

Abstract
Paranodal axoglial junctions in myelinated nerve fibers are essential for efficient action potential conduction and ion channel clustering. We show here that, in the mature CNS, a fraction of the oligodendroglial 155 kDa isoform of neurofascin (NF-155), a major constituent of paranodal junctions, has key biochemical characteristics of a lipid raft-associated protein. However, despite its robust expression, NF-155 is detergent soluble before paranodes form and in purified oligodendrocyte cell cultures. Only during its progressive localization to paranodes is NF-155 (1) associated with detergent-insoluble complexes that float at increasingly lower densities of sucrose and (2) retained in situ after detergent treatment. Finally, mutant animals with disrupted paranodal junctions, including those lacking specific myelin lipids, have significantly reduced levels of raft-associated NF-155. Together, these results suggest that trans interactions between oligodendroglial NF-155 and axonal ligands result in cross-linking, stabilization, and formation of paranodal lipid raft assemblies.
Introduction
Rapid and efficient action potential propagation in vertebrates has been achieved through the development of myelin and the clustering of ion channels at nodes of Ranvier. Traditionally, myelin has been viewed as a passive structure, facilitating conduction through increased membrane resistance and decreased membrane capacitance. However, it is now clear that myelin has active roles in electrical signaling and regulates many neuronal properties, including ion channel expression and localization (Poliak and Peles, 2003; Salzer, 2003). Although the mechanisms responsible for this regulation are not well understood, recent work suggests that paranodal axoglial junctions flanking nodes of Ranvier are important for the structural integrity of myelin–axolemmal interactions, myelin–axolemmal bidirectional signaling, and for the appropriate clustering of both voltage-gated sodium (Nav) and potassium (Kv) channels (Rasband and Trimmer, 2001; Spiegel and Peles, 2002; Menon et al., 2003; Rasband et al., 2003; Rios et al., 2003). The axonal cell adhesion molecules Caspr and contactin and the oligodendroglial 155 kDa isoform of neurofascin (NF-155) are thought to be critical components of this axoglial junction (Rios et al., 2000; Tait et al., 2000; Charles et al., 2002).
Some of the best evidence that axoglial junctions influence ion channel clustering comes from mutant mice with disrupted paranodes. For example, mice lacking either Caspr or contactin have appropriately compacted myelin but displaced Kv1 channels, broadened Nav channel clusters with reduced channel densities, and impaired junctional attachment characterized by everted paranodal loops and absent transverse bands (Bhat et al., 2001; Boyle et al., 2001; Rios et al., 2003). Interestingly, mice lacking the enzymes necessary for the synthesis of galactocerebroside and sulfatide [UDP-galactose:ceramide galactosyltransferase-null (CGT-null)] or of sulfatide alone [cerebroside sulfotransferase-null (CST-null)] have a similar phenotype (Dupree et al., 1998; Honke et al., 2002; Ishibashi et al., 2002). A careful comparison of these animals shows that both Caspr- and contactin-null mice lack axonal paranodal components but have glial paranodal NF-155, albeit at reduced levels (Bhat et al., 2001; Boyle et al., 2001; Poliak et al., 2001). In contrast, CGT-null mice have low levels of axonal paranodal Caspr and contactin but lack glial paranodal NF-155 (Poliak et al., 2001). Although the phenotypes of the Caspr- and contactin-deficient mice are easy to understand, it is less clear why loss of galactolipids results in decreased paranodal NF-155 and disrupted axoglial junctions.
Recently, the concept of “lipid rafts,” or specialized membrane lipid microdomains, has been suggested as a mechanism whereby signaling, trafficking, and the assembly of adhesion complexes takes place (Brown and London, 2000; Simons and Toomre, 2000; Harris and Siu, 2002), although dissenting views have been offered (Munro, 2003). Neuronal and myelin membranes may have several kinds of rafts, because various proteins can segregate into biochemically distinct entities depending on the experimental and physiological conditions (Madore et al., 1999; Hakomori, 2002; Taylor et al., 2002; Marta et al., 2003). Proteins that are included in raft assemblies often have lipid modifications (Melkonian et al., 1999; el-Husseini Ael and Bredt, 2002). For example, of the proteins that comprise the paranodal protein complex, contactin and neurofascin have glycosylphosphatidylinsositol (GPI) and palmitate modifications, respectively (Ren and Bennett, 1998; Faivre-Sarrailh et al., 2000). Given these considerations, one possible explanation for the phenotype of CGT-null mice is that the formation and stabilization of paranodal axoglial junctions is impaired as a result of the disruption of NF-155 partitioning into lipid rafts.
We show here that a fraction of NF-155 has key biochemical characteristics of a raft-associated protein and that its inclusion into lipid rafts occurs during paranode formation. We suggest that trans interactions between oligodendroglial NF-155 and axonal ligands may result in cross-linking, stabilization, and formation of paranodal lipid raft assemblies.
Materials and Methods
Animals. Rat and mouse brains and optic nerves were obtained by rapidly dissecting out the tissue after killing animals with halothane. Genotypes for CGT –/– and CST –/– were determined as described previously (Bansal et al., 1999). Animals were housed at the Center for Laboratory Animal Care at the University of Connecticut Health Center and in accordance with all National Institutes of Health guidelines for the humane treatment of animals.
Antibodies. The pan-NF monoclonal antibody (mAb) (clone L11A/41.6) was made by immunizing mice with a glutathione S-transferasefusion protein containing the intracellular domain of rat neurofascin, common to both the 155 and 186 kDa isoforms. This fusion protein was kindly provided by Drs. Steven Tait and Peter Brophy (University of Edinburgh, Edinburgh, UK). Preincubation of the pan-NF antibody with the immunizing peptide abolished all immunoreactivity (data not shown). Surprisingly, although this antibody was very robust against rat NF-155 and NF-186, it did not recognize the mouse forms of these proteins. Because the intracellular domains of rat and mouse NF differ by only two amino acids, the epitope for pan-NF likely includes these two residues. Polyclonal (affinity purified) and monoclonal (IgM) anti-Caspr antibodies were made against a Caspr-fusion protein kindly provided by Dr. Elior Peles (The Weizman Institute of Science, Rehovot, Israel) (Peles et al., 1997). Two different polyclonal rabbit anti-NF-155-specific antibodies were kindly provided by Drs. Peter Brophy and Hiroko Baba (Tokyo University of Pharmacy and Life Science, Hachloji, Japan). These NF-155 antibodies were generated against synthetic peptides specific to NF-155 (CLWVSQKRQQASFPGDRPR) (Tait et al., 2000). Polyclonal anti-Kv1.2 and monoclonal anti-pan Nav channel antibodies have been described previously (Rhodes et al., 1995; Rasband et al., 1999) and were kindly provided by Dr. James Trimmer (University of California, Davis, CA). Goat anti-contactin antibodies were purchased fromR&D Systems (Minneapolis, MN). The O1, O4, and MBP (myelin basic protein) antibodies have been described previously (Bansal et al., 1996). The anti-CNP (2′,3′-cyclic nucleotide 3′-phosphodiesterase) was purchased from Sternberger Monoclonals (Baltimore, MD).
