





Two Argentinean siblings (a boy and a girl) from a nonconsanguineous family presented with hypercalcemia, hypercalciuria, hypophosphatemia, low parathyroid hormone (PTH), and nephrocalcinosis.
The goal of this study was to identify genetic causes of the clinical findings in the two siblings.
Whole exome sequencing was performed to identify disease-causing mutations in the youngest sibling, and a candidate variant was screened in other family members by Sanger sequencing. In vitro experiments were conducted to determine the effects of the mutation that was identified.
Affected siblings (2 y.o. female and 10 y.o male) and their parents were included in the study. Informed consent was obtained for genetic studies.
A novel homozygous mutation in the gene encoding the renal sodium-dependent phosphate transporter SLC34A1 was identified in both siblings (c.1484G>A, p.Arg495His). In vitro studies showed that the p.Arg495His mutation resulted in decreased phosphate uptake when compared to wild-type SLC34A1.
The homozygous G>A transition that results in the substitution of histidine for arginine at position 495 of the renal sodium-dependent phosphate transporter, SLC34A1, is involved in disease pathogenesis in these patients. Our report of the second family with two mutated SLC34A1 alleles expands the known phenotype of this rare condition.
Renal phosphate absorption that occurs in the renal proximal tubules is a key process in the phosphate homeostasis pathway. Two transporters sodium dependent-phosphate transporter (NaPi-IIa) and NaPi-IIc, both of which are localized to the apical membrane of mammalian proximal tubules, mediate renal phosphate reabsorption (1–3). While in murine animal models, NaPi-IIa (SLC34A1) has been shown to be a key regulator of renal phosphate reabsorption; its role in phosphate metabolism in humans remains unclear (4). Although heterozygous SLC34A1 variants have been associated with renal phosphate leak and hypercalciuria (5), it has been argued by others that these variants might not play a causative role in the disease pathogenesis (6, 7). A recent report by Magen et al showed that a homozygous mutation (a 21 bp duplication) in SLC34A1 resulted in renal Fanconi's syndrome with proximal tubulopathy (8).
Here we report a single nucleotide change in SLC34A1 identified by whole exome sequence analysis as a disease-causing variant in two siblings with hypercalcemia, hypercalciuria, hypophosphatemia, and nephrocalcinosis. Phosphate uptake assays in HEK 293 (human embryonic kidney) cells showed the inability of this novel SLC34A1 variant to transport phosphate and thereby confirming pathogenicity of SLC34A1 sequence variant.
Subjects and Methods
Subjects.
We describe two Argentinean siblings (one boy and one girl) from a nonconsanguineous family who presented with hypercalcemia, hypercalciuria, hypophosphatemia, and nephrocalcinosis without proximal tubulopathy, renal failure nor skeletal dysplasia (see pedigree in Figure 1).
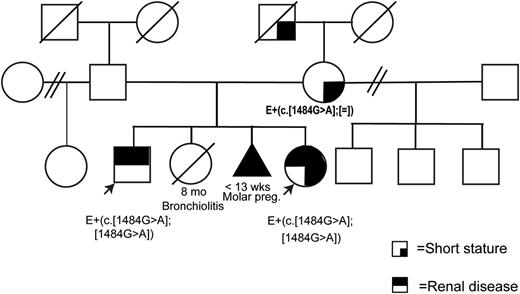
Pedigree of the family.
E+(c.[1484G>A];[1484G>A]) signifies that a DNA sample from the patient was evaluated and revealed the presence of the mutation in the homozygous state. (c.[1484G>A];=) refers to a heterozygous mutation.
Patient 1 is a male born at term with normal weight and height. Nephrocalcinosis was found at 18 months in an ultrasound performed due to repeated urinary tract infections. His clinical examination and skeletal radiographs were unremarkable. He had transient moderate hypercalcemia, which was resolved at 2 years of age, transient hypophosphatemia that required oral phosphate supplements, and has persistent hypercalciuria. Parathyroid hormone (PTH) was initially undetectable but became appropriate for serum calcium at 10 years of age.
