-treated TBI animals had persistently increased errors compared with their respective sham controls. Conversely, CCX872-treated mice exhibited no difference in the average errors compared with their respective sham operated controls but did have significantly fewer errors compared with vehicle-treated TBI mice. e–h, CCR2 antagonism abrogates TBI-induced chronic activation of proinflammatory response (e, f) while promoting increased expression of M2a mediators (g). Specifically, CD68, CD45, TNFα, CCL2, and IL-1β were all significantly downregulated in CCX872-treated TBI mice compared with vehicle-treated TBI animals, whereas CCX872 treatment induced the upregulation of several M2-associated markers at this chronic time point following injury. Notably, FIZZ1 and Arg1 were significantly increased, whereas positive trends were observed for IL-10 and Ym1, which was almost significant. All gene expression data were obtained from isolated hippocampi of WT mice following completion of RAWM. Gene expression values are relative to vehicle-sham. Data are mean ± SEM. **p < 0.01 and ***p < 0.001 for pairwise comparison between vehicle-Sham and vehicle-TBI. #p < 0.05, pairwise comparison for vehicle-TBI versus CCX872-TBI. ##p < 0.01, pairwise comparison for vehicle-TBI versus CCX872-TBI. Black bars represent sham. Red bars represent TBI. V, Vehicle; C, CCX872.
Discussion
Following neurotrauma, the innate immune response is set into rapid action such that microglia accumulate at the site of injury (Davalos et al., 2005) followed by a sequential recruitment of systemic immune cells (Morganti-Kossmann et al., 2007; Kigerl et al., 2009; Donnelly et al., 2011). However, the ability to discriminate the function of recruited CCR2+ peripheral macrophages to the injured CNS remains poorly understood as resident microglia acquire a phenotype akin to peripheral macrophages following TBI (Raivich et al., 1999; Cao et al., 2012). Therefore, a better understanding of the function of recruited macrophage response to the injured brain is required to develop targeted strategies to prevent TBI-associated secondary injury. To investigate this problem, we purposefully used the unique CX3CR1GFP/+CCR2RFP/+ reporter mice (Saederup et al., 2010) to accurately delineate the contribution of CCR2+ macrophages in the TBI neuroinflammatory response. By using these mice, we have avoided the confounding effects of developing bone marrow chimeras and radiation exposure to the CNS, which have been shown to increase infiltration of peripheral macrophages into the CNS (Mildner et al., 2009; Morganti et al., 2014). Our study demonstrates that TBI induces a robust response involving the recruitment and accumulation of peripheral CCR2+ macrophages into the injured ipsilateral parenchyma at a discrete time point. With regard to the dorsal hippocampus, a brain region involved in learning and memory, the accumulation of CCR2+ cells localized within the CA3/4 and hilar regions of the dentate gyrus. Concomitant to the cellular macrophage accumulation, we show that the polarization response of these cells was heterogeneic and overlapping, such that proinflammatory responses were occurring simultaneous to expression of anti-inflammatory and tissue repair mediators. Although the inflammatory response was heterogeneic, multivariate analysis revealed distinct correlations among macrophage cell accumulation in the ipsilateral hemisphere, inflammatory gene expression, and time after injury. Importantly, our findings show that targeting the subset of CCR2+ monocytes/macrophages with CCX872, a Phase I CCR2 antagonist, significantly reduced the accumulation of peripheral macrophages following injury while also reducing their inflammatory and neurotoxic profile in the acute term. Cumulatively, administration of CCX872 for a short duration was able to prevent TBI-induced cognitive dysfunction in the long-term.