Membrane homogenates. Brain and optic nerve membrane homogenates were prepared from freshly dissected tissues. Unless otherwise stated, all steps using membrane homogenates or its derivatives were performed at 4°C or on ice. Each brain was homogenized in ice-cold 0.32 m sucrose, 5 mm sodium phosphate, pH 7.4, and 1 mm sodium fluoride, containing 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml antipain, and 10 μg/ml benzamidine (10 ml/gm wet brain weight). Crude homogenates were then centrifuged at 600 × g for 10 min to remove debris and nuclei. The resulting supernatant was then centrifuged at 45,000 × g for 60 min. This pellet was then resuspended in 2.5 ml of ice-cold homogenization buffer per gram of brain used. For optic nerves, 24–48 nerves were pooled for each membrane preparation. Protein concentrations for each membrane preparation were determined using the BCA method (Pierce, Rockford, IL).
Detergent solubilization of membrane homogenates. Detergent-resistant membranes were isolated by solubilizing brain or optic nerve membrane homogenates in 1% Triton X-100 lysis buffer (20 mm Tris-HCl, pH 8.0, 10 mm EDTA, 0.15 m NaCl, 10 mm iodoacetamide, 0.5 mm PMSF, 10 mm sodium azide, and the same mixture of protease inhibitors as described above) at a concentration of 1 mg/ml protein for 1 hr on a rotator at 4°C or at 37°C with frequent vortexing. The resulting lysate was centrifuged at 13,000 × g for 30 min to separate the soluble and insoluble fractions. The insoluble pellet was then resuspended in lysis buffer to the original starting volume. Equal volumes of the resuspended insoluble pellet and the detergent soluble fraction were then added to 2× concentrated reducing sample buffer, boiled, and then loaded and size fractionated on SDS-polyacrylamide gels for a direct comparison of the relative amounts of protein in each fraction by immunoblotting.
Cholesterol extraction. Brain membrane homogenates were diluted to 1 mg/ml in lysis buffer (without TX-100) in the presence or absence of 0.2% saponin and placed on a rotator at 4°C for 30 min. These membranes were then centrifuged at 13,000 × g for 10 min. The saponin-treated and untreated pellets were then resuspended in 1 ml of 1% TX-100 lysis buffer and placed on a rotator at 4°C for 1 hr. Detergent-soluble and -insoluble fractions were then collected. The soluble fraction was ethanol precipitated and resuspended in 200 μl of reducing sample buffer. The detergent-insoluble pellets were resuspended in 200 μl of reducing sample buffer. Saponin-treated homogenates were then compared with untreated homogenates by SDS-PAGE and immunoblotting.
Sucrose gradient centrifugation. Preparation of sucrose gradients and isolation of rafts was performed as described by Taylor et al. (2002). Briefly, detergent-resistant membranes were resuspended to 1 ml in lysis buffer (no TX-100). The pellet suspension was then mixed with 1 ml of 2 m sucrose. The resulting mixture was then overlaid with 2 ml of 1 m sucrose and 1.5 ml of 0.2 m sucrose. Samples were then centrifuged for 19 hr at 192,000 × g, and 0.4 ml fractions were then collected from the top of the tube. Sucrose and protein concentrations were then measured for each fraction. In some cases (e.g., optic nerve gradients), proteins were concentrated by overnight ethanol precipitation. The pellets were then resuspended in 100 μl of reducing sample buffer, boiled, and analyzed by SDS-PAGE and immunoblotting.
Immunoblotting. Membranes, detergent extracts, insoluble pellets, or fractions from sucrose density gradients were separated on 7.5% SDS-polyacrylamide gels. Lauryl sulfate (Sigma, St. Louis, MO) was the SDS source used in all SDS-PAGE experiments (Shi et al., 1994). Size-fractionated proteins were transferred electrophoretically to nitrocellulose paper, and the resultant immunoblots were stained with mAb (anti-Caspr or anti-pan-NF) tissue culture supernatants (diluted 1:2–1:4) or anti-contactin, anti-NF-155, or anti-CNP antibodies (diluted 1:1000). The blots were then incubated in the appropriate secondary antibodies for 1 hr, washed, and soaked in enhanced chemiluminescence substrate (NEN, Boston, MA) for 1 min and autoradiographed on Eastman Kodak (Rochester, NY) XLS-1 film. Films were then digitized with a transparency flatbed scanner (Hewlett-Packard, Palo Alto, CA). In the case of GM1 [monosialoganglioside Gal-GalNAc-(NAN)-Gal-Glc-Cer], 1 μlof each fraction was assayed by dot blot using horseradish peroxidase-conjugated cholera toxin (1:20,000; Sigma). Homogenates from mixed primary cultures of CGT-null mice were separated on 12% SDS polyacrylamide gels, electrophoretically transferred to polyvinylidene difluoride membranes, and immunoblotted using anti-NF-155 (1:1000) and anti-CNP.
Cell culture. Mixed primary cultures of neonatal [postnatal day 2 (P2)] mice telencephala were prepared as described previously (Bansal et al., 1999). Briefly, dissociated cells were plated in 10% fetal calf serum (FCS) in DMEM at a density of 2.5 × 105 cells/cm2 into poly-l-lysine-coated (50 μg/ml; Sigma) 35 mm tissue culture plates for protein isolation. After 1 d, the cultures were changed to defined medium mN2 [DMEM with d-glucose (4.5 gm/l), human transferrin (50 μg/ml), bovine pancreatic insulin (5 μg/ml), 3,3,5-triiodo-l-thyronine (15 nm), sodium selenium (30 nm), d-biotin (10 nm), hydrocortisone (10 nm), sodium pyruvate (0.11 mg/ml), penicillin–streptomycin (10 IU/ml and 100 μg/ml, respectively), and 0.1% BSA (all ingredients from Sigma)] plus 1% FCS and 1% horse serum. Purified populations of developmentally synchronized rat oligodendrocyte lineage (OL) cells were prepared at three stages: early progenitors, late progenitor, and terminally differentiated OLs. Purity and phenotype of each population were extensively characterized by immunolabeling cells with a panel of antibodies (Bansal et al., 1996). Briefly, progenitors were obtained from mixed primary cultures from neonatal rat telencephalon by overnight shaking, followed by differential adhesion and complement lysis with anti-galactocerebroside to remove astrocytes, macrophages, and terminally differentiated OLs. Cells were plated in 5% FCS/DMEM in tissue culture plates coated with poly-d,l-ornithine (50 μg/ml; Sigma). After cell attachment for 2–3 hr, the medium was changed to serum-free, defined medium mN2 (above). Cultures were grown for 7 d to produce terminally differentiated OLs, or expanded and arrested at either the early progenitor stage by growth in PDGF-BB chain homodimer plus FGF-2 (10 ng/ml each) for 2 d, or at the late progenitor stage by growth in FGF-2. Astrocyte cultures prepared from monolayer cultures remaining after releasing OL progenitors from mixed primary cultures (see above) (Bansal et al., 1996) were 99% positive for the astrocytic marker GFAP.