Patient 2 is a girl born at term, small for gestational age (birth height −3.3 SDS) with breech presentation. Because of her brotheŕs medical history, she was evaluated at 2 months of age and was found to have nephrocalcinosis. She was under vitamin D prophylaxis (500 IU/d) with 25(OH)2D3 serum levels in the upper normal range and elevated 1,25(OH)2D3 levels. She had severe hypercalcemia with only transient response to medical treatment consisting of hyperhydration, diuretics, methylprednisone 1 mg/kg/d given twice and five IV pamidronate cycles. Hypercalcemia and low PTH resolved at 3.2 years of age. Hypophosphatemia progressively worsened and periodically required oral phosphate supplements for serum phosphate normalization. Nevertheless, hypercalciuria was persistently elevated throughout the follow-up period (there was only one normal urinary calcium determination within glucocorticoid treatment). Therefore, potassium citrate therapy was instituted to prevent nephrocalcinosis progression. Patient 1 is growing appropriately, but patient 2 has a severely short stature, without any sign of skeletal disease or dysplasia on the radiographic skeletal survey. No biochemical or imaging abnormalities were identified in their parents; the only clinical sign of importance is the mother's short stature.
Whole exome sequencing.
Exome sequencing was performed as described previously (9). Briefly, exomes were captured on Nimblegen's Baylor VCRome library (Roche NimbleGen) and sequencing was performed on the Illumina HiSeq 2000 platform (Illumina). Sequence reads were aligned to the hg18 reference human genome, SNPs were called, variants were annotated, and candidate genes were assessed using various databases to predict expression pattern, function, and potential pathogenic impact of the variants. The accession numbers for the human SLC34A1 gene are as follows: NCBI Gene: 6569; GenBank transcript: NM_003052.4; CCDS protein: 4418.1.
Plasmids.
hSLC34A1-pCMV Sport 6 plasmid was purchased from the Harvard Plasmid repository. The arginine to histidine point mutation at position 495 was generated using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) as per the manufacturer's protocol and confirmed by sequencing.
Cell culture.
HEK 293 cells were grown in DMEM containing 10% fetal bovine serum and 1% each of penicillin, streptomycin, and L-glutamine.
Real time PCR.
Equal numbers of HEK 293 cells were plated in 6-well plates. Cells were transfected with 2 μg DNA per well with 1:4 ratio of XtremeHP transfection reagent (Roche). Forty eight hours after transfection, the growth medium was aspirated and the confluent monolayer was rinsed with phosphate buffered saline. Total RNA was extracted using a Trizol reagent (Life Technologies). Samples were DNase (Roche) treated and the Superscript III First Strand RT-PCR kit (Life Technologies) was used for cDNA synthesis. The qRT-PCR was performed on a LightCycler instrument (Roche). hGAPDH was used as the internal control to normalize gene expression.
Immunofluorescence assay.
Cells were transfected with 1 μg DNA per well with the XtremeHP transfection reagent (Roche) in two-well chamber slides (Nunc). Briefly, cells were fixed with paraformaldehyde, permeabilized with triton X-100, blocked with donkey serum plus bovine serum albumin. Primary antibody (rabbit anti-Npt2a antibody, Sigma) was incubated at 1:75 dilution in 1% bovine serum albumin for 90 min. After washing, samples were incubated with both alexa fluor 488 wheat germ agglutinin and donkey-anti-rabbit conjugated with alexa fluor 594 for 1 hour. The slides were then washed and mounted with an antifade mounting reagent with 4′,6-diamidino-2-phenylindole.
Phosphate uptake assay.
Cells were transfected as described above and 48 h after transfection, then the confluent monolayer was rinsed with an incubation buffer lacking the transport solute (10). Transport was then initiated by adding 1 mL of incubation buffer containing 1 μCi/mL of 32P labeled monobasic potassium phosphate (Perkin Elmer). The plates were slowly agitated at 37°C. Cells were washed with ice-cold stop solution at 0, 5, 10, and 20 min to terminate the uptake process. Cells were then solubilized with 0.5 mL triton X-100 containing protease inhibitors. Radioactivity was measured in 150 μL of the homogenate and total protein was measured using the Biorad protein assay.
Results
Clinical characteristics of the patients
The results of clinical and biochemical evaluations of the patients compared to the 21 bp duplication described by Magen et al, are described in Table 1. The ultrasound of the female child performed at 2 months of age showed nephrocalcinosis and the radiographic images revealed rhizomelia at 4 months of age (Figure 2). At 3 years of age, the child did not show any signs of rickets or skeletal dysplasia (Figure 2D).
