

Abstract
Approaches to stimulate remyelination may lead to recovery from demyelinating injuries and protect axons. One such strategy is the activation of immune cells with clinically used medications, since a properly directed inflammatory response can have healing properties through mechanisms such as the provision of growth factors and the removal of cellular debris. We previously reported that the antifungal medication amphotericin B is an activator of circulating monocytes, and their tissue-infiltrated counterparts and macrophages, and of microglia within the CNS. Here, we describe that amphotericin B activates these cells through engaging MyD88/TRIF signaling. When mice were subjected to lysolecithin-induced demyelination of the spinal cord, systemic injections of nontoxic doses of amphotericin B and another activator, macrophage colony-stimulating factor (MCSF), further elevated the representation of microglia/macrophages at the site of injury. Treatment with amphotericin B, particularly in combination with MCSF, increased the number of oligodendrocyte precursor cells and promoted remyelination within lesions; these pro-regenerative effects were mitigated in mice treated with clodronate liposomes to reduce circulating monocytes and tissue-infiltrated macrophages. Our results have identified candidates among currently used medications as potential therapies for the repair of myelin.
Introduction
Loss of myelin, demyelination, occurs after a variety of insults to the CNS. These injuries include acute conditions such as transverse myelitis, traumatic spinal cord injury, and acute disseminated encephalomyelitis, as well as chronic conditions such as multiple sclerosis (MS) and neuromyelitis optica. In many cases, oligodendrocyte precursor cells (OPCs) are recruited into demyelinated lesions where they differentiate into oligodendrocytes that reform myelin around denuded axons, a repair process known as remyelination. Recent literature shows that the magnitude of remyelination can be extensive in neurological conditions such as MS (Patrikios et al., 2006; Goldschmidt et al., 2009), although the degree is incomplete in the majority of human subjects.
Endogenous mechanisms for remyelination thus exist in demyelinating disorders but are insufficient to reverse the demyelination completely (Franklin and ffrench-Constant, 2008; Kotter et al., 2011; Zhang et al., 2011). It would therefore be beneficial to identify medications that promote the intrinsic capacity for remyelination in the CNS, particularly by using those that are already in use in humans for other indications.
In recent years, immune cells, particularly monocytes and tissue-infiltrated macrophages, along with CNS intrinsic microglia have been found to be important for remyelination (Franklin and Kotter, 2008; Miron et al., 2013; Rawji and Yong, 2013). Specifically, the inhibition of the recruitment of monocytes and macrophages in animal models of demyelination impairs phagocytosis of myelin debris and remyelination (Kotter et al., 2001, 2005; Kondo et al., 2011; Ruckh et al., 2012). Moreover, remyelination is reduced in mice deficient for cytokines produced prominently by monocytes, macrophages, and microglia, as follows: TNF-α (Arnett et al., 2001) and IL-1β (Mason et al., 2001). When acute inflammation is promoted locally in mice by the application of the toll-like receptor (TLR)-2 and TLR-4 ligands zymosan and lipopolysaccharide (LPS), respectively (Glezer et al., 2006; Setzu et al., 2006; Schonberg et al., 2007), remyelination is enhanced. Although these yeast (zymosan) or bacterial (LPS) ligands of TLRs are potent activators of monocytes, macrophages, and microglia, they are unsafe for use in humans. It is therefore important to examine approved medications that can safely activate these cells.
In the current study, we tested the hypothesis that stimulating monocytes, macrophages, and microglia with compounds approved for human use would promote remyelination after a demyelinating event. In previous studies, we identified the antifungal medication amphotericin B (AmpB), from a 1040 medication screen as an activator of these cells in culture (Samanani et al., 2013; Sarkar et al., 2014). Here, we describe the novel findings that AmpB, particularly when administered together with another stimulator, macrophage colony-stimulating factor (MCSF), enhances the representation and activity of microglia and macrophages (henceforth also referred to as microglia/macrophages as they are not easily distinguishable from one another within lesions) in the lysolecithin model of demyelination and promotes remyelination. These results emphasize the important role of immune cells for remyelination, and they identify the judicious use of medications to promote the activity of monocytes, macrophages, and microglia for remyelination after an episode of demyelination.
Materials and Methods
Cultures.
For mouse microglia, brains from neonatal C57BL/6 pups (Charles River) were minced and vortexed vigorously for 1 min to kill neurons. Suspensions were sieved through 80- and 11-μm-pore size meshes. Cells were seeded at a density of one pup per coated T-75 flask in medium containing 10% serum. When cells were confluent, the flasks were shaken at 250 rpm for 20 min to detach the loosely adherent microglia. The purity of microglia was in excess of 95%, as determined through ionized calcium-binding adaptor molecule 1 (Iba1) immunohistochemistry (data not shown).
Mouse bone marrow-derived macrophages (BMDMs) were isolated from adult 8- to 10-week-old C57BL/6 mice, as described previously (Goncalves DaSilva and Yong, 2008), with minor modifications. In brief, the tibia and femur bones were removed and cleaned, and marrow was flushed from the bones using complete bone marrow growth medium (DMEM, 10% fetal bovine serum, 2% penicillin/streptomycin, and 10% L929 conditioned medium, which is a source of mouse MCSF). Cells were spun at 1100 rpm for 10 min, resuspended in fresh growth medium, counted, and then plated at a density of 1.2 million cells/ml in a 10 cm culture dish. Cells were incubated at 37°C at 10% CO2 for 5 d before replacing half the medium with fresh growth medium. On the seventh day, the growth medium was changed completely. On the 10th day, cells were harvested by trypsinization and plated at a density of 1.5 million/well in a six-well culture plate in the above medium but without L929-containing MCSF. After 72 h of incubation at 37°C in 10% CO2, the medium was completely replaced by fresh medium and test treatments. The following media were evaluated: AmpB (10 μm), MCSF (40 ng/ml), the combination of AmpB and MCSF, and LPS (100 ng/ml). For AmpB, we used the commercial preparation fungizone (Life Technologies/Invitrogen), which is AmpB solubilized into a colloidal dispersion with sodium deoxycholate and buffered with sodium phosphates; this mixture is similar to that used to medicate patients with severe fungal infections. Thus, the vehicle used in this study was phosphate-buffered sodium deoxycholate.
