

Abstract
Electrophysiological recordings have enabled identification of physiologically distinct yet behaviorally similar states of mammalian sleep. In contrast, sleep in nonmammals has generally been identified behaviorally and therefore regarded as a physiologically uniform state characterized by quiescence of feeding and locomotion, reduced responsiveness, and rapid reversibility. The nematode Caenorhabditis elegans displays sleep-like quiescent behavior under two conditions: developmentally timed quiescence (DTQ) occurs during larval transitions, and stress-induced quiescence (SIQ) occurs in response to exposure to cellular stressors. Behaviorally, DTQ and SIQ appear identical. Here, we use optogenetic manipulations of neuronal and muscular activity, pharmacology, and genetic perturbations to uncover circuit and molecular mechanisms of DTQ and SIQ. We find that locomotion quiescence induced by DTQ- and SIQ-associated neuropeptides occurs via their action on the nervous system, although their neuronal target(s) and/or molecular mechanisms likely differ. Feeding quiescence during DTQ results from a loss of pharyngeal muscle excitability, whereas feeding quiescence during SIQ results from a loss of excitability in the nervous system. Together these results indicate that, as in mammals, quiescence is subserved by different mechanisms during distinct sleep-like states in C. elegans.
SIGNIFICANCE STATEMENT Sleep behavior is characterized by cessation of feeding and locomotion, reduced responsiveness, and rapid reversibility. In mammals and birds, there are sleep states that have fundamentally different electrophysiology despite outwardly similar behavior. However, it is not clear whether behavioral sleep is a uniform state in animals in which electrophysiology is not readily possible. The nematode Caenorhabditis elegans displays sleep-like behavior under two conditions: during development and after exposure to environmental stressors. Here, we show that feeding and locomotion quiescence during these two sleep-like states are produced by different mechanisms. This provides the first identification of two mechanistically distinct forms of quiescence during sleep-like states in an invertebrate.
Introduction
Over the past 15 years, the study of small, nonmammalian genetic models such as zebrafish, fruit flies, and roundworms has yielded many insights into the mechanisms of sleep regulation (Crocker and Sehgal, 2010). Electrophysiological characterization of sleep states, which is routinely used to identify sleep and its substages in mammals, is difficult in these smaller organisms, so sleep in nonmammals has typically been defined as a behavioral state characterized by quiescence of feeding and locomotion with an elevated arousal threshold and rapid reversibility (Allada and Siegel, 2008; Zimmerman et al., 2008). However, electroencephalography (EEG) has revealed that, despite its apparent behavioral homogeneity, mammalian sleep is not a physiologically homogenous state (Loomis et al., 1937). For example, despite appearing behaviorally essentially identical, REM and non-REM sleep are regulated by drastically different circuit and neurochemical mechanisms (Siegel, 2005).
The presence of distinct states of mammalian sleep raises the important question of whether behaviorally indistinguishable sleep states can be physiologically distinct in animals for which EEG is not feasible. In the nematode Caenorhabditis elegans, two behaviorally indistinguishable sleep-like states have been observed. Developmentally timed quiescence (DTQ), or lethargus, occurs during larval transitions (Raizen et al., 2008), and is coupled to a larval timing mechanism involving LIN-42, a homolog of the circadian timing protein PERIOD (Jeon et al., 1999; Monsalve et al., 2011). Stress-induced quiescence (SIQ) follows exposure to conditions that induce cellular stress and requires the epidermal growth factor (EGF) LIN-3 (Hill et al., 2014). Both states are characterized by a cessation of locomotion and feeding and reduced responsiveness to weak sensory stimuli, and animals in DTQ as well as those in SIQ resume movement but not feeding in response to a strong mechanical stimulation (Cassada and Russell, 1975; Jones and Candido, 1999; Raizen et al., 2008; Hill et al., 2014). Sensory neuron Ca2+ levels are decreased both during DTQ and after overexpression of EGF (mimicking SIQ), demonstrating that the physiology immediately proximal to increased arousal threshold is similar between these states (Cho and Sternberg, 2014). The molecular genetic regulation of DTQ (Choi et al., 2013; Nelson and Raizen, 2013; Schwarz and Bringmann, 2013; Singh et al., 2014) has several similarities to the regulation of circadian-timed sleep in Drosophila (Renn et al., 1999; Hendricks et al., 2001; Joiner et al., 2006; Parisky et al., 2008; Guo et al., 2011; He et al., 2013), whereas the molecular genetic regulation of SIQ (Nelson et al., 2014) has similarities to the regulation of stress-induced sleep in Drosophila (Zimmerman et al., 2008; Lenz et al., 2015), demonstrating conserved mechanisms of quiescence regulation between C. elegans and other animals.
Recent evidence suggests that there might be differences in the underlying molecular and circuit mechanisms that regulate behavioral quiescence during DTQ and SIQ. For example, disruption of the function of the ALA interneuron results in severely defective quiescence after cellular stress (Hill et al., 2014), but only minor changes in quiescence during larval development (Van Buskirk and Sternberg, 2007), and key neurotransmitters used by the ALA interneuron to induce quiescence, the seven FMRFamide-like neuropeptides encoded by the flp-13 gene, are required for normal quiescence during SIQ but not DTQ (Nelson et al., 2014).
In this study, we aimed to elucidate differences and similarities in the circuit and molecular mechanisms underlying behavioral quiescence during DTQ and SIQ. Our results suggest that, despite the outwardly identical appearance of DTQ and SIQ, feeding and locomotion inhibition arise from fundamentally distinct circuit and molecular mechanisms during these states.
Materials and Methods
Worm strains and cultivation.
We performed all experiments with hermaphrodites. Unless otherwise specified, animals were cultivated on the surface of NGM agar in a 20°C incubator. To conditionally overexpress nlp-22 and flp-13, we used the strains NQ251 qnIs142[Phsp-16.2::nlp-22; Phsp-16.2::GFP; Pmyo-2::mCherry; unc-119(+)] (Nelson et al., 2013) and NQ570 qnIs303[Phsp-16.2::flp-13; Phsp-16.2::GFP; Prab-3::mCherry] (Nelson et al., 2014), respectively. The mutant strains that we used in our candidate screen are KG421 gsa-1(ce81gf) I (Schade et al., 2005), KG518 acy-1(ce2gf) III (Schade et al., 2005), KG744 pde-4(ce268) II (Charlie et al., 2006), KG532 kin-2(ce179) X (Schade et al., 2005), JT734 goa-1(sa734) I (Robatzek and Thomas, 2000), PS998 goa-1(sy192dn) I (Mendel et al., 1995), CG21 egl-30(tg26gf) I (Doi and Iwasaki, 2002), JT609 eat-16(sa609) I (Hajdu-Cronin et al., 1999), KP1097 dgk-1(nu62) X (Nurrish et al., 1999), NL594 gpa-12(pk322) X (Jansen et al., 1999), MJ500 tpa-1(k501) IV (Tabuse and Miwa, 1983), DA467 eat-6(ad467) V (Avery, 1993), MT6129 egl-19(n2368gf) IV (Lee et al., 1997), VC223 tom-1(ok285) I (Dybbs et al., 2005), NM1968 slo-1(js379) V (Wang et al., 2001), DR1089 unc-77(e625gf) IV (Brenner, 1974), MC339 unc-64(md130) III (Saifee et al., 1998), CB5 unc-7(e5) X (Brenner, 1974), MT9455 tbh-1(n3247) X (Alkema et al., 2005), and MT1074 egl-4(n479) IV (Trent et al., 1983). Other strains we used include N2 (Brenner, 1974), VC1669 aptf-1(gk794) II (Turek et al., 2013), NQ596 nlp-22(gk509904) X (Nelson et al., 2013), NQ602 flp-13(tm2427) IV (Nelson et al., 2014), NQ670 qnEx95[Phsp16.2::nlp-22; Pmyo-2::mCherry; unc-119(+)] (Nelson et al., 2013), NQ230 ceh-17(np1) I; qnEx95[Phsp16.2::nlp-22; Pmyo-2::mCherry; unc-119(+)] (Nelson et al., 2013), NQ777 ceh-17(np1) I; qnIs303[Phsp-16.2::flp-13; Phsp-16.2::GFP; Prab-3::mCherry] (Nelson et al., 2014), ZM3265 lin-15(n765ts) X; zxIs6[Punc-17::ChR2(H134R)::YFP; lin-15(+)] V (Liewald et al., 2008), YX11 vsIs48[Punc-17::GFP] X; zxIs6[Punc-17::ChR2(H134R)::YFP; lin-15(+)] V (Trojanowski et al., 2014), YX62 qnIs142[Phsp-16.2::nlp-22; Phsp-16.2::GFP; Pmyo-2::mCherry; unc-119(+)]; vsIs48[Punc-17::GFP] X; zxIs6[Punc-17::ChR2(H134R)::YFP; lin-15(+)] V, YX63 qnIs303[Phsp-16.2::flp-13; Phsp-16.2::GFP; Prab-3::mCherry]/+; vsIs48[Punc-17::GFP] X; zxIs6[Punc-17::ChR2(H134R)::YFP; lin-15(+)] V, SJU8 kin-2(ce179); qnEx448[Punc-17::kin-2(+); Pmyo-3::mCherry], NQ902 kin-2(ce179) X; qnIs303[Phsp-16.2::flp-13; Phsp-16.2::GFP; Prab-3::mCherry]; qnEx448[Punc-17::kin-2(+); Pmyo-3::mCherry], NQ903 kin-2(ce179) X; qnIs142[Phsp-16.2::nlp-22; Phsp-16.2::GFP; Pmyo-2::mCherry; unc-119(+)]; qnEx448[Punc-17::kin-2(+); Pmyo-3::mCherry], NQ820 qnEx390[Pmyo-2::GCaMP6s::SL2::dsRed; rol-6(d)], CX16557 kyIs5640[Pmyo-2::Chrimson; Pelt-2::his-4.4-mCherry], and NQ904 qnEx390[Pmyo-2::GCaMP6s::SL2::dsRed; rol-6(d)]; kyIs5640[Pmyo-2::Chrimson; Pelt-2::his-4.4-mCherry].