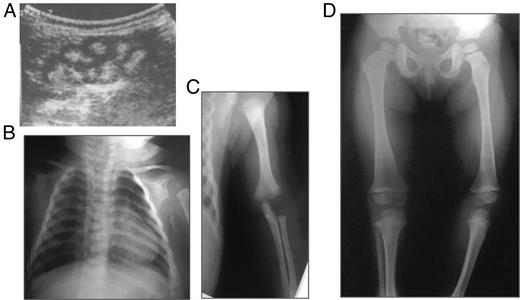
Radiographic features of the female patient.
A, Renal ultrasound at 2 months of age. B, x-ray at 3 months of age. C, rhizomelia at 4 months. D, x-ray at 3 years of age showing no signs of rickets nor skeletal dysplasia. Growth arrest lines are consistent with bisphosphonate therapy.
Clinical and Biochemical Results
| This Report | Homozygous Duplication, Magen D et al (8) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 (male) | Patient 2 (female) | Mother | Father | ||||||
| Admission | Last Visit | Admission | Last Visit | Patient 1 (male) | Patient 2 (female) | Reference Values | |||
| Age (y) | 1.5 | 10.3 | 0.24 | 3.2 | N/A | N/A | 39 | 43 | N/A |
| Height (Z-score) | −1.2 | −1.8 | −3.1 | −3.4 | −2.0 | −1.5 | −4.1 | −2.8 | N/A |
| Dysmorphisms | None | None | Narrow forehead, thin lips, low-set ears | Narrow forehead, thin lips, low-set ears | No | No | No | No | N/A |
| Hypercalciuria | Yes | Yes | Yes | Yes | No | No | In adolescence but not adulthood | In adolescence but not adulthood | N/A |
| Nephrocalcinosis | Yes | Yes | Yes | Yes | No | N/A | No | No | N/A |
| Skeletal deformities | No | No | Rhizhomelia, short neck | Rhizhomelia, short neck | No | No | Bone pain, leg deformities, osteomalacia | N/A | N/A |
| Proximal tubulopathy | No | No | No | No | No | N/A | Yes | Yes | N/A |
| BMD (L1–L4) gm/cm2 | N/A | 0.864 | N/A | 0.651 | N/A | N/A | N/A | N/A | N/A |
| Blood tests | |||||||||
| Calcium (mg/dL) | 11 | 9.7 | Between 11.5 and 14.7 | 9.8 | 9.8 | 9.3 | 9.9, previously elevated | 9.1, previously elevated | 8.5–10.8 |
| Phosphate (mg/dL) | 4.3 (Nl 4.3–5.4) | 3.9 (Nl 3.7–5.4) | 3.7 (Nl 4.3–5.4) | 4.7 (Nl 4.3–5.4) | 3.5 (Nl 2.5–4.5) | 3.2 | 2 (Nl 2.5–4.5) | 2.2 (Nl 2.5–4.5) | Varies according to age and gender |
| Magnesium (mg/dL) | 1.9 | 1.8 | 2 | 1.9 | 1.9 | 2 | N/A | N/A | 1.7–2.3 |
| Alkaline phosphatase (UI/L) | 906 | 287 | 791 | 339 | 79 | 151 | 113 | 102 | 10–270 |
| Creatinine (mg/dL) | 0.38 | 0.7 | 0.3 | 0.3 | 0.78 | 0.89 | 1.5 | 0.9 | 0.5–1.3 |
| Uric acid (mg/dL) | 2.8 | 4.5 | 7.3 | 5.7 | 4.7 | 5.9 | 1.5 | 1.9 | 2.2–5.8 |
| pH | 7.42 | 7.36 | 7.38 | 7.39 | 7.36 | N/A | 7.36 | 7.37 | 7.38–7.41 |
| Bicarbonate (mmol/L) | 22.4 | 22.5 | 23 | 24.2 | 26.2 | N/A | 24 | 25 | 22–26 |
| Intact PTH (pg/mL) | Undetectable | 19 | Undetectable | 12 | 22 | 24 | 45 | 29 | 8–54 |
| 25(OH)D3 (ng/mL) | N/A | 31 | 69 | 35.7 | 31,9 | 46.1 | 12.4 | 8.3 | >30 |
| 1,25 (OH)2D3 (pg/mL) | N/A | N/A | 213 | N/A | N/A | N/A | 21, previously elevated | 12, previously elevated | 20–71 |
| FGF23 sequencing | N/A | N/A | normal | N/A | N/A | N/A | N/A | N/A | N/A |