BMDMs were also derived from mice deficient for toll-like receptor (TLR) signaling. The TLR2-null mice were from Jackson Laboratory, while the MyD88 and TRIF double-null mice to ablate signaling from all TLRs were from Dr. Paul Kubes (University of Calgary, Calgary, Alberta, Canada). The TLR2-null mice lack TLR2 signaling in all cells, and the MyD88/TRIF double-null mice lack TLR signaling universally; our focus in this study is on BMDMs from these animals.
Quantitative real-time PCR.
BMDM cells were washed with ice-cold PBS, and 1 ml of TRIzol reagent (Life Technologies/Invitrogen) was then added. After 5 min of incubation at room temperature, RNA was extracted as per manufacturer instructions. RNA was then treated with DNase (Promega) and reverse transcribed using Superscript II reverse transcriptase (Life Technologies/Invitrogen). The resulting cDNA was used as a template for the Bio-Rad iCycler MyiQ detection system and 2× SYBR green mastermix (Qiagen). Every primer that was used was purchased from Qiagen, and details can be found on their homepage. The expression of gene transcripts was normalized against at least two housekeeping genes (i.e., GAPDH and β-actin). Relative expression levels for genes of interest were determined using the formula 2−ΔCT, where ΔCT = CT (gene of interest) − CT (housekeeping gene).
Lysolecithin demyelination.
A local ethics committee approved all animal procedures in accordance with the guidelines of the Canadian Council of Animal Care. C57BL/6 female mice between 8 and 10 weeks of age were anesthetized with a mixture of ketamine (200 mg/kg) and xylazine (10 mg/kg). After exposure of the spinal cord, a 32 ga needle was overlaid on the dorsal column between the T3 and T4 vertebrae. Unlike our previous work (Larsen et al., 2003; Gregg et al., 2007), where the needle penetrated the dorsal column, we discovered that consistent and profound demyelination could be elicited by having the needle tip press lightly onto the surface of the dorsal column without penetration, and then depositing 1.5 μl volume of 1% d-lysophosphatidylcholine (lysolecithin; Sigma) solution over 2 min. This method was used for all mice, and they were killed at 3, 5, 7, 14, and 28 d after lysolecithin application. These time points were chosen based on our previous studies that 3–7 d represents the demyelinating phase and 14–28 d represent the remyelination phase (Larsen et al., 2003; Gregg et al., 2007). Sham mice were subjected to the same insult except that PBS was deposited onto the spinal cord in place of lysolecithin. Uninjured mice were included as naive controls.
Treatment of mice.
MCSF or PBS control was injected intraperitoneally 1 and 2 d before the induction of demyelination with lysolecithin. The dose of MCSF was 40 μg/kg following a regimen that we have reported previously (Boissonneault et al., 2009). For AmpB, animals were treated daily with intraperitoneally injections on the day of surgery (day 0) until day 7 with 1 mg/kg dose; this dose was chosen after preliminary experiments that this was a nontoxic amount compared with higher doses (10 or 15 mg/kg; data not shown). In total, there were four treatment groups for the lysolecithin-demyelinated mice: deoxycholate vehicle, MCSF alone, AmpB alone, or the combination of MCSF and AmpB (AmpB/MCSF).
Tissue harvest and analyses.
After a lethal overdose of ketamine/xylazine, mice were perfused with ice-cold 4% paraformaldehyde (PFA) in PBS through the left ventricle of the heart. Spinal cord sections were dissected and post-fixed overnight in 4% PFA and cryoprotected in a 30% sucrose solution for at least 24 h. A 3 mm (1 mm caudal and 2 mm rostral of the lesion epicenter) spinal cord block was isolated, embedded in an optimum cutting temperature compound, flash frozen in a dry ice/isopentane bath, and stored at −80°C. The entire 3 mm of spinal cord tissue was cut in 10 μm coronal sections into 10 consecutive series, where each series comprised of silanized microscope slides that accommodate sections 100 μm sequentially apart from each other. These sections were stored in the presence of silica gel at −20°C, and subsequently were used for histological and immunofluorescence analyses.
To assess the integrity of myelin for demyelination and remyelination, one series of coronal spinal cord sections was stained for myelin with the Eriochrome Cyanine protocol, as previously described (Skihar et al., 2009). To evaluate the extent of demyelination in Eriochrome Cyanine-stained sections, we used the Image-Pro Plus Program (Media Cybernetics) to measure the area within each dorsal column that did not contain blue staining, and expressed that as a ratio of the area of that dorsal column. We averaged this across a mean of 30 sections, each spaced 100 μm apart, per mouse.
For immunofluorescence staining, sections were blocked for 30 min with skimmed milk, and incubated for 1 h at room temperature or at 4°C overnight with primary antibodies diluted in skimmed milk, followed by 1 h at room temperature with secondary antibodies. Sections were mounted in Mowiol (Calbiochem) and stored at 4°C in the dark. The following primary antibodies were used: anti-Iba1 (1:500, microglia/macrophages, Wako Chemicals), anti-platelet-derived growth factor α receptor (PDGFRα; 1:50; for oligodendrocyte precursor cells; a gift from Dr. Bill Stallcup, Sanford-Burnham Medical Research Institute, La Jolla, CA), anti-adenomatus polyposis coli [APC (CC1); 1:50; for oligodendrocytes, Calbiochem], and anti-myelin basic protein (MBP; 1:500; Dako). Alexa Fluor 488 (1:750; Invitrogen) or Alexa Fluor 546 (1:750; Invitrogen) secondary antibodies were used to visualize the primary antibodies. To quantitate Iba1-stained reactivity in tissue section, digital images of the Iba1-stained sections were analyzed using the morphological image analysis functions in Matlab version 7.7 (MathWorks).
For in situ hybridization, coronal sections were subjected to dual labeling of immunohistochemistry for microglia/macrophages (CD68 staining) and localization of macrophage scavenger receptor type 1 (MSR-1) mRNA using 35S-labeled cRNA probes. Riboprobe synthesis and preparation and in situ hybridization were performed according to a protocol described previously (Laflamme et al., 2001).
Finally, for quantitative real-time PCR (qRT-PCR), fresh tissue was collected and an excised 3 mm coronal block (1 mm caudal and 2 mm rostral of the lesion epicenter) was homogenized in TRIzol using, successively, 20 and 25 ga needles. RNA samples were reverse transcribed and subjected to qRT-PCR, as described above for BMDMs.
Measurement of AmpB levels in serum and tissue.
