

Abstract
Changes in membrane cholesterol content can alter protein kinase activity, however, it is not known whether kinases regulating transmitter release are sensitive to membrane cholesterol content. Here we have used the cholesterol extracting agent methyl-β-cyclodextrin to measure the effects of acute cholesterol reduction on transmitter release from cultured cerebellar neurons. Cholesterol depletion increased the frequency of spontaneous transmitter release without altering the amplitude and time course of mEPSCs. Evoked transmitter release was decreased by cholesterol extraction and the paired pulse ratio was also decreased. Alterations in synaptic transmission were not associated with failure of action potential generation or changes in presynaptic Ca2+ signaling. Both the increase in mEPSC frequency and the change in paired pulse ratio were blocked by the broad spectrum protein kinase inhibitor staurosporine. The increase in mEPSC frequency was also sensitive to selective inhibitors of PKC and PKA. Our results therefore demonstrate that the activity of presynaptic protein kinases that regulate spontaneous and evoked neurotransmitter release is sensitive to changes of membrane cholesterol content.
Introduction
Reductions in the cholesterol content of neuronal membranes have been observed under conditions that include maturation of neurons in culture (Prinetti et al., 2001) and pathological disruption of cholesterol trafficking in Niemann-Pick C1 disease (Karten et al., 2002). Studies that have used the cholesterol binding oligosaccharide methyl-β-cyclodextrin (MβCD) to acutely reduce membrane cholesterol content have demonstrated that many of the molecules and processes involved in synaptic transmission are sensitive to cholesterol levels. For instance, cholesterol regulates the activity of voltage and Ca2+ gated K+ channels that regulate action potential waveform and firing rate (Xia et al., 2004; Shmygol et al., 2007) and the function of N and P/Q type Ca2+ channels that supply the Ca2+ trigger for synaptic vesicle exocytosis at most synapses (Toselli et al., 2005; Davies et al., 2006). Cholesterol is required for the formation of synaptic vesicles (Thiele et al., 2000) and is required for t-SNARE clustering at vesicle docking sites in PC12 cells (Lang et al., 2001). Additionally, the localization or physical properties of neurotransmitter receptors including the NMDA receptor (Hering et al., 2003) are altered by changes in membrane cholesterol level.
Depletion of cholesterol from cortical synaptosomes reduces the amount of glutamate released by subsequent depolarization with elevated external K+ (Taverna et al., 2004). In the intact synapses of crayfish neuromuscular junctions (Zamir and Charlton, 2006) and of cultured mouse hippocampal neurons (Wasser et al., 2007) cholesterol extraction reduces the amplitude of evoked synaptic responses but by different mechanisms. At the crayfish neuromuscular junction cholesterol depletion causes a failure in action potential transmission to release sites; when stimulation is applied focally at the release site the postsynaptic response is increased (Zamir and Charlton, 2006). In cultured hippocampal synapses cholesterol reduces the size of the readily releasable pool of vesicles (Wasser et al., 2007). Surprisingly, cholesterol depletion at the crayfish neuromuscular junction increases the frequency of spontaneous neurotransmitter release even after presynaptic injection of the Ca2+ chelator BAPTA. Therefore, cholesterol depletion increases spontaneous transmitter release through a Ca2+ independent mechanism (Zamir and Charlton, 2006).
Cholesterol depletion can lead to activation of several protein kinases that are known regulators of neurotransmitter release (Burgos et al., 2004; Cabrera-Poch et al., 2004). Functional cerebellar impairment is associated with impaired cholesterol trafficking in Niemann-Pick C1 disease so we have used a cerebellar culture model system to determine whether the activity of kinases regulating neurotransmitter release in the cerebellum is sensitive to changes in membrane cholesterol content.
Materials and Methods
Cell culture.