Immunostaining. Optic nerves were rapidly dissected and immediately fixed in ice-cold 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.2, for 30 min. After fixation, nerves were transferred to ice-cold 20% sucrose (w/v) in 0.1 m PB until equilibrated. The tissue was then frozen in Tissue-Tek OCT mounting medium. Sections were then cut (5–10 μm), placed in 0.1 m PB, spread on gelatin-coated coverslips, and allowed to air dry. In the case of CGT –/– and wild-type (WT) mice, sections were postfixed for 1 min in Bouin's fixative, because the rabbit polyclonal anti-NF-155 only stains Bouin's-fixed tissue. Immunofluorescence staining was performed as described previously (Bansal et al., 1996; Rasband et al., 1999). Secondary antibodies were Alexa-488, Alexa-594, Cy3, or FITC conjugated to goat anti-rabbit or goat anti-mouse IgG or IgM antibodies (Molecular Probes, Eugene, OR). Digital images were collected on a Zeiss (Thornwood, NY) Axioskop 2 fluorescence microscope fitted with a Hamamatsu (Bridgewater, NJ) ORCA-ER camera. In some instances (see Figs. 1 A, B,7 A, B), z-stack of images was collected at 0.2 μm intervals, and the resulting stacks were then deconvolved by iterative restoration using the software package Volocity (Improvision, Lexington, MA). In the case of detergent treatment of whole optic nerves, we rapidly dissected the nerves and then immediately immersed them in 1% TX-100 lysis buffer at 4°Cor37°C. These nerves were then incubated for 1 hr, removed, immediately fixed in 4% paraformaldehyde at 4°C, and processed as described above.

A subpopulation of NF-155 has key biochemical characteristics of a lipid raft-associated protein. A, B, Rat optic nerve sections double labeled for pan-NF and NF-155. C–E, Analysis of rat brain membranes (RBM). C, Immunoblot showing the fractionation of NF isoforms into the 1% TX-100 detergent-insoluble fraction (or pellet) or soluble fraction at either 4°C or 37°C. Each temperature condition shows the equivalent of 10 μg of protein split between soluble and pellet fractions. The RBM lane was loaded with 10 μg of protein. All immunoblots (C–F) were performed using pan-NF antibodies. D, Previous cholesterol extraction from brain membrane homogenate by 0.2% saponin at 4°C promotes the solubility of NF-155 in 1% TX-100 at 4°C. S, 1% TX-100-soluble fraction. P, 1% TX-100-insoluble pellet. E, Sucrose density gradient analysis of the detergent-insoluble fraction immunoblotted for NF isoforms. Scale bars, 5 μm.

Raft-associated NF-155 is reduced in rodents with disrupted paranodes. A, B, Nodes of Ranvier from sciatic nerves (A) and optic nerves (B) of WT and CST-null (CST) and CGT-null (CGT) mice double labeled for NF-155 (green) and Na+ channels (red). (Note that the axons run horizontally in A and vertically in B.) Rabbit polyclonal anti-NF-155 was used in all experiments shown (A–E). C, Immunoblots of sucrose gradients using detergent-insoluble pellets from P30 WT and CGT-null mouse brains assayed for NF-155, Caspr, contactin, and GM1. D, A comparison of the NF-155 protein levels in P30 WT and CGT-null mouse brains. Each lane was loaded with 40 μg of brain membrane proteins. E, Mouse oligodendrocyte cultures from WT and CGT-null mice. NF-155 and CNP immunoreactivity from cultures from four mouse pups, two of each genotype are shown. These experiments were repeated in two independent litters of animals with identical results. F, Immunoblot of brain membranes from WT and md rats showing (1% TX-100 at 4°C) soluble (S) and insoluble (P) fractions. Immunoblots were performed using anti-pan-NF antibodies (arrow indicates NF-155). Scale bars, 5 μm.
Results
A fraction of NF-155 is present in lipid rafts
Neurofascin is present as two major splice variants in the adult CNS: a 155 kDa isoform expressed in oligodendrocytes and localized to paranodes [NF-155 (Collinson et al., 1998; Tait et al., 2000] and a 186 kDa isoform expressed in neurons and localized to nodes of Ranvier and axon initial segments [NF-186 (Davis et al., 1996)]. pan-NF antibodies detect both nodal NF-186 and paranodal NF-155 in optic nerve sections (Fig. 1A), whereas NF-155-specific antibodies label only paranodes (Fig. 1B).
To test whether NF isoforms associate with lipid rafts, we used four biochemical criteria that have been used to potentially identify raft-associated proteins (Kim and Pfeiffer, 1999; Brown and London, 2000; Simons and Toomre, 2000; Taylor et al., 2002). For criteria 1 and 2, lipid raft-associated proteins are insoluble during extraction in 1% TX-100 at 4°C but soluble in 1% TX-100 at 37°C. NF-186 was almost completely extracted from rat brain membrane homogenates in 1% TX-100 at both temperatures (Fig. 1C; S, soluble fraction; P, pellet or insoluble fraction). In contrast, ∼20% of NF-155 remained insoluble during extraction at 4°C, but this fraction was solubilized at 37°C (Fig. 1C). For criterion 3, lipid raft-associated proteins are soluble in 1% TX-100 at 4°C after cholesterol perturbation. Pretreatment of brain homogenates with 0.2% saponin at 4°C, which extracts cholesterol from membranes and perturbs lipid rafts (Schroeder et al., 1998), followed by extraction with 1% TX-100 at 4°C, resulted in the essentially complete solubilization of NF-155 (Fig. 1D, compare pellet with and without saponin treatment). In contrast, this sequential treatment did not render soluble the small amount of insoluble NF-186 (Fig. 1D). For criterion 4, lipid raft-associated proteins float at low densities on sucrose gradients. The majority of the NF-155 left insoluble after treatment with 1% TX-100 at 4°C floated to a low density during equilibrium sucrose gradient centrifugation (Fig. 1E, fractions 4–7), with the remainder coming to equilibrium at a higher density (fractions 12–13). These biochemical results suggest that, although NF-155 and NF-186 have identical intracellular and transmembrane domains, only a subpopulation of NF-155 associates with lipid rafts.
The density and detergent insolubility of NF-155 are developmentally regulated
Because NF-155 is a principle component of paranodes and may be involved in axoglial junction formation and maintenance (Tait et al., 2000), we hypothesized that the association of NF-155 with lipid rafts might be related to junction formation. Optic nerves were used to test this possibility, because they are a pure white matter tract with many paranodes, the formation of which begins at approximately P8, reaches a peak rate of formation at approximately P15, and all Na+ channel clusters are flanked by Caspr-labeled paranodes by P24 (Rasband et al., 1999). We tested the solubility of NF isoforms from rat optic nerve membrane homogenates at P12, P18, and in adult (>2 months) in 1% TX-100 at either 4°C or 37°C. As expected, the ratio of NF-155 to NF-186 in optic nerve (Fig. 2A) was much higher than that seen in brain (Fig. 1C) because some structures rich in NF-186 (e.g., axon initial segments) (Jenkins and Bennett, 2001) are not present in optic nerve. In adult optic nerve, as in brain, the fraction of NF-155 that was insoluble at 4°C was mostly soluble in 1% TX-100 at 37°C. In contrast, at P12 and P18, only approximately one-half of the insoluble NF-155 at 4°C became soluble at 37°C.