AmpB was assayed using HPLC tandem mass spectrometry (LC/MS/MS). Serum and tissue extraction of amphotericin B were performed as described previously with some modifications (Casaccia et al., 1991; Lee et al., 2001). Briefly, 5 μl of internal standard (2 μg/ml; natamycin, Sigma-Aldrich) was added to 50 μl of serum, and protein was then precipitated using 800 μl of methanol and 200 μl of 0.2% acetic acid. The mixture was kept in a freezer overnight. Denatured proteins were discarded after centrifugation, and 5 μl of supernatant was injected into the LC/MS/MS.
Tissue extraction was performed as follows: 5 μl of internal standard (natamycin 500 ng/ml) and 200 μl of lysis buffer (40 mm Tris HCl, 40 mm sodium acetate) were added to 50 mg of homogenized brain or spinal cord. The mixture was sonicated on ice three times for 5 s, each time using a sonic dismemberator (Model 100, Fisher Scientific). The sample was then diluted with 800 μl of dilution solvent (methanol/0.2% acetic acid 4:1, v/v) and kept in a freezer overnight. The precipitate was discarded after centrifugation, and 25 μl of supernatant was injected into the LC/MS/MS.
The LC/MS/MS analysis was performed with an Agilent Technologies 1100 HPLC autosampler (Agilent Technologies) coupled to an API 4000 triple-quadrupole LC/MS/MS system equipped with a Turbo-Ionspray Interface (AB Sciex). Amphotericin B was separated using a 2.1 × 50 mm, 3 μm, Eclipse plus C18 column (Agilent Technologies). The mobile phase composed of 0.2% acetic acid and acetonitrile was delivered at a flow rate of 0.3 ml/min. A binary gradient started with 85% acetic acid and 15% acetonitrile, which was then increased to 70% acetonitrile at 2 min, subsequently to 80% acetonitrile at 5 min, and ending at 90% acetonitrile at 6 min. The column was equilibrated with 15% acetonitrile for 10 min before the next sample injection. The mass spectrometer was operated in positive, multiple reaction-monitoring mode with a source temperature of 450°C, curtain gas of 15, collision gas of 4, gas1 30, gas2 of 30, and ion spray voltage of 5000 V. Quantitation of amphotericin B was achieved using the 924.6/906.6 transition with the 924.6/743.6 transition as confirmation ion. Transitions monitored for the internal standard (natamycin) were 666.4/503.3 and 666.4/485.2.
White blood cell counts and flow cytometry.
Complete blood counts were performed as previously described (Stirling and Yong, 2008). Briefly, at 7 d after injections of either 1 mg/kg AmpB or vehicle, animals (n = 5 per group) were anesthetized with a lethal dose of ketamine/xylazine, and 0.5 ml of whole blood was collected by cardiac puncture into BD Microtainer tubes with EDTA (BD Biosciences). The samples were immediately analyzed using a COULTER Gen-S Hematology Analyzer (Beckman Coulter). Since monocyte numbers tended to be low in blood, we also resolved monocyte numbers using flow cytometry of Ficoll-centrifuged blood samples. Monocytes were identified using CD11b and CD45 markers after Ficoll centrifugation of blood, so that neutrophils are absent, or with CD11b and Ly6C post-Ficoll.
We also determined the density of microglia/macrophages in the spinal cord using flow cytometry. A 3 mm coronal block (1 mm caudal and 2 mm rostral of the lesion epicenter) was mechanically dissociated, Percoll centrifuged, and then subjected to CD45 and CD11b staining and analyses as previously described (Goncalves DaSilva and Yong, 2009). Neutrophils are not present within the spinal cord at days 5–7 post-lysolecithin (unpublished observations). Because of the very localized nature of the lesion, it was necessary to pool samples from five animals to obtain a single flow analysis result.
Toluidine blue-stained semithin sections and g-ratio analyses.
For semithin cross sections, mice injected with lysolecithin or PBS were perfused with 10 ml of ice-cold PBS followed by 10 ml of 2.5% glutaraldehyde in HMSS buffer (137 mm NaCl, 5.4 mm KCl, 5.5 mm glucose, 0.04 mm phenol red, and 100 mm HEPES, pH 7.4). Spinal cord blocks were dehydrated in a graded series of alcohol and embedded in Epon Resin (Electron Microscopy Sciences). Semithin sections (1 μm) were cut and stained with 0.5% toluidine blue for g-ratio analysis.
The g-ratio is defined as the ratio between the diameter of the axon and the outer diameter of the myelin sheath plus axon. Analysis was conducted across a 1 mm2 area containing and surrounding the demyelinated lesion. In brief, an algorithm was written in Matlab that methodically selects each thresholded axon within the 1 mm2 area. The diameter of the axon was then determined automatically in Matlab using the equation D (diameter) = 2 × square root of (A (area)/π). The algorithm in Matlab also measured each myelin sheath thickness; we only accepted the g-ratio of axons where a myelin sheath was evident, as it was often not possible to accurately identify axons that were without sheaths (thus, a completely demyelinated axon was not included in our analyses). At least 100 axons were evaluated per animal by a blinded observer, and the mean g-ratio for each animal was then obtained. For electron microscopy (EM), 70 nm sections were cut on an ultramicrotome and contrasted with lead citrate. Specimens were examined with an Hitachi 7650 transmission electron microscope.
Clodronate liposome preparations and injection.
Multilamellar liposomes containing clodronate were prepared as described previously (Van Rooijen and Sanders, 1994). For depletion of circulating monocytes, mice were injected intravenously with 0.2 ml of 0.5 mg of dichloromethylene diphosphonate (clodronate)-containing liposomes or with 0.2 ml of the empty (containing only PBS) liposomes at days 1, 4, and 7 post-demyelination.
Statistical analysis.
GraphPad Instat version 3.1 software was used for the statistical analyses. A two-tailed unpaired Student's t test was used where two experimental groups with parametric scores were compared. In the case of multiple experimental groups, a one-way ANOVA with either Bonferroni or Tukey–Kramer post hoc tests were used.