Acute heat shock.
Unless otherwise indicated, we triggered stress-induced quiescence by acute heat shock, performed as described in “protocol 1” by Nelson et al. (2014). We heat-shocked day one adults at 35°C in a water bath for 30 min on standard NGM agar plates seeded with DA837. We then counted pumping rate as described previously (Raizen et al., 2012) for individual worms between 35 and 45 min after the end of heat shock.
Conditional neuropeptide overexpression.
Unless otherwise indicated, to induce neuropeptide overexpression we placed day one adults on NGM agar plates seeded with DA837 in a 33°C water bath for 30 min and allowed them to recover at room temperature for 2–3 h, at which time the effects of acute heat shock had worn off (Nelson et al., 2013, 2014). To screen for mutants with abnormal flp-13- or nlp-22-induced quiescence, we tested all of the candidate strains overexpressing one neuropeptide gene on the same day with the experimenter blinded to the genotype of the strains. We counted the number of pumps per 20 s and the number of body bends per 20 s (where one full back and forth movement of the anterior body was counted as one body bend) for each of 12–15 worms. We tripled each value to convert to pumps per minute or body bends per minute. For experiments testing the effect of ceh-17 mutation on nlp-22 and flp-13 overexpression-induced quiescence, we used NQ670 and NQ570 as the respective control strains.
Effects of 5-HT.
To test the effect of 5-HT on feeding after neuropeptide overexpression, during SIQ, or during DTQ, we immobilized worms on agarose pads containing 10 mm 5-HT as described previously (Trojanowski et al., 2014).
Single neuron optogenetics.
We performed optogenetic stimulation of single neurons after neuropeptide overexpression as described previously (Trojanowski et al., 2014, Trojanowski and Fang-Yen, 2015) except that the worms were first submitted to the conditional neuropeptide overexpression protocol, as described above. We examined worms between 2 and 3 h after heat shock.
Wide-field optogenetics.
To stimulate pharyngeal neurons during SIQ or DTQ, we grew ZM3265 worms on OP50 containing all-trans retinal (ATR) as described previously (Trojanowski et al., 2014). For SIQ, we performed acute heat shock as described above except that plates seeded with ATR-containing OP50 were used. We then illuminated these worms with blue light (using GFP optics, irradiance = 0.66 mW/mm2) from a mercury halide lamp on a Leica MZ16F stereomicroscope and quantified pump rate in 20 s intervals (Raizen et al., 2012). We tripled these values to calculate pumps per minute.
Pharyngeal muscle optogenetics.
We performed optogenetic stimulation of pharyngeal muscle during wake and DTQ similarly to stimulation of single neurons, with some modifications because Chrimson was not tagged with a fluorescent protein. First, we stimulated the entire head region of the worm instead of only the pharynx by setting the digital micro mirror device to illuminate the entire field of view. Next, we used a pulse generator (Stanford Research Systems DG535) to generate 5 V pulses of a specified duration and frequency and connected this output to the modulation input of the laser to control stimulus timing. To identify the stimulus interval on the camera, we attached a red collimated LED to the same pulse generator output via a relay and directed this light toward the objective, allowing us to detect a small increase in bright-field intensity when the laser was on. All other aspects of the experiment were unchanged.
Ca2+ imaging.
We performed experiments using GCaMP6s the same way as pharyngeal muscle optogenetics experiments with slight modifications. With the laser continuously illuminating the field of view, we recorded the Pmyo-2::GCaMP6s signal for about 30 s (1000 frames at 30 frames/s). We then identified the maximum and minimum fluorescence values in a region of the metacorpus during this time and calculated the difference between these values to determine the maximum fluorescence change during this interval. The laser power we used here was the same as that for optogenetics experiments.
Strain construction.
To rescue the kin-2 defect specifically in cholinergic neurons, we used an Eppendorf FemtoJet microinjection system on a Leica DMIRB inverted differential interference contrast microscope to inject Punc-17::kin-2::unc-54utr in CFJ151 at 25 ng/μl in combination with 5 ng/μl pCFJ104(Pmyo-3::mCherry). We created the Punc-17::kin-2::unc-54utr construct using standard Gateway cloning procedures. Briefly, we recombined Punc-17 in pDONR P4P1r and kin-2 cDNA in pDONR221 into the CFJ151 destination vector. To create qn390, we used overlap extension PCR (Nelson and Fitch, 2011) to generate Pmyo-2::GCaMP6s::SL2::dsRed. We then injected Pmyo-2::GCaMP6s::SL2::dsRed at 20 ng/μl and pRF4 at 100 ng/μl into N2. We created Pmyo-2::Chrimson by subcloning the myo-2 promoter into the pSM-Chrimson vector (Gordus et al., 2015) using FseI and AscI. To generate kyIs5640, we injected Pmyo-2::Chrimson at 0.4 ng/μl in combination with 5 ng/μl Pelt-2::his-4.4-mCherry. The resulting extrachromosomal array spontaneously integrated during the course of strain maintenance (Mello et al., 1991).
To construct the strains used to test gene mutation effects on the behavioral quiescence conferred by neuropeptide overexpression, we followed the red fluorescent reporter on the transgene array (Pmyo-2::mCherry or Prab-3::mCherry) and the visible phenotypes of the gene mutation. In cases in which the phenotype of the gene mutation was difficult to identify, we made use of balanced chromosomes marked with GFP fluorescence (Edgley et al., 2006).
Statistics.
For the candidate mutant screens, we used Dunnett's multiple-comparison tests to determine which mutants were significantly different from the control. For the single neuron optogenetics, we performed a one-way ANOVA to determine whether there was an effect of neuron stimulation on pumping rate. For stress-induced quiescence experiments, we used a Mann–Whitney test without post hoc correction to determine which mutants were significantly different from the control. See figure legends for details.