The density and detergent insolubility of NF-155 are developmentally regulated. A, Detergent extraction of NF-155 from P12, P18, and adult rat optic nerve membrane homogenates. Each temperature condition shows the equivalent of 10 μg of protein split between soluble (S) and pellet (P) fractions. Immunoblotting was performed using the pan-NF antibody. B–E, Detergent-insoluble pellets from P13, P18, P24, and adult rat optic nerve (ON) homogenates were floated on sucrose gradients. Equal volume fractions were collected from the gradients and immunoblotted using pan-NF (B), Caspr(C), or anti-CNP (D). Equal volumes from each fraction were loaded on the gels. The ganglioside GM1 was assayed by dot blot using peroxidase-conjugated cholera toxin (E). One microliter of each fraction was loaded. Pellets from the gradients (fraction 13) are not shown. F, The percentage of sucrose was measured for each fraction used in the experiments shown in B–E.
We determined the density of the 1% TX-100 (4°C) insoluble material from P13, P18, P24, and adult rat optic nerves by sucrose density gradient analysis and assayed fractions by immunoblot for the paranodal proteins NF-155 (Fig. 2B) and Caspr (Fig. 2C), and the myelin raft components CNP (Fig. 2D) (Kim and Pfeiffer, 1999; Taylor et al., 2002) and ganglioside GM1 (Fig. 2E) (Vyas et al., 2001). CNP and GM1 were present in low-density fractions (fractions 3 and 4) at all ages, becoming concentrated in fraction 4 in adult optic nerve (Fig. 2D,E). In contrast, NF-155 and Caspr were present in low-density fractions (fractions 4 and 5) during early paranode formation and myelination but floated at progressively lower densities with increasing age (Fig. 2B,C). Specifically, at P13 and P18, the NF-155 and Caspr floated up to fraction 4 (19% sucrose) (Fig. 2F), at P24 to fraction 3 (14.6% sucrose), and in adult optic nerves up to fraction 2 (10.6% sucrose). Thus, the myelin raft components CNP and GM1 achieve a common density already by P13, whereas the paranodal components NF-155 and Caspr become associated with a particularly low-density microdomain as a function of development.
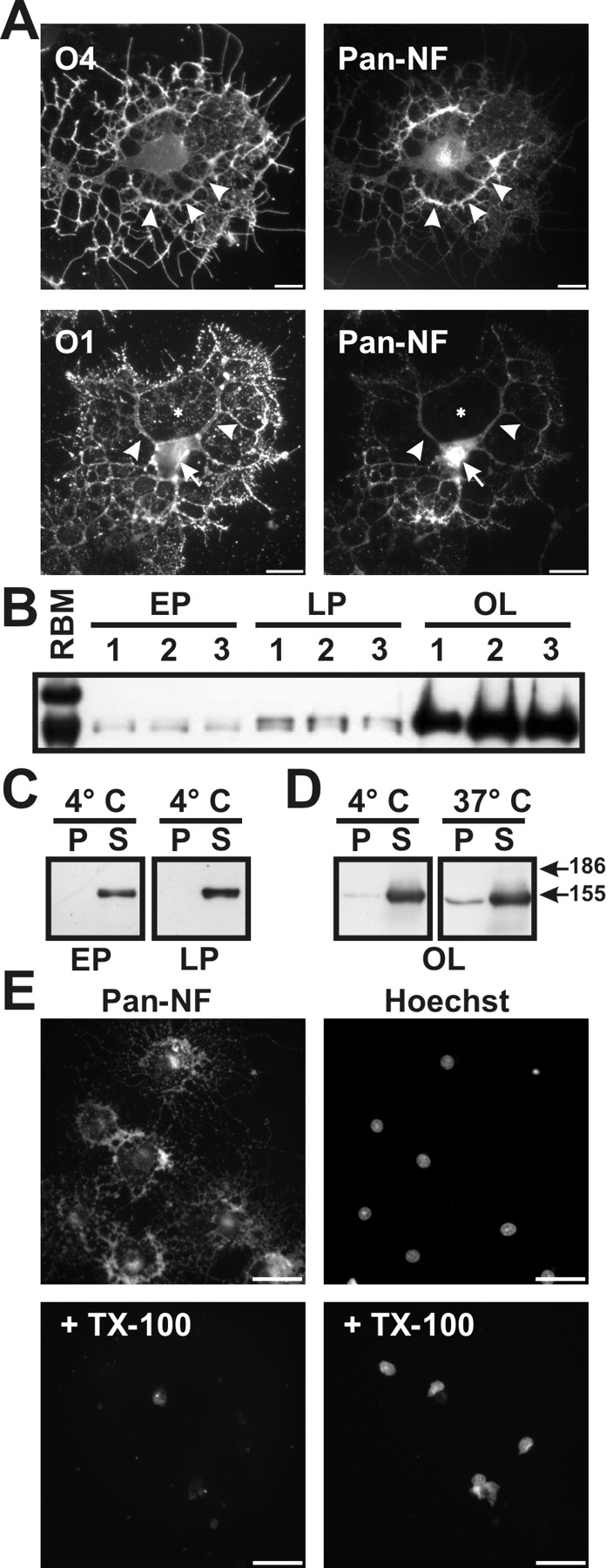
NF-155 is not associated with lipid rafts in cultured oligodendroctyes
Previous studies have shown that a variety of proteins exist in lipid rafts isolated from cultured oligodendrocytes even in the absence of compact myelin or axonal ligands (Kim et al., 1995; Kim and Pfeiffer, 2002; Baron et al., 2003), whereas others (e.g., myelin oligodendrocyte glycoprotein) require an extrinsic signal for raft inclusion (Marta et al., 2003). We used well established markers and morphology to define the developmental stages of oligodendrocyte differentiation (Pfeiffer et al., 1993; Bansal et al., 1996, 1999). In oligodendrocytes in culture, identified by immunolabeling with O4 and O1 antibodies, NF-155 was enriched in the major processes and cell body (Fig. 3A, arrowheads and arrow, respectively) but was not detected in “membrane sheets” (Fig. 3A, asterisk). Immunoblot analysis of early progenitor (EP), late progenitor (LP), and oligodendrocyte (OL) cultures showed that NF-155 was dramatically upregulated as oligodendrocytes entered terminal differentiation (Fig. 3B), whereas NF-186 was undetectable. Astrocytes in culture were devoid of NF immunoreactivity (data not shown).