| GH tests (max peak-ng/mL) | N/A | N/A | N/A | 10.1 | N/A | N/A | N/A | N/A | >6 |
| Urine tests | |||||||||
| Glucose (mg/dL) | N/A | N/A | N/A | 90 (units) | N/A | N/A | 100 | 300 | N/A |
| TRP (%) | N/A | 88 | 90 | 91.6 | N/A | N/A | 49 | 66 | 85–95 |
| Calcium: creatinine ratio | 0.55 | 0.21 | 2.2 | 0.4 | N/A | N/A | 0.19 | 0.1 | <0.2 |
| Protein (g/24 h) | N/A | 0.09 | N/A | 0.04 | N/A | N/A | 1.8 | 0.6 | <0.2 |
| Citrate | N/A | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | N/A | N/A | 375 mg/24 h (Nl > 320) | 420 mg/24 h (Nl > 320) | N/A |
| This Report | Homozygous Duplication, Magen D et al (8) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 (male) | Patient 2 (female) | Mother | Father | ||||||
| Admission | Last Visit | Admission | Last Visit | Patient 1 (male) | Patient 2 (female) | Reference Values | |||
| Age (y) | 1.5 | 10.3 | 0.24 | 3.2 | N/A | N/A | 39 | 43 | N/A |
| Height (Z-score) | −1.2 | −1.8 | −3.1 | −3.4 | −2.0 | −1.5 | −4.1 | −2.8 | N/A |
| Dysmorphisms | None | None | Narrow forehead, thin lips, low-set ears | Narrow forehead, thin lips, low-set ears | No | No | No | No | N/A |
| Hypercalciuria | Yes | Yes | Yes | Yes | No | No | In adolescence but not adulthood | In adolescence but not adulthood | N/A |
| Nephrocalcinosis | Yes | Yes | Yes | Yes | No | N/A | No | No | N/A |
| Skeletal deformities | No | No | Rhizhomelia, short neck | Rhizhomelia, short neck | No | No | Bone pain, leg deformities, osteomalacia | N/A | N/A |
| Proximal tubulopathy | No | No | No | No | No | N/A | Yes | Yes | N/A |
| BMD (L1–L4) gm/cm2 | N/A | 0.864 | N/A | 0.651 | N/A | N/A | N/A | N/A | N/A |
| Blood tests | |||||||||
| Calcium (mg/dL) | 11 | 9.7 | Between 11.5 and 14.7 | 9.8 | 9.8 | 9.3 | 9.9, previously elevated | 9.1, previously elevated | 8.5–10.8 |
| Phosphate (mg/dL) | 4.3 (Nl 4.3–5.4) | 3.9 (Nl 3.7–5.4) | 3.7 (Nl 4.3–5.4) | 4.7 (Nl 4.3–5.4) | 3.5 (Nl 2.5–4.5) | 3.2 | 2 (Nl 2.5–4.5) | 2.2 (Nl 2.5–4.5) | Varies according to age and gender |
| Magnesium (mg/dL) | 1.9 | 1.8 | 2 | 1.9 | 1.9 | 2 | N/A | N/A | 1.7–2.3 |
| Alkaline phosphatase (UI/L) | 906 | 287 | 791 | 339 | 79 | 151 | 113 | 102 | 10–270 |
| Creatinine (mg/dL) | 0.38 | 0.7 | 0.3 | 0.3 | 0.78 | 0.89 | 1.5 | 0.9 | 0.5–1.3 |
| Uric acid (mg/dL) | 2.8 | 4.5 | 7.3 | 5.7 | 4.7 | 5.9 | 1.5 | 1.9 | 2.2–5.8 |
| pH | 7.42 | 7.36 | 7.38 | 7.39 | 7.36 | N/A | 7.36 | 7.37 | 7.38–7.41 |
| Bicarbonate (mmol/L) | 22.4 | 22.5 | 23 | 24.2 | 26.2 | N/A | 24 | 25 | 22–26 |
| Intact PTH (pg/mL) | Undetectable | 19 | Undetectable | 12 | 22 | 24 | 45 | 29 | 8–54 |