Results
Feeding quiescence during SIQ does not require pathways that regulate DTQ
Two classes of neurons are known to be required for normal quiescence during DTQ: the paired RIA neurons, which release the neuropeptide NLP-22, and the RIS neuron, which requires the AP2 transcription factor APTF-1 (Nelson et al., 2013; Turek et al., 2013). The ALA neuron, which secretes the neuropeptides encoded by flp-13, is the only neuron known to be required for SIQ (Van Buskirk and Sternberg, 2007; Hill et al., 2014; Nelson et al., 2014) (Fig. 1A). Although SIQ and DTQ are behaviorally indistinguishable, it is unclear to what extent there is overlap between the mechanisms of behavioral quiescence during these states.

Quiescence during DTQ and SIQ is mediated by different neuropeptides. A, Schematic representing the known pathways that control quiescence during DTQ and SIQ. In DTQ, the RIA neurons release NLP-22, whereas the RIS neuron releases an unidentified peptide. The aptf-1 gene is required for the function of the RIS neuron and the ceh-17 gene is required for the function of the ALA neuron. During SIQ, the ALA neuron releases the FLP-13 neuropeptides. It is unknown how these neuropeptides lead to behavioral quiescence. B, Feeding quiescence during SIQ does not require pathways that regulate DTQ. Mutants defective in RIA and RIS signaling (nlp-22 and aptf-1 mutants, respectively) have normal feeding quiescence during SIQ. Each point represents an observation from one worm and the horizontal bar represents the median of each group. n = 15 for each group. Statistical significance was calculated using the Mann–Whitney test. *p < 0.05.
Although flp-13 mutants have defective feeding and locomotion quiescence after cellular stress induction, their quiescence during larval transitions is normal (Nelson et al., 2014). To further compare the mechanisms of quiescence during DTQ and SIQ, we examined stress-induced feeding quiescence in mutants with defective quiescence during larval transitions. Although we detected differences in SIQ feeding quiescence between wild-type worms and flp-13 mutants, neither nlp-22 mutants nor aptf-1 mutants displayed a defect in SIQ feeding quiescence, demonstrating that these DTQ-promoting factors are not required for quiescence during SIQ (Fig. 1B). Therefore, some factors required for quiescence during SIQ are not necessary for quiescence during DTQ and vice versa.
Overexpression of flp-13 or nlp-22 neuropeptide genes inhibits feeding and locomotion
We next sought to identify conserved pathways that regulate quiescence downstream of the ALA and RIA interneurons during SIQ and DTQ, respectively. Quiescence during SIQ requires the release of the FMRFamide-like FLP-13 neuropeptides from the ALA interneuron (Nelson et al., 2014) and quiescence during DTQ requires the release of the NLP-22 neuropeptide, which is structurally similar to the mammalian neuropeptide Neuromedin S, from the RIA interneuron. nlp-22 mRNA cycles in phase with mRNA of lin-42 (Jeon et al., 1999), the C. elegans homolog of the PERIOD gene, and an nlp-22 loss-of-function mutation decreases quiescence during DTQ (Nelson et al., 2013). Likewise, flp-13 mRNA is increased after organismal stress and a flp-13 loss-of-function mutation decreases quiescence during SIQ (Nelson et al., 2014; Fig. 1B).
We conditionally overexpressed these neuropeptides under control of the heat shock promoter (Phsp-16.2::flp-13 and Phsp-16.2::nlp-22) to robustly induce the quiescent behavioral states mimicking SIQ and DTQ (Nelson et al., 2013, 2014). This approach, similar to one recently used to study somnogenic neuropeptides in zebrafish (Woods et al., 2014), was selected for four reasons. First, in contrast to chronic loss-of-function experiments using genetic mutants, these conditional overexpression experiments are not subject to redundancy, compensation, or other developmental or physiological defects that may be part of the loss-of-function phenotype. Second, expressing the neuropeptides at supraphysiological levels is likely to activate all or nearly all of the receptors of these neuropeptides, so their effects will be limited only by the expression patterns of their receptors. Third, prolonged overexpression of the somnogenic peptides provides the experimental advantage of inducing a behavioral state lasting longer than the endogenous behavior, facilitating the identification of defects in these behaviors and characterization of downstream signaling pathways. Finally, the temporal control afforded by this conditional approach allowed us to compare quiescence induced by the two peptides in the same early adult stage, minimizing effects of developmental time on behavior. It is important to note that, whereas flp-13 and nlp-22 have been implicated in SIQ and DTQ, respectively, it is unlikely that overexpression of these neuropeptide genes faithfully recapitulates all aspects of these sleep-like states.
Since SIQ is triggered by acute activation of the ALA neuron by heat shock, it is possible that the somnogenic effects of overexpressing these neuropeptide genes using the heat-shock-inducible promoter are affected by ALA activation and the quiescence we observe does not reflect purely the somnogenic actions of the neuropeptides. Although we attempt to avoid effects of SIQ in our neuropeptide overexpression experiments by observing the animals at least 2 h after heat exposure and by inducing gene expression with a less stressful 33°C stimulus (Nelson et al., 2014), it remains possible that residual effects of SIQ influence the behavior of these animals. To test for this possibility, we overexpressed nlp-22 and flp-13 in the ceh-17 mutant background, which lacks a functional ALA neuron (Pujol et al., 2000). We found that worms with a ceh-17 mutation were indistinguishable from controls 2 h after transgene induction with respect to both locomotion (nlp-22 overexpression: control: 1.0 ± 0.2 body bends per minute (bbpm), ceh-17: 1.6 ± 0.3 bbpm, p = 0.51; flp-13 overexpression: control: 0.6 ± 0.2 bbpm, ceh-17: 1.0 ± 0.2 bbpm, p = 0.54; mean ± SEM, n = 15) and feeding (nlp-22 overexpression: control: 30.6 ± 4 pumps per minute (ppm), ceh-17: 15.2 ± 1.9 ppm, p = 0.26; flp-13 overexpression: control: 20.4 ± 4.1 ppm, ceh-17: 16.2 ± 2.1 ppm, p = 0.72; mean ± SEM, n = 15), demonstrating that ALA activation is not required for the quiescence observed after flp-13 or nlp-22 overexpression.
Activation of the Gαq or Gαs pathways inhibits both flp-13- and nlp-22-induced locomotion quiescence
To identify conserved genes that regulate locomotion quiescence induced by overexpression of flp-13 or nlp-22, we crossed strains overexpressing these neuropeptides into strains containing mutations in candidate genes with vertebrate homologs. Candidate genes were those that cause increased neurotransmitter release, increased membrane excitability, hyperactive locomotion, defective behavioral quiescence, or resistance to anesthetics. We focused in particular on strains with increased neurotransmitter release due to increased Gαq and Gαs signaling (Fig. 2, adapted from Perez-Mansilla and Nurrish, 2009), since these mutants show hyperactive locomotion and some have defects in DTQ locomotion quiescence (Belfer et al., 2013; Iwanir et al., 2013; Schwarz and Bringmann, 2013; Singh et al., 2014), and therefore they might be resistant to the effects of somnogenic neuropeptides.