NF-155 is expressed at high levels in cultured oligodendrocytes and is soluble in 1% TX-100. A, Immunolabeling of purified oligodendrocytes using pan-NF and O4 or O1 antibodies. Oligodendrocytes expressed NF-155 in the cell body (arrow) and major processes (arrowheads), but NF-155 was excluded from the membrane sheets (asterisk). B, Homogenates from three separate populations of early progenitors (EP), late progenitors (LP), and oligodendrocytes (OL) immunoblotted using pan-NF; NF-186 was not detected. C, D, 1% TX-100 solubilization of oligodendrocytes and its progenitors at either 4°C or 37°C. E, Cultured oligodendrocytes stained for NF-155 and Hoechst with and without previous detergent extraction of live cells in 1% TX-100 at 4°C. Scale bars, 20 μm.
Treatment of oligodendrocyte progenitors (EP, LP) with 1% TX-100 at 4°C effectively solubilized all of the NF-155 (Fig. 3C). In oligodendrocytes (OL), similar to the situation for optic nerve at P12 and P18, a small amount of NF-155 remained insoluble at both 4°C and 37°C (Fig. 3D); this insoluble material may represent intracellular NF-155 (Fig. 3A, arrow). Finally, treatment of live oligodendrocytes with 1% TX-100 at 4°C eliminated all detectable NF-155 immunoreactivity (Fig. 3E). These results suggest that, in cultured, premyelinating oligodendrocytes, NF-155 is not associated with lipid rafts.
NF localization during early myelination and node of Ranvier formation in the CNS
At the onset of myelination in the optic nerve (P6), NF immunoreactivity was mainly in oligodendrocyte cell bodies and major branches (Fig. 4A, arrowhead). By P8, when many axons become ensheathed by MBP-positive oligodendrocyte membrane (see below), NF was present also as diffuse immunoreactivity in the sheaths (Fig. 4B) as well as at high levels in the soma (Fig. 4B, arrowhead). In contrast to early myelination, adult optic nerves had intense NF immunoreactivity only at or near nodes of Ranvier (Fig. 4C, arrowhead).
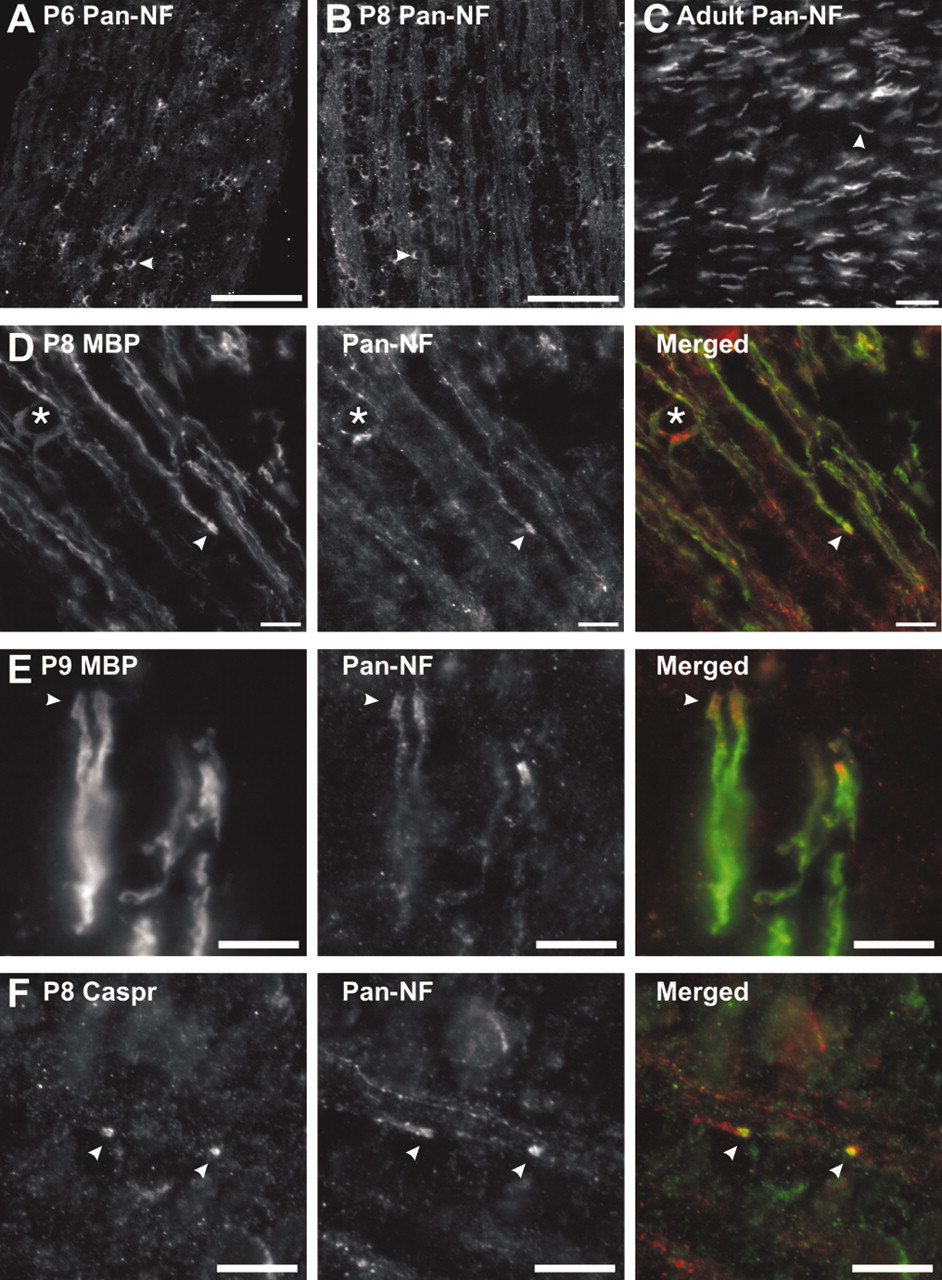
NF-155 expression and localization during myelination. A, B, pan-NF immunolabeling of P6 and P8 rat optic nerve. Pronounced NF-155 staining can be seen in the cell bodies of oligodendrocytes (arrowheads). C, Adult optic nerve labeled with pan-NF. Immunostaining is restricted to nodes and paranodes (arrowhead). D, Double labeling of rat optic nerves at P8 with antibodies against MBP (green) and pan-NF (red). NF-155 is localized at the ends of MBP-labeled oligodendrocyte processes (arrowheads). An oligodendrocyte cell body is identified by the asterisk. E, Double labeling of rat optic nerves at P9 with antibodies against MBP (green) and pan-NF (red). NF-155 is nested within MBP staining and is localized at the ends of the MBP-labeled oligodendrocyte processes (arrowheads). F, Double labeling of rat optic nerve at P8 using antibodies against Caspr (green) and pan-NF (red). Staining for NF-155 and Caspr is colocalized at forming paranodes (arrowheads). Scale bars: A, B, 100 μm; C, D, 10 μm; E, F, 5 μm.