All procedures were performed in accordance with animal welfare guidelines at the University of Toronto and were approved by the institutional animal care and use committee. Cerebellar cultures were prepared from embryonic day 17–18 mice according to a published protocol (Furuya et al.,1998) with modifications. Dissected cerebella were digested in 0.25% trypsin/EDTA (Invitrogen) for 15 min at 37°C then dissociated, filtered through a cell strainer, centrifuged at 240 × g for 5 min, resuspended in plating solution (DMEM/F12, 10% fetal bovine serum, 1× l-glutamine, 1× Penn/Strep) and plated at a density of 2500 cells/mm2 on 12 mm coverslips coated with poly-d-lysine (70–150 kDa; Sigma). Cells were incubated at 37°C for 3 h then 450 μl of culture media (DMEM/F12, 2% B27 supplements, 2 mm l-glutamine, 1× Penn/Strep; all Invitrogen) was added per well. Experiments were performed on cells after 10–16 d in vitro.
Confocal fluorescence microscopy and cholesterol quantitation.
Cells on coverslips were treated as indicated in the figure legend, rinsed in PBS then fixed in 4% formaldehyde, permeabilized and stained sequentially with anti-calbindin D28K (Millipore) and Alexa 594 anti-rabbit (Invitrogen) antibodies. For cholesterol staining, the D4 domain of Perfringolysin O (PfO) toxin (Ohno-Iwashita et al., 1990) and emerald-GFP were amplified and ligated behind GST into modified pGEX-KG vector then transfected into competent BL21(DE3) E. coli; bacterial growth, induction and protein purification were performed using standard procedures. The recombinant protein was applied at 250 μg/ml for 10 min following fixation without a permeabilization step. Coverslips were mounted on microscope slides in ProLong Gold reagent (Invitrogen), images were captured with a Leica TCS SL confocal microscope. Biochemical measurement of cholesterol was performed using the Amplex Red Cholesterol Assay Kit (Invitrogen) according to the manufacturer's instructions, without cholesterol esterase treatment. Resorufin product was imaged in 96 well plates with a CCD-based fluorescent scanner (Ettan DIGE Imager; GE Healthcare) and quantified with ImageJ software. Protein concentration of lysates was measured spectrophotometrically using Bradford Reagent (Sigma) with BSA standards.
Electrophysiology.
Experiments were performed in HEPES-buffered saline (HBS) containing the following (in mm): 140 NaCl, 10 glucose, 3–5 KCl, 2 CaCl2, 1 MgCl2, 5 sucrose, 10 HEPES, pH 7.35, at 25°C, and cells were allowed to equilibrate to this buffer for at least 30 min before the start of experiments. Coverslips with adherent cultured neurons were placed in a perfusion chamber on an upright microscope and viewed under a 20×, 0.4 numerical aperture (NA) water-immersion objective. Solutions were exchanged by gravitational flow at a rate of 0.5–1.0 ml/min. For recording of synaptic currents, Purkinje cells were visually identified on the criteria of large soma, highly branched processes and prominent nucleus and were placed under whole-cell voltage clamp at holding potential of −75 mV using 2.5–3.5 MΩ electrodes containing the following (in mm): 140 CsCl, 10 HEPES, 5 EGTA, 2 MgATP [pH adjusted to 7.35 with CsOH; corrected for liquid junction potential (LJP) of 6 mV]. Picrotoxin, 50 μm, was present for all experiments and 0.5 μm tetrodotoxin (Alomone Labs) was present for mEPSC recordings. Recording instrumentation consisted of an AM Systems model 2400 patch-clamp amplifier and a National Instruments PCI 6024E digital I/O board under the control of an Electronic Data Recorder or Whole Cell Program (Strathclyde Electrophysiology Software). Output current signals were low pass filtered at 5 kHz and digitized at 10 kHz. Initial series resistance values were between 8 and 20 MΩ and were checked at 5 min intervals during recording; series resistance was not compensated. Recordings were excluded from analysis if holding current exceeded 250 pA or series resistance varied by >30% during the experiment. A saline filled patch electrode or θ-glass electrode with the tip placed immediately above the soma of a granule neuron was used for extracellular stimulation. Cell pairs were selected for responses displaying facilitation to paired pulses at 20 Hz. Current-clamp recordings from granule neurons were performed with electrodes of 5–7 MΩ and intracellular buffer containing the following (in mm): 140 K-gluconate, 10 KCl, 10 HEPES, 5 EGTA, 2 MgATP (pH adjusted to 7.35 with KOH; recordings compensated for LJP of 15 mV).