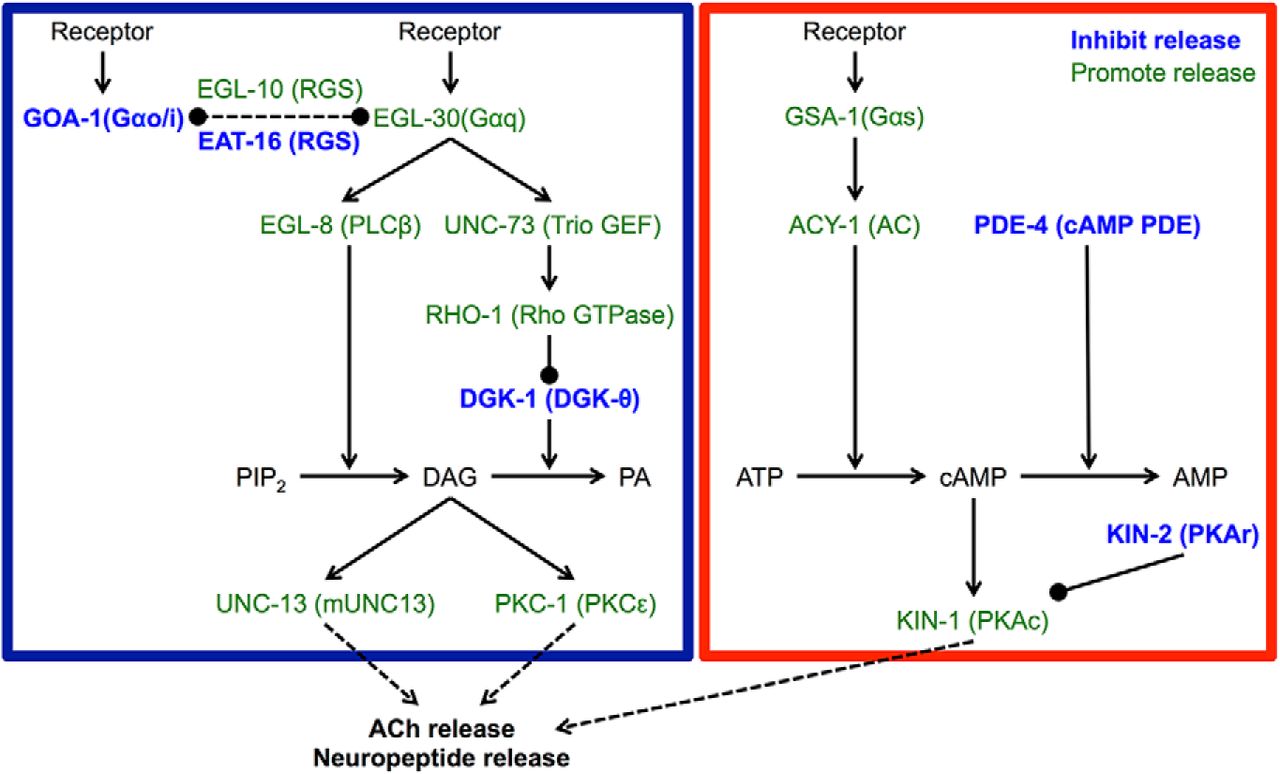
Schematic representation of the Gαq and Gαs pathways that regulate neurotransmitter secretion in C. elegans. Gαq signaling causes increased DAG levels, whereas the Gαs pathway causes increased cAMP levels. C. elegans protein names are shown in uppercase letters and mammalian homolog names are shown in parentheses. The blue box surrounds the Gαq pathway and the red box surrounds the Gαs pathway. Proteins labeled in blue inhibit neurotransmitter release and those in green promote release. Lines ending in arrows are positive regulation; lines ending in balls are negative regulation. Figure modeled after Perez-Mansilla and Nurrish (2009).
The Gαq signaling pathway acts antagonistically to Gαo signaling in C. elegans and promotes neurotransmitter release by increasing diacylglycerol (DAG) levels (Miller et al., 1999), whereas the Gαs pathway acts downstream of DAG to increase neurotransmitter release (Reynolds et al., 2005). Hyperactivation of the Gαq signaling pathway can be achieved either directly, using a gain-of-function mutation in the Gαq gene egl-30 (Doi and Iwasaki, 2002), or indirectly, using loss-of-function mutations in the Gαo gene goa-1 (Miller et al., 1996), the regulator of G-protein signaling gene eat-16 (Hajdu-Cronin et al., 1999), or the diacylglycerol kinase theta gene dgk-1 (Miller et al., 1999). Hyperactivation of the Gαs signaling pathway can also be achieved either directly, using gain-of-function mutations in the Gαs gene gsa-1 (Schade et al., 2005) or the adenylate cyclase type IX gene acy-1 (Schade et al., 2005), or indirectly, using loss-of-function mutations in the phosphodiesterase-4 gene pde-4 (Charlie et al., 2006) or the cAMP-dependent protein kinase (PKA) regulatory subunit gene kin-2 (Schade et al., 2005).
We also examined strains with mutations that increased neurotransmitter release by other means, including loss-of-function mutations in the tomosyn gene tom-1 (Dybbs et al., 2005) and the BK channel gene slo-1 (Wang et al., 2001), as well as mutations that generally increased membrane excitability, including a gain-of-function mutation in the l-type Ca2+ channel gene egl-19 (Lee et al., 1997) and a loss-of-function mutation in the Na+/K+ transporter α-subunit gene eat-6 (Davis et al., 1995). We also tested strains with mutations that confer resistance to anesthesia, including a gain-of-function mutation in the NALCN channel subunit gene unc-77 (Humphrey et al., 2007; Morgan et al., 2007; Yeh et al., 2008), a loss-of-function mutation in the innexin gene unc-7 (Starich et al., 1996), and a neomorphic mutation affecting the syntaxin gene unc-64 (van Swinderen et al., 1999). Finally, we tested strains with mutations that caused defects in various types of behavioral quiescence, including loss-of-function mutations in the cGMP-dependent protein kinase (PKG) gene egl-4 (Raizen et al., 2008), the Gα12 gene gpa-12 (van der Linden et al., 2003), the protein kinase C epsilon (PKC-ε) gene tpa-1 (van der Linden et al., 2003), and the tyramine β-hydroxylase gene tbh-1 (Alkema et al., 2005). Table 1 lists all mutants tested and their effects on relevant signaling pathways and cell physiology.
Mutations that increase Gαq or Gαs signaling suppressed the locomotion quiescence induced by overexpression of either flp-13 or nlp-22 (Fig. 3A,B, Table 2). However, whereas overexpression of nlp-22 in mutants with activated Gαs signaling caused qualitatively normal locomotion, overexpression of flp-13 in the same mutants caused aberrant locomotion characterized by uncoordinated twitches and accordion-like contractions. Gαq pathway genes are expressed in neurons, but not in body wall muscles (Nurrish et al., 1999; Bastiani et al., 2003), and cholinergic neuron stimulation during DTQ causes contraction of body wall muscle (Dabbish and Raizen, 2011), suggesting that FLP-13 and NLP-22 neuropeptides both act on neurons to inhibit locomotion.

Activation of Gαq or Gαs pathways impairs locomotion quiescence caused by flp-13 or nlp-22 overexpression. A, Mutations that increase Gαq or Gαs signaling impair locomotion quiescence after flp-13 overexpression. B, Mutations that increase Gαq or Gαs signaling impair locomotion quiescence after nlp-22 overexpression. A and B, Each bar represents the mean ± SEM of body bends for 12–15 worms during a 20 s window. Each bar represents the data obtained for a different mutant strain containing the designated overexpression transgene. For detailed genotypes and data, see Table 2. gf, Gain-of-function mutation; dn, dominant-negative mutation; the others are loss-of-function mutations. Statistical significance was calculated using one-way ANOVA followed by Dunnett's multiple-comparison tests. *p < 0.05, †p < 0.01, ‡p < 0.001. C, Rescuing kin-2 function in cholinergic neurons using the unc-17 promoter rescues the effects of the kin-2 mutation on locomotion quiescence after flp-13 or nlp-22 overexpression. Con, flp-13 overexpression and nlp-22 overexpression control strains NQ570 or NQ251, respectively. n = 13–15. Each bar represents mean ± SEM. Statistical significance was calculated using one-way ANOVA followed by Dunnett's multiple-comparison tests. *p < 0.05, **p < 0.01, ***p < 0.001.
To test whether these neuropeptides act via cholinergic neurons to regulate locomotion, we rescued the function of kin-2, the PKA regulatory subunit, in cholinergic neurons using the unc-17 promoter and assessed the effect of neuropeptide overexpression on locomotion. We found that, after restoring kin-2 function in cholinergic neurons, locomotion was strongly inhibited after overexpression of either neuropeptide gene (Fig. 3C), demonstrating that decreased PKA signaling in cholinergic neurons is important for inhibiting locomotion downstream of these neuropeptides. However, because we observed different locomotion phenotypes after overexpressing flp-13 or nlp-22 in backgrounds with activated Gαs signaling, these neuropeptides likely act on different molecular targets and/or different subsets of cholinergic neurons.