Results
Activation of macrophages and microglia by AmpB
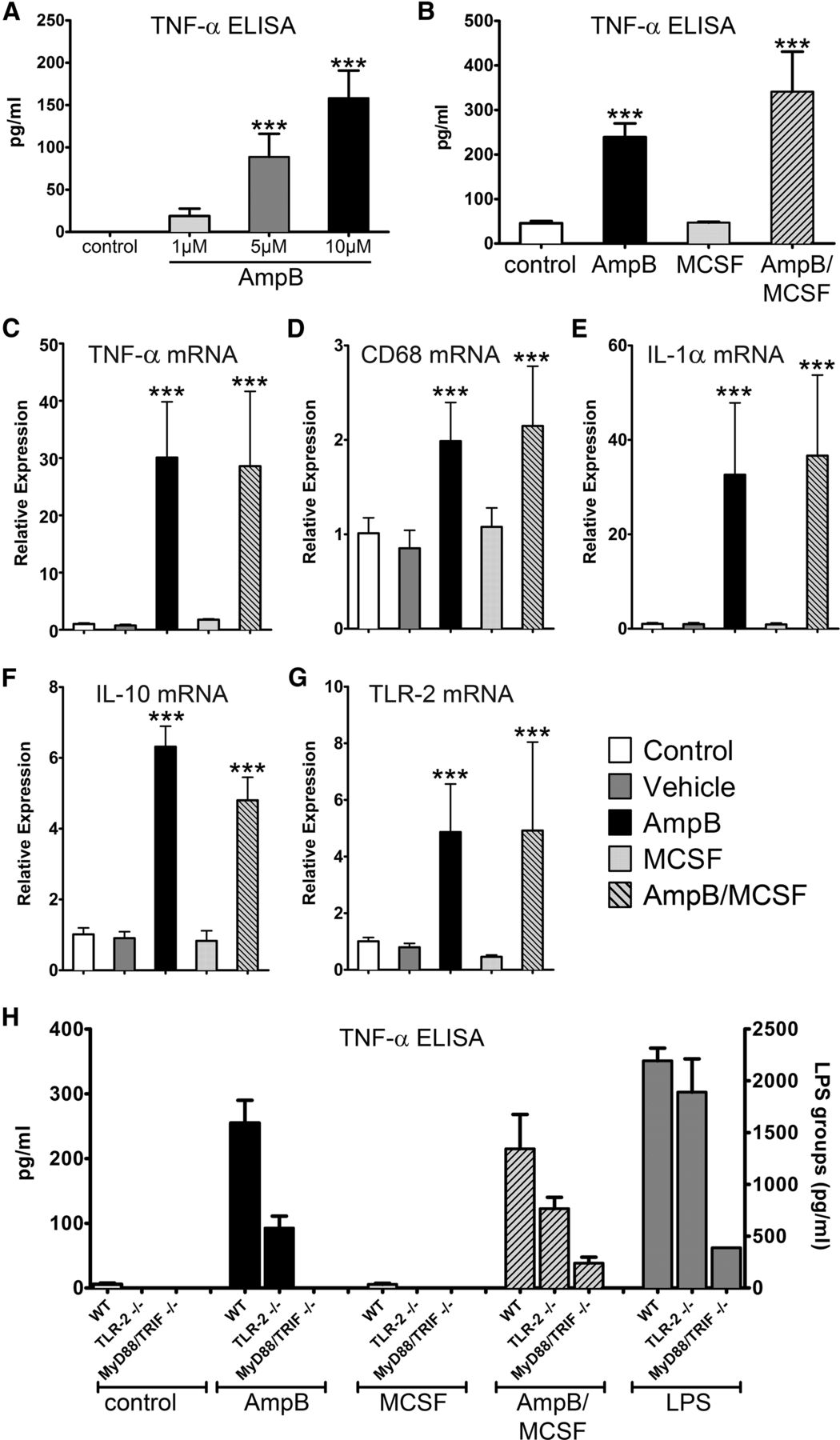
We previously screened the National Institute of Neurological Disorders and Stroke Custom Collection II (also referred to as the U.S. Drug Collection) library of 1040 compounds, the majority of which are Food and Drug Administration (FDA)-approved medicines, and identified AmpB as an activator of human microglia, monocytes, and macrophages (Samanani et al., 2013; Sarkar et al., 2014). In the current study, we evaluated the impact of AmpB on mouse microglia and BMDMs. Figure 1A shows that AmpB stimulates cultured mouse microglia in a concentration-dependent manner, using TNF-α production as an index of cellular activation. We found that AmpB also stimulates BMDMs (Fig. 1B).

Mouse microglia and macrophages are activated by AmpB in vitro through a MyD88/TRIF-dependent mechanism. A, B, TNF-α protein detected in the conditioned medium of mouse microglia (A), and similar detection in BMDM I (B). C–G, Relative expression of transcripts from BMDMs, whereby the AmpB-induced increase in transcripts was not augmented by the addition of MCSF. In H, the increase in TNF-α levels resulting from AmpB or AmpB/MCSF was reduced in TLR-2-null cells and abrogated in MyD88/TRIF-null cells. LPS was used as a positive control, and values for LPS are displayed on a scale with different magnitude. Values are reported as the mean ± SD of quadruplicate analyses, and results have been reproduced in at least one other set of cultures. ***p < 0.001 compared with control or respective wild types.
To maximize cellular activation in vivo, we considered whether AmpB could be combined with MCSF. In vivo, MCSF increases the number of circulating monocytes by stimulating the proliferation, differentiation, and survival of their precursor cells in the bone marrow (Stanley et al., 1997; Auffray et al., 2009; Hashimoto et al., 2011). In vitro, MCSF exposure for several days (see Materials and Methods) is a routine requirement to derive an enriched population of macrophages from mouse bone marrow-derived cells. Using these MCSF pre-exposed BMDMs, we found that additional incubation with MCSF did not further elevate TNF-α protein levels; moreover, MCSF did not augment the elevation of TNF-α produced by AmpB, although a trend was observed (Fig. 1B). As the cultured BMDMs have been previously exposed to MCSF, these data did not discourage plans to use MCSF and AmpB in combination in vivo (see below) in naive mice that have not been pre-exposed to MCSF.
Next, we analyzed the transcripts of several macrophage activation markers in BMDMs by qRT-PCR. Concurrent with the findings of the ELISA measurements, we detected a significant elevation of TNF-α mRNA expression after 6 h of treatment with AmpB (Fig. 1C). Increases were also detected for CD68 (Fig. 1D), and for IL-1α (Fig. 1E), and IL-10 (Fig. 1F), which are representative of proinflammatory and anti-inflammatory/regulatory cytokines, respectively. Finally, mRNA expression of TLR-2, another marker of activation, was also significantly increased after AmpB treatment (Fig. 1G). We did not find MCSF to alter levels of the above molecules alone or in combination with AmpB.