Ca2+ imaging.
Coverslips were incubated in control saline or in saline containing 5 mm MβCD for 30 min. Granule neurons were then voltage-clamped at −70 mV using whole-cell electrodes containing (in mm) 140 K-gluconate, 10 KCl, 10 HEPES, 2 MgATP, and 0.2 fura-2, and dialyzed for 10 min to allow indicator diffusion along the proximal axon segment. Imaging equipment consisted of a Polychrome V monochromator (TILL Photonics), Ixon DU897 EM-CCD camera (Andor Technology) operated in conventional readout mode with 2 × 2 pixel binning (0.8 μm × 0.8 μm) and an upright Nikon microscope with 40×, 0.7 NA Olympus water dipping lens. Image acquisition was controlled with WinFluor software (Strathclyde Electrophysiology Software). Ratio pairs (360/380 nm excitation) were acquired at 2 Hz while an action potential-like stimulus train consisting of 1 ms voltage steps to +30 mV, repeated 20 times at 20 Hz, was applied. Ratio images were generated using a cutoff of 20 intensity levels above background at both wavelengths. Presynaptic ROIs were readily identifiable from cellular morphology (see Fig. 3D) and consisted of 2–8 contiguous pixels. Mean ratio values from these ROIs were converted to [Ca2+]i values with Rmin and Rmax values calculated from in vitro calibration solutions containing 0 or 39.8 μm free Ca2+ (Invitrogen), a viscosity correction factor of 0.8 and an assumed Kd of 224 nm.
Data analysis.
Measurement of EPSC peak amplitude and timing was performed with Clampfit software (Molecular Devices); AP and afterhyperpolarization (AHP) amplitude were defined as the maximum and minimum amplitude changes occurring within 10 ms of the visually identified onset of the AP. mEPSC amplitude and frequency were measured with Mini Analysis software (Synaptosoft). Graphing and statistical analysis of results was performed with GraphPad Prism software.
Results
Acute depletion of cholesterol from cultured cerebellar neurons
The synapses formed between granule and Purkinje neurons in vitro allow a defined, physiologically relevant synaptic connection to be studied in a dissociated cell monolayer (Hirano et al., 1986). To visualize the effect of MβCD treatment on cholesterol distribution in this preparation we used the cholesterol binding D4 domain of Perfringolysin O. (PfO) toxin, a marker of lipid raft domains (Waheed et al., 2001). Under basal conditions D4 domain staining revealed a pattern of bright spots and more diffuse membrane staining (Fig. 1A). Treatment with 5 mm MβCD caused a progressive reduction in the intensity of D4 domain staining (Fig. 1B), demonstrating that lipid rafts were dispersed by MβCD. To quantitate the amount of cholesterol extracted we used the Amplex Red cholesterol assay; this demonstrated that MβCD treatment caused a progressive decline in cholesterol content to <50% of the initial value after 30 min (Fig. 1C). We also investigated the possibility that cholesterol depletion compromised membrane integrity. Results in Figure 1D demonstrate that this was not the case as the morphology of Purkinje cells visualized by Calbindin D28K staining was similar between control treatment and 20 min treatment with 5 mm MβCD.