Feeding quiescence induced by flp-13 overexpression, but not nlp-22 overexpression, is suppressed by activation of the Gαq or Gαs pathways
C. elegans feeds by rhythmic contraction of its pharynx, a neuromuscular pump possessing 20 neurons of 14 types (Albertson and Thomson, 1976). To identify conserved signaling pathways that regulate feeding quiescence downstream of flp-13 and nlp-22, we overexpressed these neuropeptide genes in the mutant backgrounds described above and in Table 1. As with locomotion quiescence, feeding quiescence induced by flp-13 overexpression was strongly suppressed by mutations that increase neurotransmitter release by activating either the Gαq or Gαs signaling pathways (Fig. 4A, Table 2), which are present in all or nearly all neurons (Ségalat et al., 1995; Korswagen et al., 1997; Bastiani et al., 2003). In these mutants, feeding rates were substantially higher than the control feeding rate, even though in the absence of neuropeptide overexpression, many of these strains feed at rates similar to that of the control (Song and Avery, 2012). In addition, some mutations that increase neurotransmitter release by other mechanisms or confer resistance to anesthesia also suppressed the feeding quiescence caused by flp-13 overexpression (Fig. 4A). In contrast, none of the mutants tested suppressed the feeding quiescence induced by nlp-22 overexpression (Fig. 4B). These results indicate that FLP-13 and NLP-22 neuropeptides promote feeding quiescence via distinct molecular mechanisms.

Activation of Gαq or Gαs pathways impairs feeding quiescence caused by flp-13 overexpression, but not that caused by nlp-22 overexpression. A, Mutations that increase Gαq or Gαs signaling impair feeding quiescence after flp-13 overexpression relative to the control flp-13 overexpression strain. B, Mutations that increase Gαq or Gαs signaling do not affect feeding rate after nlp-22 overexpression relative to the control nlp-22 overexpression strain. gf, Gain-of-function mutation; dn, dominant-negative mutation; the others are loss-of-function mutations. Each bar represents the mean ± SEM of pharyngeal pumps for 12–15 worms during a 20 s window for a different mutant strain containing the designated overexpression transgene. Statistical significance was calculated using a one-way ANOVA followed by Dunnett's multiple-comparison tests. *p < 0.05, †p < 0.01, ‡p < 0.001.
FLP-13 inhibits feeding by acting on neurons; NLP-22 inhibits feeding downstream of neurons
Because flp-13-induced feeding quiescence is suppressed by mutations that increase neurotransmitter release, but nlp-22-induced feeding quiescence is not, we hypothesized that the FLP-13 neuropeptides promote feeding quiescence by acting on the nervous system and that the NLP-22 neuropeptide promotes feeding quiescence by acting directly on the pharyngeal muscle. To test this hypothesis, we rescued the function of kin-2, the PKA regulatory subunit, in cholinergic neurons and assessed the effect of neuropeptide overexpression on feeding rate. We found that restoring kin-2 function in cholinergic neurons restored pumping levels after flp-13 overexpression to that of the control, whereas restoring kin-2 function in cholinergic neurons after nlp-22 overexpression did not affect feeding rate (Fig. 5A).

FLP-13 inhibits pharyngeal pumping by acting on neurons, but NLP-22 inhibits feeding downstream of motor neuron excitation. A, Rescuing kin-2 function in cholinergic neurons using the unc-17 promoter rescues the effect of the kin-2 mutation on feeding quiescence induced by flp-13 overexpression, but does not affect feeding quiescence induced by nlp-22 overexpression. Con, flp-13 overexpression and nlp-22 overexpression control strains NQ570 or NQ251 respectively. n = 13–15. Each bar represents mean ± SEM. Statistical significance was calculated using one-way ANOVA followed by Dunnett's multiple-comparison tests. B, In immobilized worms, 5-HT blocks the inhibitory effect of flp-13 overexpression on feeding, but not that of nlp-22 overexpression on feeding. n = 10. Each bar represents mean ± SEM. Statistical significance was calculated using Student's t test. C, flp-13 overexpression and nlp-22 overexpression cause similar effects on feeding in worms in the presence of food when overexpressed to similar degrees. n = 9–15. Each point represents mean ± SEM. Statistical significance was calculated using a two-way ANOVA. D, The difference in the effects of 5-HT on feeding rate in immobilized worms after flp-13 overexpression and nlp-22 overexpression is not affected by the degree of neuropeptide overexpression. n = 9–15. Each point represents mean ± SEM. Statistical significance was calculated using a two-way ANOVA. E, Schematic of the excitatory cholinergic pharyngeal neurons and SER-7 5-HT receptor. F, Optogenetic excitation of the pharyngeal cholinergic motor neurons MC, M2, and M4 stimulates pumping in immobilized worms after flp-13 overexpression, but not after nlp-22 overexpression. n = 10. Each bar represents mean ± SEM. Statistical significance was calculated using a one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001.
Three classes of pharyngeal cholinergic motor neurons, the paired MC and M2 neurons and the single M4 neuron, stimulate pharyngeal pumping (Avery and Horvitz, 1989; Raizen et al., 1995; Trojanowski et al., 2014). Based on the above results, we predicted that flp-13-induced feeding quiescence, but not nlp-22-induced feeding quiescence, could be overcome by perturbations that excite pharyngeal cholinergic motor neurons. To test this hypothesis, we used both pharmacological and optogenetic approaches.
First, we tested the effects of the neuromodulator serotonin (5-hydroytryptamine or 5-HT) on feeding quiescence induced by flp-13 or nlp-22 overexpression. 5-HT stimulates pharyngeal pumping primarily via the SER-7 5-HT receptor and downstream Gαs signaling in pharyngeal cholinergic motor neurons (Hobson et al., 2006; Song and Avery, 2012). Therefore, the effects of neuropeptides that promote feeding quiescence by acting on or upstream of pharyngeal motor neurons should be suppressed by 5-HT, whereas the effects of neuropeptides that act downstream of the motor neurons should be unaffected by 5-HT. We found that nlp-22-induced feeding quiescence was not suppressed by the excitatory effects of 5-HT, consistent with the NLP-22 acting downstream of the pharyngeal motor neurons (Fig. 5B). In contrast, flp-13-induced feeding quiescence was fully suppressed by 5-HT, consistent with the FLP-13 neuropeptides acting on or upstream of the pharyngeal motor neurons (Fig. 5B).
An alternative explanation for these differential effects that would be consistent with both neuropeptides suppressing feeding by the same mechanisms is that nlp-22 is overexpressed at higher levels than flp-13 and that 5-HT can overcome mild overexpression, but not strong overexpression. We tested this possibility two ways, both making use of the fact that the transgenes expressing the neuropeptides were under the control of a heat-inducible promoter and we could thus vary the degree of transgene overexpression. First, we induced different degrees of expression of each neuropeptide transgene by varying the duration of the animals' exposure to 33°C. In the presence of food, an environmental stimulant to feeding rate, but in the absence of exogenous 5-HT, overexpression of either neuropeptide gene caused a similar reduction in feeding rate 2–3 h after heat exposure that was a function of the duration of prior heat exposure (and thus a function of neuropeptide expression; Fig. 5C). In contrast, in the presence of exogenous 5-HT, but not food, overexpression of flp-13 and nlp-22 produced different effects on feeding rate. Similar to its effect in the presence of food, nlp-22 overexpression caused a dose-dependent reduction in feeding rate in the presence of 5-HT. In contrast, even strong overexpression of flp-13 failed to inhibit feeding in the presence of 5-HT (Fig. 5D).