AmpB stimulates macrophages through toll-like receptor signaling
The mechanism by which AmpB stimulates cells to produce cytokines is unclear. Since TLRs are associated with the activation of monocytes, macrophages, and microglia, and TLR signaling requires the downstream intermediary of either the MyD88 and/or TRIF pathways, we used BMDMs isolated from MyD88/TRIF double-null mice. BMDMs from TLR-2-null mice were also used. We determined that TNF-α produced by AmpB or AmpB plus MCSF (henceforth referred as AmpB/MCSF)-stimulated macrophages was attenuated in the absence of TLR-2, and almost completely abrogated when both MyD88 and TRIF were absent (Fig. 1H). While these results did not address which of the 13 known mammalian TLRs are involved in AmpB signaling, they illustrate that MyD88/TRIF signaling is required for AmpB stimulation of macrophages.
The lysolecithin model of demyelination has a robust increase of microglia/macrophages activity after injury, and this is promoted by the combination of AmpB and MCSF
Previous studies (Gregg et al., 2007; Lau et al., 2012) noted that lysolecithin produced early demyelination (within the first 7 d) followed by spontaneous remyelination thereafter. Thus, we killed mice at day 3 after lysolecithin administration and confirmed that significant demyelination was present in the dorsal column, as evaluated by lost or disrupted MBP immunoreactivity (Fig. 2A,B); we observed that fragmented myelin yielded a very strong immunofluorescence, likely due to additional exposed epitopes for the MBP antibody. Corresponding with demyelination, there was intense Iba1 immunoreactivity of activated microglia/macrophages. In contrast, at a region removed from the lesion (Fig. 2A, ventral column), normal profiles of myelin ring and Iba1-stained ramified microglia were observed.

Activation of macrophages and microglia is extensive after lysolecithin demyelination in the spinal cords of mice, and this is further promoted by AmpB/MCSF. A, The dorsal column of the spinal cord 3 d after lysolecithin administration shows extensive accumulation of Iba1+ microglia/macrophages (red; top two panels, and also bottom left with the green overlay of the top left panel removed) in the vicinity of injury. Demyelination, as evident by intense MBP reactivity (green), reveals disrupted myelin structures in the midst of amoeba-shaped Iba1+ (red) cells (top right panel, which is magnified from the area depicted by arrow in the top left panel). In contrast, at a region remote from the injury and depicted by the box in the top left panel, normal myelin ring profiles and ramified shaped microglia are observed. B, At the interface (dotted line, top) of an area of demyelination with its adjacent unaffected area 3 d after lysolecithin, the MBP profiles are disrupted and highly fluorescent (right of dotted line) in the demyelinated area compared with less fluorescent ring profiles in the unaffected region. B, Bottom, In the demyelinated area, myelin profiles (green) within Iba1+ microglia/macrophages (red) indicative of phagocytosis of myelin can be observed (an example is featured at the intersection of the two white lines, and where the z-stack is displayed at the side and bottom edge of the panel). NY, Nuclear yellow dye to detect nuclei (stained blue). C–J, qRT-PCR of the spinal cord indicates that several molecules were elevated 3 d after demyelination compared with naive or sham-injured animals (mean ± SD, n = 5). Alone, AmpB and MCSF did not further elevate transcripts above that of injury, but the AmpB/MSCF combination resulted in significant elevations of transcripts encoding inflammatory cytokines (TNF-α and IL-1α), cellular markers (CD68 and TLR-2), scavenger receptors (MSR-1 and Fcrl), chemokine receptor (CX3CR-1), and chemokine (CCL-2). *p < 0.05, **p < 0.01, ***p < 0.001 compared with injury control. K, L, In situ hybridization of CD68+ cells (brown) shows that the transcripts for MSR-1 are on microglia/macrophages in the demyelinated dorsal column (K); optical density (OD; n = 5 each) denotes that AmpB/MCSF resulted in significant elevation of MSR-1 transcripts (L). *p < 0.05 compared with control.
To quantitate the activation of microglia/macrophages across the groups, qRT-PCR was used. Figure 2C–J denotes the significant increase of several markers of microglia/macrophages at the lesion site by 3 d after lysolecithin injury, compared with naive (uninjured) or sham (PBS deposition into the dorsal column) controls. These markers include TNF-α, IL-1α, CD68, TLR-2, scavenger receptors MSR-1 and Fcrl, the chemokine receptor CX3CR-1, and the macrophage chemoattractant chemokine CCL-2.
Compared with the amounts found in mice subjected to lysolecithin injury, where robust increases in microglia/macrophages markers were apparent from naive or sham controls, we determined that AmpB or MCSF alone was not sufficient to increase further the markers of microglia/macrophages activity. However, the double treatment with AmpB/MCSF elevated transcripts above the already substantial elevation that occurred after lysolecithin injury (Fig. 2C–J).
The capacity of microglia/macrophages to stimulate remyelination is likely related to the faster clearance of myelin debris through elevated phagocytotic activity. Focusing on the scavenger receptor MSR-1, we addressed whether its expression was predominantly on microglia/macrophages around the lesion site. Performing in situ hybridization on spinal cord section from mice 3 d after insult, coupled with a cell type-specific marker, we found that MSR-1 was predominantly on CD68-stained microglia/macrophages at the lesion site (Fig. 2K). Moreover, quantitative analyses found a significant increase of MSR-1 hybridization signal over CD68-positive cells in the AmpB/MCSF-treated animals compared with controls (Fig. 2L).
To further evaluate the activation of microglia/macrophages by AmpB/MCSF, sections collected 5 d after injury were stained for Iba1, and its normalized intensity was compared across groups of mice. Figure 3, A and B, shows that the localized increase in Iba1 intensity at the lesion site was elevated further in the combination AmpB/MCSF animals, supporting the contention that there is elevated activity of microglia/macrophages in these animals.

AmpB/MCSF treatment increased the representation of microglia/macrophages at the lesion site. A, Intense Iba1 immunoperoxidase staining in the dorsal column 5 d after lysolecithin demyelination (top left; magnified view of boxed area in top right), and low immunoreactivity outside of the lesion (ventral column, bottom left; magnified view of the boxed area in the bottom right). B, Iba1 intensity of demyelinated mice 5 d after lysolecithin administration was measured across sections of three mice per treatment group; data are reported as the mean ± SD. *p < 0.05 compared with controls. C, Flow cytometry was used to quantitate the number of microglia/macrophages (y-axis: CD45+; x-axis: CD11b+; ellipse), and representative profiles are shown per pool of five mice. The highest number of microglia/macrophages occurred in response to AmpB/MCSF treatment; the trend was reproduced in another experiment.