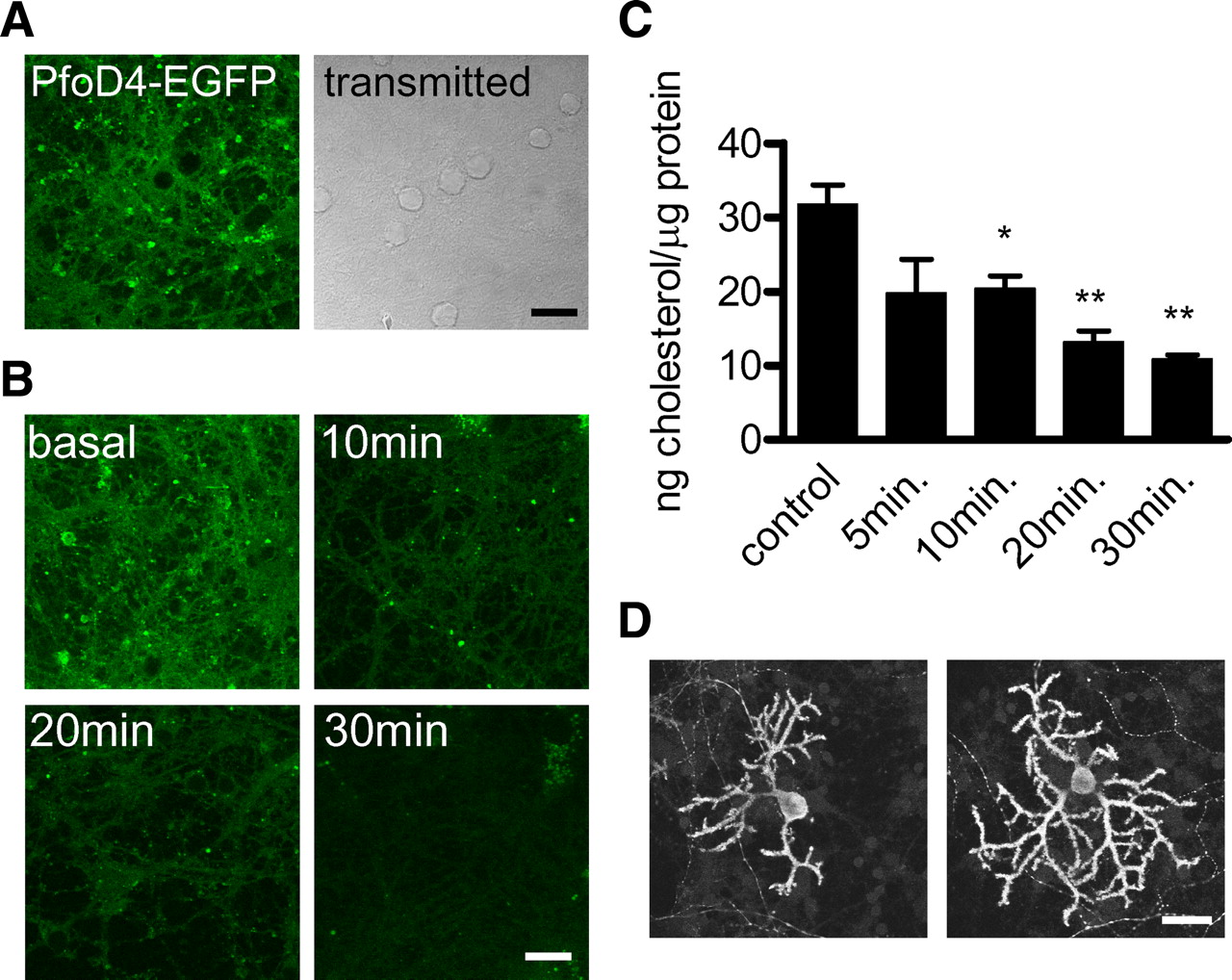
Treatment with MβCD causes extensive reduction of membrane cholesterol content. A, Distribution of lipid rafts in cultured cerebellar neurons labeled with PfO-D4-EGFP (left); transmitted light image (right). B, Cholesterol depletion following exposure to 5 mm MβCD for the indicated durations is demonstrated by reduced PfO-D4-EGFP binding. Scale bars: A, B, 10 μm. C, Cholesterol content of cell lysates quantified by Amplex Red cholesterol assay under control conditions and following MβCD treatment for the indicated periods; data were normalized to the lysate protein concentration. Bars show the mean and SEM of experiments performed in triplicate; *p < 0.05, **p < 0.01 versus control by t test. D, Calbindin D28K staining of cultured Purkinje cells under control conditions (left) and following exposure to 5 mm MβCD for 20 min (right). Scale bar, 20 μm.