As a second way of testing if the different effects of 5-HT after overexpressing nlp-22 or flp-13 were due to differential overexpression of transgenes, we exposed the animals to 29°C instead of 33°C for 30 min to induce a lower level of transgene expression. This lower induction temperature had the additional advantage of being less likely to trigger quiescence on the basis of acute heat exposure (Nelson et al., 2014). Animals carrying transgenes with the heat shock promoter driving either flp-13 or nlp-22 overexpression showed feeding quiescence 2 h after this 30 min 29°C heat exposure (Phsp-16.2::flp-13: 182.1 ± 8.7 ppm before heat, 20.1 ± 8.6 ppm after heat, p < 0.001; Phsp-16.2::nlp-22: 145.2 ± 5.2 ppm before heat, 9.3 ± 3.6 ppm after heat, p < 0.001; mean ± SEM, n = 10), demonstrating that both transgenes were expressed at sufficiently high levels to induce quiescence even at this milder activation temperature. At an earlier time point (35 min) after the same 30 min 29°C heat exposure, worms overexpressing flp-13, but not worms overexpressing nlp-22, showed feeding quiescence (Phsp-16.2::flp-13: 181.2 ± 8.7 ppm before heat, 11.4 ± 5.2 ppm 35 min after heat, p < 0.001; Phsp-16.2::nlp-22: 145.2 ± 5.2 ppm before heat, 136.2 ± 16.3 ppm 35 min after heat, p = 0.61; mean ± SEM, n = 10). These observations suggest that the differential effects of 5-HT on feeding quiescence are not explained by reduced activation of the flp-13 transgene relative to the nlp-22 transgene.
Next, we used an optogenetic approach to test where the NLP-22 and FLP-13 neuropeptides act in relation to depolarization of cholinergic pharyngeal motor neurons (Fig. 5E). Although 5-HT activates pumping via these neurons, optogenetic stimulation via the light-sensitive cation channel Channelrhodopsin-2 (ChR2) of individual cholinergic motor neurons in the presence of 5-HT induces an even greater increase in feeding rate during wake (Trojanowski et al., 2014). We stimulated single pharyngeal motor neurons and monitored resulting changes in feeding rate. We found that stimulation of any of the excitatory cholinergic neurons MC, M2, or M4 caused an increase in feeding rate after flp-13 overexpression, but not after nlp-22 overexpression (Fig. 5F). These results further support the hypothesis that the FLP-13 neuropeptides act on or upstream of the pharyngeal cholinergic neurons, whereas the NLP-22 neuropeptide acts downstream of cholinergic neuron excitation, likely on the pharyngeal muscle.
Feeding quiescence during SIQ is abolished by activation of the Gαq, but not the Gαs, pathway
Having gained insight into the mechanisms through which the NLP-22 and FLP-13 neuropeptides control feeding, we next investigated whether feeding is regulated by similar mechanisms in their associated sleep-like states. Because we found that mutations that increase Gαs or Gαq signaling suppressed flp-13-induced feeding quiescence, we hypothesized that these mutations would also suppress feeding quiescence during SIQ. We found that mutations that increased Gαs signaling did not affect feeding quiescence during SIQ, whereas mutations that increased Gαq signaling did suppress feeding quiescence during SIQ (Fig. 6). Therefore, activation of Gαs signaling suppresses feeding quiescence induced by flp-13 overexpression, but does not suppress the feeding quiescence observed during SIQ, whereas activation of Gαq signaling suppresses feeding quiescence after either flp-13 overexpression or during SIQ. These results suggest that other neurotransmitters released by ALA affect feeding quiescence during SIQ, perhaps by acting downstream of Gαs signaling.

Activation of Gαq, but not Gαs, pathway impairs feeding quiescence during SIQ. Mutants with increased DAG levels due to activation of the Gαq pathway have increased feeding during SIQ, but mutants with an activated Gαs pathway do not. In addition, an egl-4 mutation impairs feeding quiescence during SIQ. Each point represents an observation from one worm and the horizontal bar represents the median of each group. n = 15 for each group. Statistical significance was calculated using the Mann–Whitney test. *p < 0.05, **p < 0.01.
We also found that a loss-of-function mutation in egl-4 (PKG) suppressed feeding quiescence during SIQ (Fig. 6), suggesting that EGL-4 acts downstream of or in parallel to ALA activation. Because the egl-4 mutation did not suppress feeding quiescence in response to flp-13 overexpression (Fig. 4A, Table 2), egl-4 may be acting downstream of or in parallel to a neurotransmitter distinct from FLP-13 that is released from ALA. Alternatively, FLP-13 released by flp-13 overexpression under the heat shock promoter may act on receptors that are not engaged by FLP-13 released from ALA. These possibilities are not mutually exclusive.
The pharyngeal nervous system can excite feeding during SIQ but not DTQ
Based on our results from animals overexpressing either flp-13 or nlp-22, we hypothesized that feeding is inhibited at the level of the pharyngeal motor neurons during SIQ, whereas during DTQ feeding is inhibited downstream of motor neuron excitation. To test this hypothesis, we again used both pharmacological and optogenetic approaches. First, we placed worms in either SIQ or DTQ on agarose pads containing 5-HT to determine whether excitation of pharyngeal neurons with 5-HT could stimulate feeding during these states. We found that worms in SIQ, but not DTQ, pumped in the presence of 5-HT, suggesting that feeding quiescence during SIQ occurs at the level of or upstream of pharyngeal cholinergic motor neurons, whereas feeding is inhibited at a level downstream of pharyngeal cholinergic motor neuron during DTQ (Fig. 7A).

The pharyngeal nervous system can excite feeding during SIQ, but not during DTQ. A, 10 mm 5-HT stimulates feeding during SIQ, but not during DTQ. n = 8–10. B, Optogenetic excitation of all cholinergic pharyngeal neurons (AChN) by wide-field fluorescence stimulates feeding in worms on bacterially seeded agar plates during SIQ, but not during DTQ. n = 13 for each group. C, Optogenetic excitation of pharyngeal muscle stimulates feeding in the presence of 10 mm 5-HT during wake, but not during DTQ. n = 10. D, Ca2+ transients are absent from pharyngeal muscle during DTQ and cannot be stimulated by muscle excitation. The difference between maximum and minimum GCaMP6s intensity of a region of the metacorpus of the pharynx during an ∼30 s interval was calculated for each condition to measure the magnitude of Ca2+ transients. n = 10. E, GCaMP6s fluorescence from a region of the metacorpus from one representative worm during wake. F, GCaMP6s fluorescence from a region of the metacorpus from one representative worm during DTQ. a.u., Arbitrary units in D–F. Statistical significance was calculated using Student's t test. *p < 0.05, **p < 0.01, ***p < 0.001.
Absence of feeding induction by 5-HT during DTQ could be explained by decreased 5-HT responsiveness during this state. Alternatively, it could be explained by reduced excitability of the motor neurons or by reduced excitability of pharyngeal muscle downstream of motor neuron excitation. To further delineate the circuit mechanism of feeding cessation during SIQ and DTQ, we optogenetically depolarized pharyngeal cholinergic motor neurons during these states. To minimize the effects of animal immobilization on behavior and to provide as strong an excitatory input to pumping as possible, we used wide-field blue light illumination to stimulate ChR2 in all cholinergic neurons in worms on bacterially seeded agar plates. As with nlp-22 overexpression, stimulation of cholinergic neurons did not result in feeding during DTQ (Fig. 7B). However, as with flp-13 overexpression, cholinergic neuron stimulation caused feeding during SIQ. These results are consistent with NLP-22 inhibiting pharyngeal muscle during DTQ and FLP-13 inhibiting pharyngeal cholinergic motor neurons during SIQ.
Pharyngeal muscle excitability is altered during DTQ
Our result that depolarization of pharyngeal neurons during DTQ did not stimulate feeding suggests that feeding is inhibited at the level of the muscle during this state. However, another possible explanation is that neurotransmitter release is blocked during DTQ. To test directly whether feeding was inhibited at the level of the muscle during DTQ, we attempted to stimulate pharyngeal muscle optogenetically. We had difficulty expressing ChR2 in pharyngeal muscle, so instead we used the light-sensitive cation channel Chrimson (Klapoetke et al., 2014), which expressed well. The peak excitation wavelength of Chrimson is ∼590 nm, but it retains adequate sensitivity (about 25% of peak) to 473 nm blue light (Klapoetke et al., 2014), so we used the same laser and power to stimulate Chrimson as we did for ChR2. We found that optogenetic stimulation of pharyngeal muscle increased feeding rate during wake, but this effect was abolished during DTQ (Fig. 7C). Thus, even when pharyngeal muscle was optogenetically depolarized, it did not contract, suggesting that either the coupling between excitation and contraction is altered during DTQ or that Ca2+ levels in the muscle are not rising sufficiently to generate contractions.