We next quantitated the increase in microglia/macrophages cell density in the spinal cord using flow cytometry. Unlike mice afflicted with experimental autoimmune encephalomyelitis (Goncalves DaSilva and Yong, 2009), we could not clearly separate microglia (CD11b+CD45intermediate) from infiltrated macrophages (CD11b+CD45high) in the spinal cord of lysolecithin-demyelinated mice (Fig. 3C), likely due to the fact that the injured dorsal column is a small proportion of the 3 mm coronal spinal cord block taken for analyses. Thus, we quantitated the pooled CD45+ and CD11b+ populations (Fig. 3C, ellipse) and determined that the combined AmpB and MCSF animals had the highest representation of microglia/macrophages in the spinal cord of lysolecithin-demyelinated mice compared with all other treatment groups.
Together, the combination treatment of AmpB and MCSF increased significantly the injury-induced representation of microglia/macrophages in the demyelinated spinal cord.
Treatment with AmpB does not induce obvious side effects in animals
When AmpB is given at high doses in humans, the toxicity includes nephrotoxicity, leucopenia, and other complications. Peak serum concentrations attained in humans given these high doses of AmpB to treat life-threatening fungal infections ranged ∼0.5–2 μg/ml (www.drugs.com). We addressed the levels attained in the serum of mice injected with AmpB at 1 mg/kg. After a single intraperitoneal dose, levels at 1 and 2 h were ∼125 ng/ml, and these decreased thereafter (Fig. 4A). Thus, these peak concentrations in mice were at most 25% of those found in humans injected with high doses of AmpB. Interestingly, we find AmpB to accumulate steadily in spinal cord and brain (Fig. 4B,C), which may bode well for targeting its actions within the CNS.

Amphotericin concentrations in blood and brain, and the absence of cytotoxic effects in mice. A–C, After a single injection of 1 mg/kg AmpB in naive mice, blood (A), spinal cord (B), and brain (C) were obtained at 1, 2, 4, and 24 h for measurement of AmpB by HPLC. Other mice received daily injections of 1 mg/kg AmpB for 7 d, and were killed 4 h after the last injection. Values are reported as the mean ± SEM of five animals per time point. D, E, After 7 d of daily treatment with AmpB, blood counts (number of cells per liter) indicate that the total number of white blood cells (WBC), neutrophils, lymphocytes, and platelets was comparable to those in vehicle-treated animals when animals were naive (D) or demyelinated with lysolecithin (E; n = 5 animals per group; data are reported as the mean ± SEM). Results were reproduced in a different experiment, and comparable results were observed in naive mice treated with AmpB/MCSF.
In mice injected daily with 1 mg/kg AmpB for 7 d, we encountered no obvious behavioral deficits. Blood taken from these mice after 7 daily injections of AmpB showed no loss of total white blood cell counts, neutrophils, lymphocytes, and platelets, whether in mice that were naive or in lysolecithin-demyelinated mice (Fig. 4D,E). Various other parameters of a standardized clinical complete blood cell count were also unaffected by AmpB (data not shown). Thus, the dose regimen of AmpB used here in mice appeared safe, and the serum and brain levels of the medication may be used to guide potential future studies in humans.
Treatment with AmpB and MCSF increases the number of oligodendrocyte lineage cells and promotes remyelination
To examine the postinjury treatment effects on repair, we first compared the effect of vehicle or drug treatment on the initial extent of demyelination. Eriochrome-stained sections from mice at 3 d after lysolecithin demyelination were subjected to lesion volume analyses. Figure 5A shows that all lysolecithin-administered groups, regardless of subsequent treatment, displayed extensive and comparable amounts of demyelination, and this was confirmed by lesion volume analyses (Fig. 5B).

AmpB increases indices of remyelination in mice. A, B, Three days after lysolecithin application, the size of demyelination in the dorsal column was comparable across all groups, as determined using Eriochrome cyanine myelin staining (A) and quantitative analysis (B) (n = 6 mice per group). C, D, However, at day 28 the amount of remaining demyelination appears minimal in the AmpB/MSCF group (C), and this is validated through quantitative analysis (D; n = 9–10 mice/group). Note that the amount of remaining demyelination (percentage of the dorsal column not containing myelin) displayed is the average across 30 sections per mouse, with each section spaced 100 μm apart from the other. E, F, Detection of oligodendrocyte precursor cells using an antibody to PDGFRα (E) shows that this is elevated 7 d after injury in the AmpB/MCSF group (n = 4–5 mice per group; F). All values are reported as the mean ± SEM. *p < 0.05 compared with controls.
Since the process of remyelination evolves over time, mice that were treated for the first 7 d were killed at day 28 and the spinal cord tissue was analyzed by Eriochrome cyanine staining to determine whether the significant demyelination at earlier time points was being repaired, as manifested by a reduction in the amount of remaining demyelination. Figure 5C shows qualitatively that the lesion size was still significant in the control and MCSF groups, but was reduced in the AmpB and AmpB/MCSF combination animals. When lesion volume was quantified across the entire lesion (average of 30 sections per animal) of individual mice, we found that the group treated with the AmpB/MCSF combination had a significant reduction in remaining demyelination (i.e., suggestive of remyelination) compared with the control demyelinated animals; the single treatment with AmpB showed a trend toward a reduced lesion size, but this was not significantly different from controls (Fig. 5D).
Oligodendrocyte precursor cells are required for remyelination. We stained spinal cord tissue after lysolecithin administration for PDGFRα, a marker of oligodendrocyte precursor cells. Tissue collected at day 7 was used, as this is a time point where the number of oligodendrocyte precursor cells increases as an injury-induced attempt at repair (Watanabe et al., 2002; Skihar et al., 2009). We focused on the coronal section that contains the lesion epicenter (defined as the section with the largest area of demyelination for a particular mouse when all Eriochrome-stained sections from that mouse were evaluated) and counted the number of precursor cells within the lesion. We found an increase in PDGFRα-positive cells in the lesion area of the dorsal column of mice treated with the combination AmpB/MCSF (Fig. 5E,F).