Effect of cholesterol depletion on synaptic transmission
We recorded synaptic responses from Purkinje cells as cholesterol was extracted with 5 mm MβCD. This treatment caused a progressive increase in the frequency of mEPSCs over the course of MβCD exposure (Fig. 2A,B). Treatment with 5 mm MβCD precomplexed with 0.25 mm cholesterol did not increase mEPSC frequency, demonstrating a requirement for cholesterol extraction (Fig. 2B). The mean amplitude, time course and cumulative amplitude distribution of mEPSCs were unchanged by MβCD treatment (Fig. 2C,D) demonstrating that quantal size was unaffected under the conditions we used. It is therefore unlikely that there were major postsynaptic effects in our experiments but there was a significant increase in spontaneous vesicular release probability.

Cholesterol depletion alters synaptic transmission. A, Representative whole-cell recording of mEPSCs from a Purkinje neuron under basal conditions and after 25 min exposure to 5 mm MβCD. B, Relative mEPSC frequency during exposure to 5 mm MβCD (n = 5 cells, closed circles) or 5 mm cholesterol-MβCD complex (n = 5 cells, open circles). mEPSC frequency was calculated for 5 min bins for 5 min before and 30 min after exposure and then normalized to the basal frequency during the first 5 min of recording from each cell (n = 5 cells). C, Cumulative amplitude distribution of mEPSCs in 5 pA bins. The fraction of events falling within each amplitude bin was determined for the cells from the experiment shown in B during a 5 min basal period (open circles) or a 5 min period between 25 and 30 min of exposure to 5 mm MβCD (closed circles). Fractions were plotted as mean and SE of the 5 cells. D, Average of all mEPSCs recorded from a single experiment during a 5 min basal period (solid line) and a 5 min period following 25 min exposure to 5 mm MβCD (dotted line). E, EPSCs recorded from a granule neuron-Purkinje neuron pair under basal conditions and following 25 min of perfusion with 5 mm MβCD. Stimulus artifacts were removed for clarity. F, Time course of the change in peak amplitude of the EPSC during exposure to 5 mm MβCD (closed circles, n = 6) or 5 mm cholesterol-MβCD (open circles, n = 5) from 5 min of recording onwards. Data points represent the mean and SE of the normalized EPSP amplitude. *p < 0.05, **p < 0.01 versus control by t test.
To determine the effect of reducing membrane cholesterol content on evoked synaptic transmission we recorded EPSCs from Purkinje cells generated by extracellular stimulation of granule neurons (Fig. 2E). Stimulus pulses were delivered at 0.05 Hz, EPSCs had mean basal amplitude of 203 ± 38 pA and the mean amplitude of EPSCs was stable in normal saline. Treatment with 5 mm MβCD reduced EPSC amplitude (Fig. 2E,F) and as the mEPSC amplitude was unchanged, this result demonstrates that the number of vesicles released per stimulus was reduced by cholesterol depletion. However, when 5 mm MβCD/0.25 mm cholesterol complex was applied there was no change in EPSP amplitude. This indicated that MβCD had minimal nonspecific effects on EPSP amplitude. EPSC kinetics were not significantly different in these experiments, the delay from stimulus onset to EPSC peak was 3.46 ± 0.12 ms under basal conditions and 3.83 ± 0.25 ms following treatment with MβCD (p = 0.098 by paired t test). The decay time constant of single exponentials fitted to the falling portion of the EPSC was 1.41 ± 0.19 ms under basal conditions and 1.52 ± 0.23 ms following MβCD treatment (p = 0.60 by paired t test).