To test whether this uncoupling between muscle depolarization and contraction is upstream or downstream of increased Ca2+ levels, we imaged Ca2+ levels in the metacorpus region of the pharyngeal muscle (Avery and You, 2012) using the genetically encoded Ca2+ sensor GCaMP6s (Chen et al., 2013). During wake, we detected fluctuations in GCaMP6s fluorescence in the metacorpus (Fig. 7D,E); such Ca2+ transients are associated with pharyngeal pumps (Kerr et al., 2000; Akerboom et al., 2013). These fluctuations in GCaMP6s fluorescence were absent during DTQ, demonstrating that Ca2+ levels in the muscle do not oscillate during DTQ (Fig. 7D,F). Ca2+ transients during DTQ were undetectable even when pharyngeal muscle was optogenetically depolarized (Fig. 7D), demonstrating that pharyngeal excitability is fundamentally altered during DTQ such that Ca2+ levels cannot rise and trigger muscle contraction.
Discussion
Our results show that, despite the behavioral similarities of DTQ and SIQ, the quiescence-inducing mechanisms downstream of neuropeptide release are distinct in these states. Overexpression of the DTQ-associated neuropeptide gene nlp-22 inhibits feeding via action on pharyngeal muscle and acts on the nervous system to inhibit locomotion. In contrast, the SIQ-associated FLP-13 neuropeptides inhibit feeding via the pharyngeal nervous system and inhibit locomotion by acting on the nervous system through a different mechanism from that of NLP-22 (Fig. 8). Further, we found that stimulation of pharyngeal motor neurons excites feeding during SIQ but not during DTQ, and even direct stimulation of pharyngeal muscle does not excite feeding during DTQ. It is important to note that we have focused on two particular behavioral programs observed during sleep in all animals: quiescence of feeding and locomotion. Other aspects of sleep behavior, such as an elevated sensory arousal threshold, were not studied here and may be regulated by mechanisms different from those regulating feeding and locomotion quiescence.

Model for quiescence regulation during SIQ and DTQ. Green ovals represent somatic interneurons and blue text represents molecular mechanisms. Solid lines with arrowheads represent positive regulation and solid lines with balls on the end represent negative regulation. Dotted lines represent conceptual rather than molecular connections. A, During DTQ, an unidentified larval timer causes the RIA interneurons to release NLP-22. NLP-22 inhibits feeding by acting directly on pharyngeal muscles and inhibits locomotion via cholinergic neurons. The RIS interneuron also regulates feeding and locomotion quiescence during DTQ. B, Cellular stress triggers release of LIN-3/EGF from an unknown source, which then activates the ALA interneuron, causing release of FLP-13 and other neurotransmitter(s). FLP-13 inhibits feeding and locomotion by acting on pharyngeal and somatic (nonpharyngeal) cholinergic neurons, respectively.
Nevertheless, to our knowledge this is the first in vivo demonstration that a nonmammalian animal can express mechanistically distinct types of quiescence during sleep-like states. These results raise the possibility that in other animals (e.g., fruit flies, zebrafish), quiescence during sleep under different conditions may be regulated by different mechanisms despite behavioral similarities. Indeed, there are already suggestions that this is the case. For example, mechanisms of regulation of locomotion quiescence in young Drosophila adults are partially distinct from mechanisms in older Drosophila adults (Kayser et al., 2014).
Could the effects of neuropeptide overexpression be due to altered temporal dynamics of SIQ?
In our neuropeptide overexpression experiments, we used heat to induce somatic transcription of the NLP-22 or FLP-13 neuropeptides. However, as demonstrated here and previously, heat exposure can also trigger behavioral quiescence directly via induction of cellular stress (Jones and Candido, 1999; Hill et al., 2014). Therefore, it is possible that the quiescence-inducing effects of neuropeptide overexpression are confounded by SIQ. To minimize this possibility, we used a lower temperature, 33°C versus 35°C, and a later analysis time point, 2–3 h versus 35–45 min after heat exposure, to examine quiescence in response to neuropeptide overexpression. Acute feeding quiescence is less severe after 33°C exposure than after 35°C exposure (Nelson et al., 2014) and the behavioral effects of a 30 min heat shock at 33°C dissipate fully by 2 h after heat exposure (Nelson et al., 2013, 2014). We also repeated the neuropeptide overexpression experiments in ceh-17 mutants, which have defective SIQ (Hill et al., 2014; Nelson et al., 2014), and found no changes in the quiescence observed in response to neuropeptide overexpression. Finally, we found that 35 min after a mild heat shock at 29°C, worms overexpressing flp-13 were quiescent while worms overexpressing nlp-22 were not. This suggests that the behavioral effects of nlp-22 overexpression are not due to an interaction between neuropeptide overexpression and recovery of cellular stress. Because we did observe acute feeding quiescence using the milder 29°C exposure in animals carrying the Phsp-16.2::flp-13 transgene, but not in wild-type animals (Nelson et al., 2014), it is possible that overexpression of flp-13 may amplify the acute stress response, affirming the important role of flp-13 in SIQ.
Feeding quiescence during DTQ is due to a change in pharyngeal muscle excitability
Our genetic, pharmacologic, and optogenetic experiments indicate that pharyngeal muscle is not excitable during DTQ. Even with direct optogenetic stimulation of pharyngeal muscle, no Ca2+ increase was observed during DTQ, suggesting that Ca2+ entry is impaired during this state. Because pharyngeal Ca2+ increase during feeding occurs primarily via the l-type voltage-gated Ca2+ channel EGL-19 (Lee et al., 1997; Shtonda and Avery, 2005), our results suggest that the EGL-19 Ca2+ current is decreased during DTQ. Transcriptional expression of the egl-19 gene is unchanged during DTQ (George-Raizen et al., 2014), so the reduction in the EGL-19 current is likely due to a posttranscriptional change. This change could occur directly, via modulation of EGL-19 protein expression or function, or indirectly, via an increase in inhibitory currents carried by potassium and/or chloride channels. Our data also suggest that NLP-22 inhibits pharyngeal muscle directly during DTQ, although it must act in parallel to other mechanisms because feeding quiescence is not abolished in nlp-22 mutants (Nelson et al., 2013). NLP-22, like other neuropeptides, may act through a G-protein-coupled receptor to inhibit feeding. Alternatively, like certain small peptides such as Drosophila SLEEPLESS (Koh et al., 2008), it may act like a toxin and interact with ion channels directly (Wu et al., 2010; Dean et al., 2011; Wu et al., 2014). The receptor for NLP-22 is unknown.
Why is quiescence during DTQ and SIQ engaged differently?
Although feeding and locomotion quiescence are both characteristics of DTQ, recent data from many investigators support our conclusion that feeding and locomotion are inhibited at different levels. Several mutants have been described with impaired locomotion quiescence throughout DTQ (Raizen et al., 2008; Singh et al., 2011; Belfer et al., 2013; Choi et al., 2013; Nelson et al., 2013; Schwarz and Bringmann, 2013; Turek et al., 2013; Singh et al., 2014) and DTQ locomotion quiescence can be reduced by mechanical stimulation (Raizen et al., 2008; Driver et al., 2013; Nagy et al., 2014). However, mutations that suppress locomotion quiescence do not appear to affect feeding quiescence because no mutant has been described to feed throughout DTQ. It is possible that feeding quiescence during DTQ is essential for viability.