To determine the time point at which NF-155 becomes localized to the ends of oligodendrocyte processes, we double-labeled optic nerves during early myelination and paranode formation (P8–P9) (Rasband et al., 1999), using anti-pan-NF and either anti-MBP or anti-Caspr. At P8 (Fig. 4D,F) and P9 (Fig. 4E), NF began to accumulate at the distal ends of myelinating oligodendrocyte processes (Fig. 4D,E, arrowheads) and colocalized with MBP (Fig. 4E, arrowhead) and Caspr (Fig. 4F, arrowheads; the site shown may represent two adjacent heminodes that define a nascent node of Ranvier). These results indicate that, in the CNS, one of the first events associated with node of Ranvier formation is the concomitant accumulation of NF-155 and Caspr at the ends of oligodendrocyte processes.
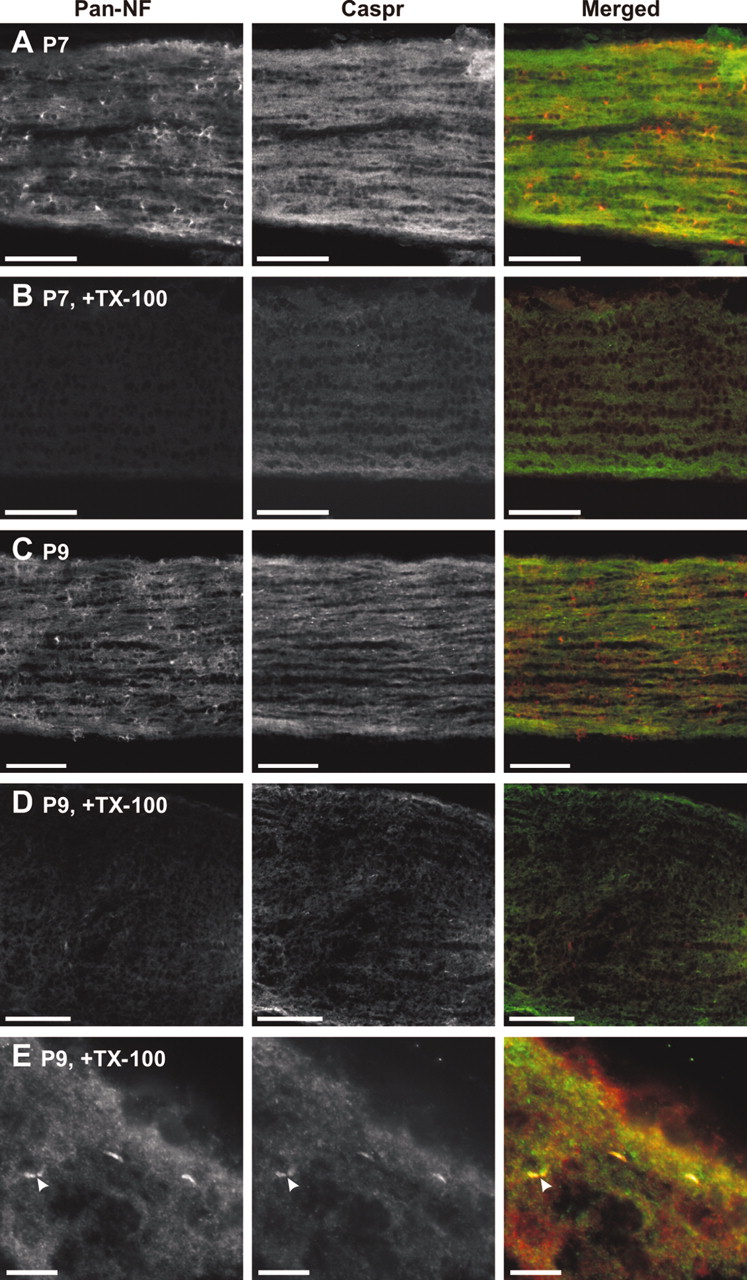
Detergent treatment of whole optic nerves reveals that paranodal proteins are resistant to extraction
We tested the solubility of NF-155 during early stages of myelination in whole P7 and P9 optic nerves by examining immunolabeled sections for retained Caspr and NF-155 after detergent treatment of nerves with 1% TX-100 at 4°C. At P7, NF-155 is expressed at high levels in oligodendroctyes and their processes, whereas Caspr immunoreactivity is still diffusely localized along axons (Fig. 5A). Detergent treatment at 4°C eliminated the NF immunoreactivity (Fig. 5B) and dramatically attenuated Caspr staining (Fig. 5B). At P9, when paranodes are just starting to form (P8–P9) (Fig. 4D–F), compared with untreated sections (Fig. 5C), there was an overall reduction in staining after detergent treatment (Fig. 5D). However, at higher magnification (Fig. 5E), it could be seen that clustered NF-155 and Caspr, localized at nascent paranodes, was not extracted from the nerve (arrowheads). Thus, before paranode formation and NF-155 clustering at these sites, NF-155 in situ is soluble in 1% TX-100 at 4°C. However, as Caspr and NF-155 accumulate at forming paranodes, these proteins become resistant to detergent extraction from the membrane.

NF-155 is readily extracted from optic nerves before paranode formation. Sections were double labeled using pan-NF (red) and antibodies against Caspr (green). At early stages of myelination, TX-100 penetrated completely through the nerve because of the fact that the tissue is much less dense than in adults and the nerves are much smaller. A, P7 optic nerve. B, P7 optic nerve after detergent treatment in 1% TX-100 at 4°C. C, P9 optic nerve. D, P9 optic nerve after detergent treatment in 1% TX-100 at 4°C. E, Detergent treatment of P9 rat optic nerve showed that NF-155 and Caspr immunoreactivity was retained at forming paranodes (arrowheads). Scale bars: A–D, 100 μm; E,10 μm.
Our biochemical results showed that, whereas NF-186 is soluble in 1% TX-100 at 4°C, a fraction of NF-155 is insoluble (Fig. 1C). We hypothesized that this reflects solubility differences in membrane proteins found in distinct domains of the node of Ranvier. To test this directly, whole adult rat optic nerves were treated for 1 hr with 1% TX-100 at either 4°C or 37°C and then immediately fixed, sectioned, and stained with antibodies against either nodal, paranodal, or juxtaparanodal proteins. Because of the density of the adult optic nerve, the detergent penetrated only a short distance into the nerve; this was apparent by the change in the opacity of the tissue. Staining for nodal Na+ channels and juxtaparanodal Kv1 K+ channels showed that both were completely extracted by 1% TX-100 at 4°C, because all immunoreactivity was lost in regions in which detergent had penetrated (Fig. 6A,B, arrows). Note that these same channels stained well in their appropriate locations in regions in which detergent had not penetrated (arrowheads). In contrast, staining for Caspr and pan-NF showed that, whereas the paranodal NF was not extracted (Fig. 6C, double arrowheads), the nodal immunoreactivity associated with NF-186 was completely eliminated by the detergent treatment, i.e., where detergent had not penetrated pan-NF immunoreactivity extended through the node (Fig. 6C, arrow). Detergent treatment of tissue at 37°C yielded poor quality of tissue preservation, but it appeared that NF and Caspr were completely extracted where the detergent had penetrated (Fig. 6D). These results indicate that NF-155 is associated with a unique, detergent-resistant membrane domain present at the paranodes.