Presynaptic action potentials and Ca2+ signaling
As discussed in the introduction, the activity of many K+ and Ca2+ channels is sensitive to membrane cholesterol levels so we investigated the possibility that changes in action potential transmission or in presynaptic Ca2+ influx caused the changes in synaptic currents we observed following cholesterol depletion. We used the whole-cell current-clamp configuration to record action potentials in cultured granule neurons. Holding current was applied to maintain cells at −85 mV and brief (50 ms) pulses of depolarizing current injection of sufficient amplitude (15–30 pA) to induce single action potentials were applied at 20 s intervals (Fig. 3A). MβCD treatment caused an increase in AP amplitude and loss of afterhyperpolarization (Fig. 3B,C). Resting membrane potential was not affected by MβCD treatment (−86.2 ± 1.6 mV basal; −86.5 ± 1.0 mV 20 min MβCD; n = 4 each). Somatic action potential generation was therefore not blocked by cholesterol depletion. To test the possibility that action potential propagation or presynaptic Ca2+ influx were altered we measured residual Ca2+ accumulation in presynaptic terminals generated in response to somatic action potential-like voltage pulses by ratiometric imaging of fura-2 (Fig. 3D). Presynaptic terminals were visually identified as swellings along the thin axon and displayed large Ca2+ increases in response to a train of 20 voltage pulses (Fig. 3D–F). No difference in the amplitude or kinetics of these responses was detected when measurements were made from cells that had been treated for 30 min with 5 mm MβCD (Fig. 3G). These results demonstrate that the decrease in EPSC amplitude observed following MβCD treatment is not a consequence of reduction in presynaptic Ca2+ influx. As basal [Ca2+] was not different in the terminals of cholesterol depleted cells, the increase in mEPSC frequency observed following cholesterol depletion occurred by a Ca2+ independent mechanism.

Alterations to presynaptic excitability and Ca2+ signaling do not explain the effects of cholesterol depletion on synaptic transmission. A, Representative action potential recordings from a granule neuron subjected to depolarizing current injection (+20 pA, 50 ms) under basal conditions and following 25 min exposure to 5 mm MβCD. B, Alteration in AP amplitude for neurons held in 5 mm MβCD for 25 min (black bars) or held in control saline for 25 min (open bars). C, Same as B but with measurement of AHP amplitude. D, Granule neuron under whole-cell voltage clamp with intracellular solution containing 200 μm fura-2 imaged with 380 nm excitation light. Scale bar, 10 μm. E, Ratio images of [Ca2+]i in a granule cell stimulated with 20 pulses at 20 Hz. F, [Ca2+]i was measured from areas corresponding to the presynaptic boutons labeled 1–3 in E. G, Mean and SE of responses measured under control conditions (n = 5 cells) or from neurons pretreated with 5 mm MβCD for 30 min (n = 3 cells).
Cholesterol depletion and presynaptic kinase activity
In many synapses spontaneous transmitter release is controlled in part by the activity of various protein kinases and the activity of some of these kinases might be sensitive to changes of membrane cholesterol content. We used a broad-spectrum protein kinase inhibitor, staurosporine, to determine whether kinases regulating neurotransmitter release were activated by cholesterol depletion. Pretreatment with staurosporine for 10 min prevented the increase of mEPSC frequency caused by subsequent MβCD treatment (Fig. 4A,C). Treatment with staurosporine by itself did not cause a significant decrease in mEPSC frequency (Fig. 4B) suggesting that basal phosphorylation turnover rate is low and the effect of MβCD is to increase the activity of a staurosporine-sensitive kinase rather than to block a phosphatase. To determine the identity of kinases mediating the increase in mEPSC frequency we assessed the ability of selective PKC and PKA inhibitors to block the mEPSC frequency increase observed following cholesterol depletion. Pretreatment with the PKA selective inhibitor Rp-cAMPS (100 μm) or with the PKC selective inhibitor GF109203X (1 μm) significantly reduced the extent of the increase in mEPSC frequency observed following cholesterol extraction when compared with control experiments performed in parallel (Fig. 4D,E). Kinase activation can also influence evoked release and can change the paired pulse ratio at parallel fiber Purkinje cell synapses in cerebellar slices (Salin et al., 1996). We investigated the effect of staurosporine pretreatment on paired pulse ratio using paired stimulation at 20 Hz. The first response was still inhibited by MβCD in cultures pretreated with staurosporine (Fig. 4F,G). The reduction in paired pulse ratio was not observed when staurosporine pretreated cultures were subject to cholesterol depletion with MβCD (Fig. 4H), demonstrating that this change was also a consequence of increased kinase activity following cholesterol depletion. Our results therefore demonstrate that kinases regulating both spontaneous and evoked neurotransmitter release are sensitive to changes in membrane cholesterol content and that the effects of cholesterol depletion on synaptic transmission can be partly explained by increased presynaptic kinase activity.