Using a comprehensive time course following TBI, we demonstrated that the TBI-induced accumulation of CCR2+ macrophages was delayed to a temporally restricted window following injury. These data are in agreement with the onset of CD45hi macrophage accumulation shown to occur by 1 d after CCI injury (Jin et al., 2012; Hsieh et al., 2013); however, our data indicate that this accumulation is discreet and disappears compared with a previous report of protracted accumulation (Jin et al., 2012). Discrepancies between these findings may be due to the differentiation of CCR2+ macrophages in favor of CX3CR1 expression, which we showed persisted for several days following injury. Previous work has shown that macrophage expression of CCR2, upon engraftment in the brain parenchyma, is downregulated in favor of CX3CR1 (Saederup et al., 2010). In this context, CCR2-expressing cells, regardless of their acquired CX3CR1 expression, significantly accumulated between 12 and 24 h and persisted through 7 d after injury. Parallel to the accumulation of peripheral CCR2 macrophages, our data show a similar expression peak of serum CCL2, which remained chronically elevated following injury. CCL2 is the strongest chemoattractant for inflammatory monocytes (Takahashi et al., 2009). Notably, CCL2 has been repeatedly shown to be rapidly elevated following various brain injury models (Glabinski et al., 1996; Rancan et al., 2001); and more importantly, CCL2 is significantly elevated in the CSF of human survivors from TBI (Semple et al., 2010). Combined, our findings in tandem with previous reports implicate circulating levels of CCL2 as a clinical biomarker for the infiltration of CCR2+ cells to injured brain parenchyma following TBI.
In the current study, our data indicate a sequential induction of inflammatory mediators spanning the M1-M2 continuum. To this end, we show that there was not a defined time after injury that could be labeled exclusively within the linear constraints of being M1, M2a, or M2c. Comparatively, FACS sorting of accumulated peripheral macrophages from the injured spinal cord also demonstrated similar heterogeneity and inflammatory overlap among gene expression responses (Shechter et al., 2013). However, we did observe a sequential inflammatory profile wherein M1 preceded M2a, which preceded M2c. These findings are similar to wound-healing responses in tissue outside the CNS, such that proinflammatory and proteolytic macrophages are the first to accumulate, followed by macrophages that promote wound-healing and anti-inflammatory characteristics (Deonarine et al., 2007). Although there was a resolution of the substantial induction of the majority of proinflammatory mediators, several persisted in the chronic phases of injury, akin to similar data published in spinal cord injury (Kigerl et al., 2009). The inability of the CNS' innate response to injury to return to basal levels is a hallmark of brain injury as chronic activation of these systems is present for years after the initial insult (Ramlackhansingh et al., 2011; Johnson et al., 2013). Our current data would suggest that a contingent of CX3CR1+ microglia/macrophages remains activated during these chronic time points and is responsible for the persistent M1 phenotype. However, an important limitation present in this study is the inability to distinguish resident CX3CR1+ macrophages from those that originated peripherally regardless of native antigenic expression of CX3CR1+ or CCR2+. Given that peripheral Ly6CloCX3CR1+ monocyte-derived macrophages are antigenically and phenotypically similar to CNS resident microglia/macrophages, it is difficult to delineate resident versus peripheral function of these two populations of cells in this model. Moreover, our data would suggest that a certain number of CCR2+ macrophages differentiated after engraftment into the injured parenchyma to more closely resemble CX3CR1+ resident microglia/macrophages as has previously been shown (Saederup et al., 2010). Accumulation of Ly6CloCX3CR1+ monocytes into the CNS may be disease-model specific as some models (Huang et al., 2006) are noticeably absent of this phenomenon. However, elegant work in a rodent spinal cord injury model has shown that Ly6CloCX3CR1+ monocytes invade and persist at protracted time points in the injured spinal cord where they are responsible for propagating neuronal damage at chronic time points (Donnelly et al., 2011).