To provide further evidence that AmpB/MCSF was promoting remyelination as suggested by lesion volume analyses, we analyzed toluidine blue-stained Epon sections and electron micrographs (Fig. 6) from mice at days 7 and 28 after injury. EM analysis showed demyelinated axons at day 7 (Fig. 6B) and several remyelinated axons at day 28 (Fig. 6E). At 7 d after lysolecithin administration, and using toluidine blue sections and an automated algorithm written in Matlab, we found no difference in g-ratios across the groups (Fig. 6C); this indicates that the initial demyelination was comparable across treatment groups. At day 28, however, mice with AmpB/MCSF combination, or AmpB alone, had a statistically significant reduction in g-ratio compared with vehicle controls (Fig. 6F). MCSF administration by itself did not improve remyelination. Finally, when axonal caliber was segregated, we determined that the pro-remyelinating effect of AmpB or AmpB/MCSF could be discerned across small-, medium-, and large-caliber axons (Fig. 6G). Together, the treatment of demyelinated mice with AmpB increased the capacity for endogenous remyelination, and this is enhanced further with the addition of MCSF.

The g-ratio analyses show that AmpB and AmpB/MCSF promote remyelination at day 28. A–C, Day 7 results. A, An Eriochrome cyanine-stained section; the boxed area shows where Epon blocks were taken for toluidine blue staining and subsequent EM analyses. B, A representative EM image (original magnification, 15,000×) from red boxed area shows several demyelinated axons (*); a normal myelin profile is indicated by the arrow. C, The g-ratio analyses show the comparable amount of demyelination at day 7 across all groups. D, E, Day 28: the boxed area with shades of myelin stain (D, where the pale blue area is suggestive of remyelination) is enlarged at the EM level (original magnification, 4000×) in E where remyelinated axons (arrows) are present. F, The lower g-ratios in AmpB and AmpB/MCSF groups are indicative of remyelination. *p < 0.05; **p < 0.01 compared with control. All controls in this figure refers to lysolecithin mice treated with vehicle. C, F, Approximately 100 axons per mouse were analyzed for g-ratio, and each histogram is the mean ± SEM of 5–9 mice. G, Separation of g-ratios (mean ± SEM) based on axonal caliber demonstrates that the efficacy of AmpB/MCSF was evident across axons of all calibers.
To establish the requirement of microglia/macrophages in the therapeutic efficacy of AmpB/MCSF, mice were treated with clodronate liposomes on days 1, 4, and 7 after lysolecithin demyelination. Figure 7A shows that the clodronate liposome treatment reduced the number of circulating monocytes in blood by >50% compared with mice injected with PBS-containing control liposomes; we did not determine the corresponding density of microglia/macrophages in the spinal cord. While AmpB/MCSF mice given control liposomes promoted remyelination at day 28 compared with vehicle-treated demyelinated mice, this effect was abrogated in demyelinated mice treated with clodronate liposomes (Fig. 7B). These results show that monocytes and microglia/macrophages are necessary for AmpB/MCSF to produce remyelination.

The depletion of circulating monocytes by clodronate liposomes reverses the capacity of AmpB/MCSF to promote remyelination. A, Seven days after the initiation of clodronate liposome and AmpB/MCSF treatments, the number of CD11b+Ly6C+ monocytes in blood was reduced compared with mice treated with control liposomes (n = 4–5 mice/group). B, At day 28 in these mice, the effectiveness of AmpB/MCSF to promote remyelination was abrogated by treatment with clodronate liposomes (n = 10 mice/group). All mice were lysolecithin demyelinated, and the controls without AmpB/MCSF were administered vehicle. Data are reported as the mean ± SEM. **p < 0.01, ***p < 0.001 compared with controls without AmpB/MCSF.
Discussion
Demyelination is a feature of chronic conditions such as MS and neuromyelitis optica, and acute CNS insults such as spinal cord injury, transverse myelitis, acute hemorrhagic leukoencephalopathy, and acute disseminated encephalomyelitis. The reformation of myelin around demyelinated axons is desirable not only for the restoration of anatomic structures or for re-establishment of saltatory conduction of impulses, but it is especially important to protect denuded axons from undergoing degeneration as the axon–myelin unit provides metabolic support for one another (Yin et al., 2006). Thus, the enhancement of remyelination is a therapeutic strategy to prevent the loss of axons and the acquisition of disability in demyelinating disorders.
The role of inflammation in promoting neural repair is gaining increasing recognition. Products of macrophages, including oncomodulin, facilitate the regeneration of axons (David et al., 1990; Yin et al., 2006). Monocytes and macrophages are also necessary to enable remyelination in animal models of demyelination as their deficiency retards the process of remyelination (Kotter et al., 2001, 2005; Kondo et al., 2011). The benefits of monocytes and macrophages, as well as their CNS counterpart, microglia, may be attributed to their being required to help clear myelin debris after a demyelinating episode, as well as their provision of a variety of growth factors into the injured CNS (Yong, 2010; Miron et al., 2013; Rawji and Yong, 2013). Thus, therapeutic medications that can stimulate these cells can be of benefit to help repair demyelinated lesions.
We sought FDA-approved medications that could stimulate monocytes, macrophages, and microglia, and found that one such medication was AmpB (Samanani et al., 2013; Sarkar et al., 2014). AmpB is used clinically as an antifungal medication through a mechanism that involves its polyene structure forming complexes with sterols such as cholesterol enriched in cell membranes, thereby forming pores in cell membrane of fungi to produce fungicidal activity (Baginski et al., 1997; Lemke et al., 2005). However, we found that the stimulation of macrophages by AmpB to produce TNF-α is critically dependent on MyD88 and TRIF signaling (Fig. 1H), implicating for the first time the activation of toll-like receptors by AmpB.
To enhance the therapeutic potential of stimulating microglia/macrophages in vivo, we combined AmpB with MCSF. We did not find MCSF to directly stimulate these cells in culture (Fig. 1). Nonetheless, we reasoned that AmpB/MCSF would still be an effective combination in vivo since MCSF mobilizes monocytes from the bone marrow, and promotes the proliferation, differentiation, and survival of monocytes, macrophages, and bone marrow progenitor cells (Stanley et al., 1997; Auffray et al., 2009; Hashimoto et al., 2011); thus, the combination would provide the two-pronged approach of increasing monocyte and macrophage numbers with MCSF, and obtaining a more activated population with AmpB. Indeed, likely due to the massive upregulation of microglia/macrophage representation and activity at the lesion that occurs after demyelination, the further detectable increase of their activity within the injured spinal cord required the combination of AmpB with MCSF (Figs. 2, 3). The combination treatment produced an increase in the number of oligodendrocyte precursor cells within the lesion. We did not resolve whether there was an elevation of oligodendrocyte precursor cells in the white matter outside of the lesion area, an oversight that should be addressed in future experiments. The elevation of oligodendrocyte precursor cells elicited by AmpB and MCSF appears biologically relevant, as this was correlated with an enhancement of remyelination, as noted by the analysis of the remaining lesion at day 28 using Eriochrome cyanine staining or by g-ratio analyses (Figs. 5, 6). We detected also enhanced remyelination in animals treated with AmpB alone, while MCSF alone did not improve repair. Finally, we determined that the mechanism of the combination of AmpB and MCSF required monocytes and tissue-infiltrated macrophages as their depletion by clodronate liposomes abrogated their effect (Fig. 7). While the toxicity of AmpB in human use must be considered, these results highlight the proof of principle that stimulating monocytes and macrophages can have clinical relevance in demyelinating conditions, particularly in combination with MCSF.