The differing mechanisms for behavioral quiescence during DTQ and SIQ may reflect the relative importance of the different types of quiescence for survival. DTQ is accompanied by a molt (Singh and Sulston, 1978) and occurs at the end of each of the four larval stages, when the worm has not yet reached reproductive maturity. The completion of each molt is essential for survival and reproduction (Frand et al., 2005), so there is strong selection for worms that can molt successfully. The correct replacement of the pharyngeal cuticle appears to be a vital part of DTQ, as defective pharyngeal molting can be lethal (Singh and Sulston, 1978; George-Raizen et al., 2014). By inhibiting feeding at the level of muscle excitability, the worm increases the likelihood that the pharyngeal cuticle will form properly because no stray neural impulses or neuromodulation could trigger muscle contraction that might disrupt cuticular assembly. Interestingly, mutations that severely decrease quiescence during SIQ can also have a small effect on quiescence during DTQ (Van Buskirk and Sternberg, 2007), suggesting that molting may be stressful and weakly stimulate SIQ.
In contrast to the precise timing of DTQ, the environmental stresses that trigger SIQ can happen at any point in the life of a worm, and no SIQ-associated structural or morphological changes have been identified. Failure to engage proper SIQ is rarely lethal in the first 24 h after stress (Hill et al., 2014), so pharyngeal contraction during this state may not be as detrimental to survival. By inducing quiescence at the level of the nervous system, the worm retains the ability to use other neuromodulators such as 5-HT to alter feeding during SIQ. This implies that, although quiescence increases the likelihood of survival in response to cellular stressors (Hill et al., 2014), there may be conditions under which feeding during this state is beneficial.
DTQ and SIQ may be functionally conserved and are regulated by evolutionarily conserved signaling pathways
There is no evidence to suggest that DTQ and SIQ represent evolutionary forms of subtypes of mammalian sleep. However, DTQ and SIQ may serve functions similar to mammalian sleep: DTQ, which occurs in phase with cycling of the C. elegans homolog of the PERIOD gene (Monsalve et al., 2011), has been implicated in synaptic plasticity (Dabbish and Raizen, 2011) and synthetic metabolism (Frand et al., 2005; Driver et al., 2013), whereas SIQ is important for survival after physiological stressors (Hill et al., 2014). Likewise, mammalian sleep has been implicated in synaptic plasticity (Tononi and Cirelli, 2014), anabolic metabolism (Mackiewicz et al., 2007), and stress responses (Toth and Krueger, 1988; Rampin et al., 1991).
The signaling pathways investigated here, as well as other previously identified regulators of DTQ (Raizen et al., 2008; Singh et al., 2011; Belfer et al., 2013; Choi et al., 2013; Driver et al., 2013; Iwanir et al., 2013; Nagy et al., 2013; Nelson et al., 2013; Schwarz and Bringmann, 2013; Turek et al., 2013; Nagy et al., 2014; Singh et al., 2014; Choi et al., 2015) and SIQ (Hill et al., 2014; Nelson et al., 2014), are found in many cell and neuron types and are highly conserved. These pathways have been implicated in sleep regulation in a variety of species (Allada and Siegel, 2008; Zimmerman et al., 2008; Crocker and Sehgal, 2010), but their ubiquitous expression patterns have made identification of specific cellular and circuit functions for these pathways challenging (but see Crocker et al., 2010). By identifying how these genes affect circuits that regulate different sleep-like states, we will gain insight into the mechanisms and functions of sleep across all species.
Footnotes
Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (Grant P40 OD010440). N.F.T. was supported by the NIH (Grant T32-HL-07953). S.W.F. was supported by the Howard Hughes Medical Institute. C.F.-Y. was supported by an Alfred P. Sloan Research Fellowship and by the NIH (Grants R01-NS-084835, R01-NS-088432, and R21-NS-091500). D.M.R. was supported by the NIH (Grants R01-NS-064030, R01-NS-088432, and R21-NS-091500).
- Correspondence should be addressed to either of the following: Christopher Fang-Yen, Department of Bioengineering, 210 S. 33rd Street, 240 Skirkanich Hall, University of Pennsylvania, Philadelphia, PA 19104, fangyen{at}seas.upenn.edu; or David M. Raizen, Department of Neurology, 210 S. 33rd Street, 240 Skirkanich Hall, University of Pennsylvania, Philadelphia, PA 19104, raizen{at}mail.med.upenn.edu
References
- Akerboom et al., 2013.↵
- Albertson and Thomson, 1976.↵
- Alkema et al., 2005.↵
- Allada and Siegel, 2008.↵
- Avery, 1993.↵
- Avery and Horvitz, 1989.↵
- Avery and You, 2012.↵
- Bastiani et al., 2003.↵
- Belfer et al., 2013.↵
- Brenner, 1974.↵
- Cassada and Russell, 1975.↵
- Charlie et al., 2006.↵
- Chen et al., 2013.↵
- Cho and Sternberg, 2014.↵
- Choi et al., 2013.↵
- Choi et al., 2015.↵
- Crocker and Sehgal, 2010.↵
- Crocker et al., 2010.↵
- Dabbish and Raizen, 2011.↵
- Davis et al., 1995.↵
- Dean et al., 2011.↵
- Doi and Iwasaki, 2002.↵
- Driver et al., 2013.↵
- Dybbs et al., 2005.↵
- Edgley et al., 2006.↵
- Frand et al., 2005.↵
- George-Raizen et al., 2014.↵
- Gordus et al., 2015.↵
- Guo et al., 2011.↵
- Hajdu-Cronin et al., 1999.↵
- Hawasli et al., 2004.
- He et al., 2013.↵
- Hendricks et al., 2001.↵
- Hill et al., 2014.↵
- Hobson et al., 2006.↵
- Humphrey et al., 2007.↵
- Iwanir et al., 2013.↵
- Jansen et al., 1999.↵
- Jeon et al., 1999.↵
- Joiner et al., 2006.↵
- Jones and Candido, 1999.↵
- Kayser et al., 2014.↵
- Kerr et al., 2000.↵
- Klapoetke et al., 2014.↵
- Koh et al., 2008.↵
- Korswagen et al., 1997.↵
- Lee et al., 1997.↵
- Lenz et al., 2015.↵
- Liewald et al., 2008.↵
- Loomis et al., 1937.↵
- Mackiewicz et al., 2007.↵
- Mello et al., 1991.↵
- Mendel et al., 1995.↵
- Miller et al., 1996.↵
- Miller et al., 1999.↵
- Monsalve et al., 2011.↵
- Morgan et al., 1990.
- Morgan et al., 2007.↵
- Nagy et al., 2013.↵
- Nagy et al., 2014.↵
- Nelson and Fitch, 2011.↵
- Nelson and Raizen, 2013.↵
- Nelson et al., 2013.↵
- Nelson et al., 2014.↵
- Nurrish et al., 1999.↵
- Parisky et al., 2008.↵
- Perez-Mansilla and Nurrish, 2009.↵
- Pujol et al., 2000.↵
- Raizen et al., 1995.↵
- Raizen et al., 2008.↵
- Raizen et al., 2012.↵
- Rampin et al., 1991.↵
- Renn et al., 1999.↵
- Reynolds et al., 2005.↵
- Robatzek and Thomas, 2000.↵
- Saifee et al., 1998.↵
- Saifee et al., 2011.
- Schade et al., 2005.↵
- Schwarz and Bringmann, 2013.↵
- Ségalat et al., 1995.↵
- Shtonda and Avery, 2005.↵
- Siegel, 2005.↵
- Singh et al., 2011.↵
- Singh et al., 2014.↵
- Singh and Sulston, 1978.↵
- Song and Avery, 2012.↵
- Starich et al., 1996.↵
- Tabuse and Miwa, 1983.↵
- Tononi and Cirelli, 2014.↵
- Toth and Krueger, 1988.↵
- Trent et al., 1983.↵
- Trojanowski and Fang-Yen, 2015.↵
- Trojanowski et al., 2014.↵
- Turek et al., 2013.↵
- Van Buskirk and Sternberg, 2007.↵
- van der Linden et al., 2003.↵
- van Swinderen et al., 1999.↵
- van Swinderen et al., 2001.
- van Swinderen et al., 2002.
- Wang et al., 2001.↵
- Wragg et al., 2007.
- Woods et al., 2014.↵
- Wu et al., 2010.↵
- Wu et al., 2014.↵
- Yeh et al., 2008.↵
- Zimmerman et al., 2008.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}