Blocking CCR2+ macrophage accumulation in neurodegenerative disease remains controversial as both sparing (Mildner et al., 2009; Semple et al., 2010) and exacerbating (Gliem et al., 2012; Shechter et al., 2013) effects can be found. These divergent findings portend the diverse role of these systemic cells in various animal models recapitulating neurological disease. In the current study, we show that CCR2+ macrophages are associated with both neurotoxic and tissue repair gene expression profiles depending upon the time they are examined after insult. However, in our model, the overarching effect from the accumulation of these cells into the brain after TBI is that of a proinflammatory and neurotoxic mediator. These findings are analogous to a recent report that found similar neurotoxic properties in CCR2+ infiltrating macrophages in a mouse model of stroke (Hammond et al., 2014). Importantly, although some T-cell populations (Mack et al., 2001) as well as endothelial cells (Dzenko et al., 2001) have been shown to express CCR2 to varying degrees, recent work using bone marrow chimeras has convincingly shown the neurotoxic actions of CCR2-expressing cells are predominantly associated with the infiltration of CCR2+ monocytes into the injured CNS (Hammond et al., 2014). The neurotoxic response associated with CCR2+ macrophages in our study is further substantiated by our data from the administration of the CCR2 antagonist CCX872. Acutely, administration of CCX872 blunted the ingress of inflammatory macrophages, which was paralleled by reduced inflammatory and neurotoxic response compared with vehicle-treated TBI animals. Similarly, preclinical administration of CCX872 has been shown to reduce inflammatory macrophage accumulation in obesity-associated steatohepatitis (Parker et al., 2014). Although statistically insignificant in our model, there were visible trends in gene expression changes due to CCX872 treatment alone. In both the acute and chronic studies, there were visible trends for CCX872 treatment alone to induce the expression of polarization markers associated within the M1/M2 polarization spectra. Given that CCX872 showed poor penetrance in the uninjured brain compared with circulating levels, these gene expression changes may suggest that circulating CCR2+ monocytes may have intrinsic signaling capabilities at the level of the CNS endothelium/microvasculature, even during nonpathological conditions. As such, these findings warrant further examination into the homeostatic roles of circulating CCR2+ monocytes on CNS function.
Clinically, TBI is one of the most powerful environmental risk factors for the development of Alzheimer's disease and dementia (Fleminger et al., 2003; Sivanandam and Thakur, 2012), which may be linked to chronic activation of the CNS' innate response (Ramlackhansingh et al., 2011). Unfortunately, pharmacological interventions using broad-spectrum approaches to inhibit innate immune function after TBI are largely inconclusive both experimentally (Bye et al., 2007; Ng et al., 2012) and clinically (Beauchamp et al., 2008). Previous studies have shown that chronic neuroinflammation disrupts the macromolecular synthesis implicated in synaptic plasticity and memory (Rosi et al., 2005) and alters information processing (Rosi et al., 2009), which is linked with hippocampal-dependent cognitive function (Belarbi et al., 2012). Given the link between homeostatic neuronal dysfunction and inflammation, we sought to determine whether pharmacologically blocking CCR2+ cell accumulation in the brain could preserve hippocampal cognitive function disrupted by TBI. Herein, we provide novel evidence that targeting CCR2+ monocytes mitigates TBI-induced hippocampal-dependent cognitive dysfunction as animals that received CCX872 in conjunction with TBI performed significantly better in the RAWM compared with TBI animals given vehicle. Importantly, the sparing of cognitive deficits following TBI in this model is presumably due to the reduced neuroinflammatory and neurotoxic profiles afforded by limiting the injury-induced migration of CCR2+ macrophages following injury, which preferentially accumulate in the hippocampus.
In conclusion, our results demonstrate that CCR2+ macrophages contribute to the long-term cognitive decline associated with a controlled cortical impact model of TBI. These findings are consistent with previous reports, which show that mobilization, and accumulation of CCR2+ macrophages in mice, promotes degenerative neuropathology (King et al., 2009; Mildner et al., 2009). Together, our proof-of-concept model demonstrates the viability of selectively targeting CCR2+ circulating monocytes to prevent TBI-induced neuroinflammatory-mediated sequelae.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke and National Institute on Aging of the National Institutes of Health Awards F32NS090805 to J.M.M., R21NS087458 to S.R., R21AG042016 to S.R., and R01CA133216 to S.R., and Alzheimer's Association IIRG-11-202064 to S.R. CCX872 was a kind gift from Chemocentryx. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Alzheimer's Association.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Dr. Susanna Rosi or Dr. Josh M. Morganti, Brain and Spinal Injury Center, University of California San Francisco, 1001 Potrero Avenue, Bldg 1, Room 101, San Francisco, CA 94110. rosis{at}ptrehab.ucsf.edu. or josh.morganti{at}ucsf.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}