Monocytes, macrophages, and microglia can be subdivided into an M1 proinflammatory subset, and an M2 regulatory/anti-inflammatory subset that is thought to promote reparative outcomes (Kigerl et al., 2009; David and Kroner, 2011). After lysolecithin-induced demyelination, M1 cells are first recruited and contribute to the proliferation of OPCs; a switch to an M2 milieu then occurs and appears to be required for the differentiation of OPCs into myelin-forming oligodendrocytes (Miron et al., 2013). Both subsets are promoted by AmpB (Sarkar et al., 2014). We limited the injections of AmpB to the first 7 d with the knowledge that myelin debris was largely cleared by microglia/macrophage cell activity during this period. Whether longer treatment would have more beneficial or adverse outcomes remains to be determined.
The mechanism of monocytes, macrophages, and microglia to promote remyelination is thought to be related to their capacity to produce and release a range of neurotrophic factors, such as nerve growth factor and brain-derived neurotrophic factor, as well as by their phagocytotic activity to remove inhibitory myelin debris (Auffray et al., 2009; David and Kroner, 2011; Miron et al., 2013). Other mechanisms may involve the detoxification of injurious molecules such as free radicals or the provision of proteases for remodeling of the extracellular matrix for repair (Larsen et al., 2003; Lau et al., 2013). These multiple effects of activated monocytes, macrophages, and microglia likely work in concert to mediate the therapeutic efficacy of AmpB/MCSF reported in this study.
When used in humans at high doses to treat life-threatening fungal infections, AmpB is associated with toxic effects such as nephrotoxicity and leukopenia. In the current study, we did not observe any obvious toxicity in culture or in mice, and hematologic cell counts and other blood parameters were normal in AmpB-treated animals (Fig. 4). This could be due to the low dose of AmpB that was used, which generated peak serum levels of AmpB that were at least fivefold lower than that observed in patients treated with high-dose AmpB. While caution is required, these results suggest the safety of AmpB at low doses to stimulate cellular activity to promote remyelination. In a previous study (Sarkar et al., 2014) involving glioma cells growing in the brain of mice, we determined that the daily treatment of mice with AmpB for up to 5 months was safe; the dose used in that study was 0.1 mg/kg, and we did not test the higher dose of 1 mg/kg used in the current study. We note that less toxic formulations of AmpB are being researched for human use (Hullard-Pulstinger et al., 2011; Singh et al., 2011), and that other means of delivering AmpB, such as by nanosuspensions, are being considered (Das and Suresh, 2011; Singodia et al., 2011). Alone or in combination with MCSF, these are potential medications to stimulate monocyte, macrophage, and microglia activity to promote remyelination.
In chronic demyelinating conditions such as MS, the persistence of neuroinflammation, including activated microglia/macrophages, is thought to be detrimental on balance (Prineas et al., 2001; Yong, 2010). The use of activators of monocytes, macrophages, or microglia in these chronic disorders would be challenging as it could promote detrimental inflammation further. Moreover, conditions such as MS have multifocal lesions that are at various stages of demyelination and remyelination, and it would be difficult to predict how individual lesions would respond to activators. We suggest the potential use of AmpB and MCSF only in acute demyelinating conditions, since the age and stage of lesion formation is more predictable, and the medications could be administered for short durations during the phase of attempted repair. These acute conditions may include spinal cord injury, transverse myelitis, acute hemorrhagic leukoencephalopathy, and acute disseminated encephalomyelitis.
Since the lesion resulting from lysolecithin remyelinates over time, our data reflect an increase in the rate of remyelination induced by AmpB/MCSF rather than the overall capacity of remyelination. It would be instructive to compare the extent of remyelination at different intervals, such as on days 14–21 after injury and in response to AmpB/MCSF, and this remains to be performed in future studies. We note that our injury method resulted in many axons that remained demyelinated at day 28. This contrasts with the pioneering work of Jeffery and Blakemore (1995) that showed that CNS remyelination was complete by day 23 after lysolecithin injury to the dorsal column. The discrepancy may be due to differences in experimental protocols, such as the use of C57BL/6 mice in our study, and OLA/MF1 and ICI strains in the Jeffery and Blakemore (1995) study. Indeed, in our hands, the C57BL/6 mice remyelinated comparatively less well after the same lysolecithin insult than another strain that we have previously used, 129/SvEv (Lau et al., 2012).
In conclusion, this study emphasizes the importance of enhancing the cellular activity of monocytes, macrophages, and microglia to promote remyelination after a demyelinating insult. In particular, our work describes potential therapeutic regimens, AmpB alone or in combination with MSCF, to facilitate the repair of demyelinated axons. As AmpB is already available for human use, albeit with significant toxicity, its translation as a therapeutic agent to stimulate remyelination may be expedited.
Footnotes
This study was supported by operating grants from NeuroScience Canada (Brain Repair Program) and the Scientific Foundation of the Multiple Sclerosis Society of Canada (MSSC). A.D., S.S., and M.M. received postdoctoral salary support from Alberta Innovates-Health Solutions (AIHS), and A.D. and S.S. received additional postdoctoral stipend from the MSSC. L.L. received studentship awards from the AIHS and MSSC. We thank Dr. Paul Kubes for the provision of MyD88/TRIF double-null mice, and Dr. Nico van Rooijen for clodronate liposomes. We are grateful for the skilled technical assistance of Janet Wang, Yan Fan, Brooke Verhaeghe, Hollie Mowbray, Fiona Yong, and Claudia Silva.
The authors declare no competing financial interests.
- Correspondence should be addressed to V. Wee Yong, Professor, Hotchkiss Brain Institute and the University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada. vyong{at}ucalgary.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}