Paranodal NF-155 and Caspr are resistant to detergent extraction from whole optic nerves. A, B, Detergent-treated rat optic nerves immunolabeled for voltage-dependent Na+ channels (red, Pan NaCh) or Kv1.2 K+ channels (red) and Caspr (green). Black arrowheads above the panels indicate the edge of the nerve. The line through each panel approximates the depth to which the detergent penetrated and the color of the fluorophore used. In regions in which detergent penetrated Na channels and Kv1.2 were efficiently extracted from nodes (A, arrow) and juxtaparanodes (B, arrow), respectively. Both were retained in regions in which detergent did not penetrate (A, B, arrowhead). C, Double labeling of detergent-treated optic nerves using antibodies against Caspr (green) and pan-NF (red). Paranodal NF-155 and Caspr are retained after detergent treatment (double arrowheads), but nodal NF-186 is extracted. In regions in which detergent did not penetrate, nodal NF-186 is retained (arrow). D, Detergent treatment at 37°C, followed by double labeling for neurofascin (red) and Caspr (green). Scale bars: A, B,10 μm; C, 5 μm; D, 20 μm.
Paranodal and raft-associated NF-155 is reduced in animals with paranodal abnormalities
Mice that are deficient in galactocerebroside and/or sulfatide (CGT-null and CST-null, respectively) exhibit alterations in paranodal architecture, including reduced Caspr clustering and everted paranodal loops (Dupree et al., 1999; Poliak et al., 2001; Ishibashi et al., 2002). Immunofluorescence staining of sciatic nerve (Fig. 7A) and optic nerve (Fig. 7B) showed that paranodal NF-155 is also reduced in CST-null mice or completely eliminated in CGT-null mice. We hypothesized that, in these mutants, the normal retention and/or partitioning of paranodal proteins into lipid rafts may be disrupted. To test this, detergent-insoluble material (1% TX-100 at 4°C) from WT and CGT-null mouse brains was examined on sucrose density gradients for NF-155, Caspr, contactin, and GM1. Compared with WT, in CGT-null mice, there was a reduction in the amount of NF-155 that floated to low densities (Fig. 7C, fractions 4–6). Furthermore, Caspr and contactin floated one fraction lower (Fig. 7C, compare fractions 3, 4). In contrast, the distribution of GM1 was unaltered. Although previous studies noted a reduction in the amount of NF-155 in myelin purified from CGT-null mice compared with WT (Menon et al., 2003), immunoblot of whole brain homogenates showed no difference (Fig. 7D). Similarly, WT and CGT-null oligodendrocytes in culture had comparable amounts of NF-155 per cell (Fig. 7E, note that cultures of CGT-null mouse brain yield a greater number of mature oligodendrocytes than WT cultures, as indicated by the increased level of CNP) (Bansal et al., 1999). Therefore, the reduction in raft-associated NF-155 is a consequence of the perturbation of myelin lipids rather than a reduction in the total amount of NF-155.
Another example of an animal with paranodal abnormalities and disrupted partitioning of NF-155 into lipid rafts is the myelin-deficient (md) rat, which has a mutation in the proteolipid protein (Plp) gene, resulting in dysmyelination and CNS-specific paranodal abnormalities that include the absence of clustered NF-155 and transverse bands (Rosenbluth, 1987; Grinspan et al., 1996; Gow et al., 1998; Arroyo et al., 2002). Immunoblot analysis of brain membrane homogenates from P18 md and WT rats showed that NF-186 was soluble in 1% TX-100 at 4°C (Fig. 7F). In contrast, whereas WT animals had an insoluble fraction of NF-155, the protein was completely soluble in the md rat. Thus, paranodal clustering of NF-155 and the association of NF-155 with lipid rafts are mechanistically related.
Discussion
How do paranodes form? The answer to this question is essential to understand myelination and node of Ranvier formation and maintenance. On the basis of our results, we propose a model (Fig. 8) that depends not only on protein–protein interactions but also on the formation of paranodal lipid rafts. During early myelination, NF-155, Caspr, and contactin are primarily unclustered in their respective membranes and are readily solubilized by 1% TX-100 at 4°C (Fig. 8A). During close contact between the axon and the oligodendrocyte at the forming paranodes, interaction between NF-155 and Caspr/contactin reciprocally stabilize these proteins and the lipid environment in which they reside (Fig. 8B). Thus, the initial binding of NF-155 and its axonal ligand(s) provides a “nucleation” site for the formation of a lipid raft protein adhesion complex. Recruitment of additional NF-155, Caspr, and contactin then occurs through thermodynamically favorable inclusion into lipid rafts, followed by further stabilization through trans interactions. Finally, NF-155 becomes phosphorylated (Jenkins et al., 2001) and the axonal Caspr/contactin complex becomes linked to the cytoskeleton through the binding of protein 4.1B (Fig. 8C) (Denisenko-Nehrbass et al., 2003). We propose that it is the sum of these protein–protein interactions within this lipid environment, rather than individual protein–protein interactions per se, that provides the basis for the strong adhesion complex at the paranode.

Model for lipid raft-dependent paranode formation. A, Caspr, contactin, and NF-155 are initially unclustered, not present in rafts, and readily solubilized by 1% TX-100. B, During binding to its axonal ligand in trans, NF-155 and its associated lipid environment are stabilized and become detergent insoluble. Additional NF-155 is recruited through thermodynamically favorable inclusion into the lipid environment surrounding the trans stabilized NF-155. C, Binding of additional Caspr/contactin (or another axonal ligand) to NF-155 then results in a stable paranodal structure with distinct lipid environments in both the axon and oligodendrocyte. This paranodal structure may then be involved in cytoskeletal interactions through protein 4.1B and signaling through phosphorylation of NF-155 (pTyr). GalC, Galactocerebroside.
Previous studies in cell lines revealed that NF is singly palmitoylated at a cytoplasmic cysteine near the transmembrane domain. Mutation of this cysteine (and the subsequent lack of palmitoylation) results in a shift in NF toward higher-density fractions when analyzed on density gradients (Ren and Bennett, 1998). This is consistent with the idea that the ability of NF-155 to function at the paranode might be coupled closely to its palmitoylation and association with lipid rafts. However, a single palmitate modification on proteins is thought to be insufficient for raft association (Melkonian et al., 1999). Palmitoylation is a reversible protein modification (el-Husseini Ael and Bredt, 2002). For example, palmitoylation of the synaptic scaffolding protein PSD-95 (postsynaptic density 95), a protein present in lipid rafts (Hering et al., 2003), “cycles” in response to glutamate receptor activity, thereby regulating its retention at synapses and synaptic strength (el-Husseini Ael et al., 2002). Thus, palmitoylation can occur at the cell membrane in response to local stimuli. With this in mind, an alternative explanation for the solubility of NF-155 before paranode formation may be that NF-155 does not become palmitoylated until after binding its axonal ligand(s). Future experiments designed to test this possibility should clarify the mechanism(s) responsible for NF-155 palmitoylation.