Protein kinase activation mediates some of the effects of cholesterol depletion. A, mEPSC recordings from cells before and after MβCD treatment under control conditions (top) or with 1 μm staurosporine pretreatment (bottom). B, Effect of 1 μm staurosporine addition on relative mEPSC frequency (open circles, n = 4) and control recordings with no drug addition (closed circles, n = 4). C, Mean and SE of the relative mEPSC frequency during MβCD wash in under control conditions (filled circles, n = 6) or following pretreatment with 1 μm staurosporine for at least 10 min before the start of recording (open circles, n = 5). D, Relative mEPSC frequency during MβCD treatment under control conditions or with 100 μm Rp-cAMPS pretreatment (n = 4 each). E, Relative mEPSC frequency during MβCD treatment under control conditions (filled circles n = 6) or with 1 μm GF109203X pretreatment (open circles, n = 6). F, Paired EPSCs recorded from Purkinje cells before and after MβCD treatment under control conditions (top panel) and with 1 μm staurosporine pretreatment (bottom panel). G, Effect of MβCD treatment on EPSC amplitude with or without treatment with 1 μm staurosporine 10 min before application of MβCD (n = 5 each). H, Effect of MβCD treatment on paired pulse ratio with or without staurosporine pretreatment (n = 5 each; *p < 0.05 vs control by t test in all panels).
Discussion
We have shown that cholesterol content controls synaptic transmission between cultured cerebellar neurons. The rate of spontaneous neurotransmitter release is increased following cholesterol depletion by a mechanism requiring activation of presynaptic protein kinases. The amplitude of evoked synaptic responses is decreased consistent with a decrease in the size of the readily releasable pool of vesicles following cholesterol depletion. Short term synaptic plasticity is also altered by cholesterol depletion, again by a kinase-dependent mechanism.
Cholesterol depletion is known to increase the activity of several staurosporine-sensitive kinases including PKA, PKC and src (Burgos et al., 2004; Cabrera-Poch et al., 2004). The mechanisms linking cholesterol levels to kinase activity are not fully understood. In MDCK cells MβCD treatment activates PKA without increasing cAMP levels, possibly by disrupting inhibitory complexes localized to lipid rafts (Burgos et al., 2004). Activation of PKCε and src in MβCD-treated PC12 cells is also thought to be a consequence of alterations to localized lipid domain interactions (Cabrera-Poch et al., 2004). In addition to direct effects on kinase activity, the activity of PP2A phosphatase is sensitive to cholesterol depletion (Wang et al., 2005), therefore phosphatase inactivation may also contribute to an increase in protein phosphorylation following MβCD treatment. Activation of both PKC and PKA can increase mEPSC frequency (Carroll et al., 1998), similar kinases may mediate the effect of MβCD on PPR as PKA activation decreases PPR at parallel fiber synapses in cerebellar slices (Salin et al., 1996). The precise nature of the phosphorylation event(s) linking cholesterol depletion to increased mEPSC frequency and decreased PPR are beyond the scope of this article but the most plausible mechanistic explanation is that lipid raft disruption allows active kinases access to some component of the release apparatus and subsequent phosphorylation increases release probability.