Why is ∼80% of the NF-155 from adult brain membranes soluble in 1% TX-100 at 4°C? The soluble NF-155 may be distributed diffusely in oligodendrocytes (not interacting with Caspr/contactin at paranodes) and at a concentration too low for detection by fluorescence microscopy. This would be analogous to the situation for Nav channels. Nav channels at nodes represent only ∼4–5% of the total pool within an axon, and the remainder are present in internodal regions at low densities undetectable by immunocytochemical methods (Shrager, 1989). Alternatively, NF-155 may be in a particularly “fragile” raft that is readily disrupted by 1% TX-100 at 4°C (Hakomori, 2002; Taylor et al., 2002).
Galactolipids, dysmyelination, and disrupted lipid rafts
Galactocerebroside and sulfatide comprise approximately onethird of myelin lipids. In addition to their passive role in myelin as “insulation,” galactolipids are important for oligodendrocyte differentiation (Bansal et al., 1999). Here we present a third role for galactolipids: to stabilize axoglial contacts by promoting the formation of NF-155-containing lipid rafts. This conclusion is consistent with the observation that loss of galactolipids results in disrupted paranodal junctions (Dupree et al., 1998; Poliak et al., 2001; Ishibashi et al., 2002; Menon et al., 2003) and a concomitant reduction in the NF-155 that associates with rafts. However, the precise role of galactolipids and how they interact with NF-155 to promote the formation of paranodal lipid rafts remains unknown.
The jimpy mouse and the md rat both have mutations in Plp, resulting in dysmyelination and early death. Interestingly, as for the contactin-, Caspr-, CGT-, and CST-null mice, both the jimpy mouse and md rat lack transverse bands and clustered paranodal proteins in the CNS (Rosenbluth, 1987; Arroyo et al., 2002; Jenkins and Bennett, 2002). How might this phenotype be explained in light of the results presented here? Recent work suggests that excess Plp results in dysregulation of lipid rafts and cholesterol accumulation in intracellular compartments of oligodendrocytes (Simons et al., 2002). Thus, one explanation for the similarities between the Plp mutants and the galactolipid-deficient mice might be that, when lipid rafts are disrupted, NF-155 cannot be stabilized and paranodal axoglial junctions cannot form.
The paranodal lipid–protein complex
Can the biochemical properties of NF-155 be explained by interactions with the cytoskeleton and/or axonal paranodal proteins? If the detergent insolubility of NF-155 were a result of interactions with cytoskeletal elements, then NF-155 should not float at very low densities of sucrose or be sensitive to extraction in 1% TX-100 at 4°C after pretreatment with saponin. Furthermore, the observation that, during development, NF-155 floats at progressively lower sucrose densities is exactly opposite to what would happen if detergent insolubility were a consequence of cytoskeletal interactions established during paranode formation. This does not mean that the cytoskeleton is irrelevant for paranode formation, just that it is not the basis of detergent insolubility. Indeed, a recent study by Marta et al. (2003) showed that an intact cytoskeleton was essential for partitioning of myelin proteins into lipid rafts.
Contactin, Caspr, and NF-155 colocalize at paranodes (Rios et al., 2000; Tait et al., 2000) and apparently interact (Charles et al., 2002). However, a recent study challenges this view and suggests that only NF-155 and contactin interact at paranodes and that Caspr inhibits their association (Gollan et al., 2003). One reason for the confusion surrounding the nature of the protein complex at paranodes may be that a direct demonstration that NF-155 interacts with Caspr and contactin in the brain, or with Contactin alone (for example by coimmunoprecipitation), has not been shown. This may be attributable to the fact that the relevant paranodal proteins are detergent insoluble, and more stringent solubilizing conditions disrupt the interactions. It is also possible that NF-155 interacts with some other as yet unidentified partner.
Interaction between NF-155 with GPI-anchored contactin (Rios et al., 2000) alone cannot explain our results because contactin and Caspr are still found at paranodes in CGT-null mice, albeit at reduced levels, whereas paranodal concentrations of NF-155 are not observed (Fig. 7A,B) (Poliak et al., 2001). Thus, both trans protein interactions and cis lipid interactions are required for association of NF-155 with paranodal lipid rafts. In support of the conclusion that paranodes are sites of trans interactions, including NF-155, Caspr, and contactin, we showed recently that it is possible to fractionate distinct domains of myelinated nerve fibers using a rigorous procedure to purify myelin (Menon et al., 2003). In these experiments, Caspr, contactin, and NF-155 all cofractionated at similar densities. Extending these results to CGT-null mice revealed a biochemical redistribution of these same proteins in a manner consistent with their delocalization in vivo. Interestingly, a dramatic reduction in the total amount of NF-155 after myelin fractionation was also shown. Although we found no difference in total NF-155 levels in brain homogenates or in oligodendrocytes in the experiments reported here, we do see reduced amounts of NF-155 on our sucrose gradients, a reduction that parallels our previous study (Menon et al., 2003). The reduction in NF-155 may be attributed to partitioning into detergent-soluble fractions in the CGT-null mouse. In the experiments of Menon et al. (2003), the reduction in NF-155 may be related to a specific redistribution of NF-155 into fractions that were not included in the final analysis of CGT-null myelin membranes.
Lipid rafts and node of Ranvier formation
A hallmark of the node of Ranvier is the specificity with which membrane proteins are localized and restricted to distinct domains. For example, Nav channels, Caspr, and Kv1 channels are all found within the axolemma clustered in very high densities but reside adjacent to one another in sharply defined, non-overlapping regions (Rasband and Trimmer, 2001). How is this accomplished? It is now clear that paranodes are important in this process (Dupree et al., 1999; Bhat et al., 2001; Boyle et al., 2001; Ishibashi et al., 2002). Current models place macromolecular sieves and diffusion barriers at the paranode, consisting mainly of cell adhesion molecules and scaffolding proteins, which restrict channels within domains (Rosenbluth, 1976; Pedraza et al., 2001; Salzer, 2003). Besides axolemmal diffusion barriers, some of these same investigators have postulated passive barriers that restrict the localization of proteins in the glial membrane. Our results are consistent with these ideas and are significant because they support the notion that the lipid environment at the paranode (in both the glial and axonal membranes) also contributes to this passive barrier, preventing the diffusion of membrane proteins laterally. Thus, ion channels and cell adhesion molecules may be maintained in their respective domains at and near the node of Ranvier not only through protein–protein interactions but also by virtue of the lipid environments from which they are excluded or in which they reside.
Footnotes
This work was supported by National Institutes of Health Grants NS44916 (M.N.R.), NS38878 (R.B.), NS10861 (S.E.P.), and NS41078 (S.E.P.). M.N.R. is a Young Investigator of the Wadsworth Foundation. We thank Drs. James Trimmer, Hiroko Baba, and Peter Brophy for antibodies and fusion proteins. We thank Drs. Koichi Honke and Brian Popko for CST-null and CGT-null mice, respectively. We thank Drs. Steve Scherer and Judith Grinspan for md rat brains. We thank Susan Winkler and Melissa Bryant for technical assistance.
Correspondence should be addressed to Dr. Matthew N. Rasband, Department of Neuroscience, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030. E-mail: rasband{at}uchc.edu.
DOI:10.1523/JNEUROSCI.5427-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243176-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}