The reduction in EPSC amplitude observed following MβCD treatment could be considered paradoxical given the effects on mEPSC frequency and PPR; however, cholesterol depletion has effects on vesicle recycling and release site structure that could explain this observation. Cholesterol depletion inhibits several forms of endocytosis (Parton and Richards, 2003) and biogenesis of synaptic like microvesicles in PC12 cells is more readily blocked by cholesterol depletion than bulk endocytosis (Thiele et al., 2000). Cholesterol is also required for clustering of the t-SNARE syntaxin in lipid raft membrane domains in PC12 cells; disruption of these domains with MβCD inhibits dense core vesicle docking and subsequent exocytosis (Lang et al., 2001; Zhang et al., 2009). Consistent with these observations, cholesterol depletion decreases the number of synaptic vesicles and the size of the readily releasable pool of vesicles in cultured hippocampal neurons (Wasser et al., 2007). Cholesterol depletion also alters the kinetics of release of dense core vesicles from PC12 cells and may be important for fusion pore expansion during exocytosis of large vesicles (Zhang et al., 2009). We found that mEPSC amplitude and time course were unchanged following cholesterol extraction suggesting that the rate of release of glutamate from small synaptic vesicles is not sensitive to changes in membrane cholesterol content. Our results also suggest that the alterations in synaptic responses we observed were mainly caused by presynaptic effects; however, we cannot rule out the possibility that cholesterol depletion may have caused a subset of synapses to become postsynaptically silenced in addition to any presynaptic effects.
We investigated the possibility that alterations in action potential transmission or presynaptic Ca2+ influx contributed to the reduction of EPSC amplitude but found no significant differences in the amplitude or time course of Ca2+ transients in presynaptic boutons from MβCD-treated cells. The increased amplitude of somatic action potentials and reduction in the afterhyperpolarization amplitude are consistent with studies demonstrating regulation of voltage- and Ca2+-gated K+ channels by the membrane lipid environment in general and by cholesterol specifically (Xia et al., 2004; Shmygol et al., 2007). These results demonstrate that changes in membrane cholesterol levels have effects on somatic excitability but that action potential transmission and presynaptic Ca2+ signaling are relatively resistant to cholesterol depletion.
The lipid composition of neuronal membranes is altered during cellular maturation and in some pathological conditions. In cerebellar neurons cultured from 8 d postnatal rats the molar ratio of cholesterol to glycerophospholipids decreases from 0.146 to 0.108 between 8 and 17 DIV (Prinetti et al., 2001). Conversely the cholesterol content of hippocampal neuronal membranes increases with time in culture (Nicholson and Ferreira, 2009). Our results suggest that changes in membrane cholesterol content could lead to alterations in presynaptic kinase signaling as neurons develop in vitro. Changes in cholesterol trafficking or metabolism have been implicated in the pathogenesis of several neurodegenerative conditions, most notably Niemann-Pick C1 disease which is caused by mutations of a gene involved in intracellular cholesterol trafficking; this causes cerebellar neurodegeneration (Sarna et al., 2003). A reduction in axonal cholesterol is observed in sympathetic neurons derived from mice deficient for the Niemann-Pick C1 (NPC1) protein (Karten et al., 2002). Interestingly, NPC1 pathology is associated with cytoskeletal damage and hyperphosphorylation of the microtubule binding protein tau (Bu et al., 2002); kinase activation caused by lipid raft disruption has been suggested as a cause of tau hyperphosphorylation (Sawamura et al., 2003). Our results demonstrate that similar mechanisms linking perturbations of membrane cholesterol content to kinase activation could contribute to synaptic dysfunction in neurodegenerative disease.
Footnotes
This work was supported by Canadian Institutes of Health Research Operating Grants MOP-82827 to M.P.C. and MOP-57825 to S.S. We thank Dr. Rodney Tweten (University of Oklahoma, Oklahoma City, OK) and Dr. Bulent Matus (University of Windsor, Windsor, ON, Canada) for providing Perfringolysin O toxin D4 domain cDNA. We also thank Drs. L.Y. Wang, J.S. Dason, and R.G. Tsushima for critically reading the manuscript.
- Correspondence should be addressed to Milton P. Charlton, Department of Physiology, MSB3308 1 King's College Circle, Toronto, Ontario M5S 1A8, Canada. milton.charlton{at}utoronto.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}