| 25(OH)D3 (ng/mL) | N/A | 31 | 69 | 35.7 | 31,9 | 46.1 | 12.4 | 8.3 | >30 |
| 1,25 (OH)2D3 (pg/mL) | N/A | N/A | 213 | N/A | N/A | N/A | 21, previously elevated | 12, previously elevated | 20–71 |
| FGF23 sequencing | N/A | N/A | normal | N/A | N/A | N/A | N/A | N/A | N/A |
| GH tests (max peak-ng/mL) | N/A | N/A | N/A | 10.1 | N/A | N/A | N/A | N/A | >6 |
| Urine tests | |||||||||
| Glucose (mg/dL) | N/A | N/A | N/A | 90 (units) | N/A | N/A | 100 | 300 | N/A |
| TRP (%) | N/A | 88 | 90 | 91.6 | N/A | N/A | 49 | 66 | 85–95 |
| Calcium: creatinine ratio | 0.55 | 0.21 | 2.2 | 0.4 | N/A | N/A | 0.19 | 0.1 | <0.2 |
| Protein (g/24 h) | N/A | 0.09 | N/A | 0.04 | N/A | N/A | 1.8 | 0.6 | <0.2 |
| Citrate | N/A | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | N/A | N/A | 375 mg/24 h (Nl > 320) | 420 mg/24 h (Nl > 320) | N/A |
Abbreviation: N/A, not applicable.
Clinical and Biochemical Results
| This Report | Homozygous Duplication, Magen D et al (8) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 (male) | Patient 2 (female) | Mother | Father | ||||||
| Admission | Last Visit | Admission | Last Visit | Patient 1 (male) | Patient 2 (female) | Reference Values | |||
| Age (y) | 1.5 | 10.3 | 0.24 | 3.2 | N/A | N/A | 39 | 43 | N/A |
| Height (Z-score) | −1.2 | −1.8 | −3.1 | −3.4 | −2.0 | −1.5 | −4.1 | −2.8 | N/A |
| Dysmorphisms | None | None | Narrow forehead, thin lips, low-set ears | Narrow forehead, thin lips, low-set ears | No | No | No | No | N/A |
| Hypercalciuria | Yes | Yes | Yes | Yes | No | No | In adolescence but not adulthood | In adolescence but not adulthood | N/A |
| Nephrocalcinosis | Yes | Yes | Yes | Yes | No | N/A | No | No | N/A |
| Skeletal deformities | No | No | Rhizhomelia, short neck | Rhizhomelia, short neck | No | No | Bone pain, leg deformities, osteomalacia | N/A | N/A |
| Proximal tubulopathy | No | No | No | No | No | N/A | Yes | Yes | N/A |
| BMD (L1–L4) gm/cm2 | N/A | 0.864 | N/A | 0.651 | N/A | N/A | N/A | N/A | N/A |
| Blood tests | |||||||||
| Calcium (mg/dL) | 11 | 9.7 | Between 11.5 and 14.7 | 9.8 | 9.8 | 9.3 | 9.9, previously elevated | 9.1, previously elevated | 8.5–10.8 |
| Phosphate (mg/dL) | 4.3 (Nl 4.3–5.4) | 3.9 (Nl 3.7–5.4) | 3.7 (Nl 4.3–5.4) | 4.7 (Nl 4.3–5.4) | 3.5 (Nl 2.5–4.5) | 3.2 | 2 (Nl 2.5–4.5) | 2.2 (Nl 2.5–4.5) | Varies according to age and gender |
| Magnesium (mg/dL) | 1.9 | 1.8 | 2 | 1.9 | 1.9 | 2 | N/A | N/A | 1.7–2.3 |
| Alkaline phosphatase (UI/L) | 906 | 287 | 791 | 339 | 79 | 151 | 113 | 102 | 10–270 |
| Creatinine (mg/dL) | 0.38 | 0.7 | 0.3 | 0.3 | 0.78 | 0.89 | 1.5 | 0.9 | 0.5–1.3 |
| Uric acid (mg/dL) | 2.8 | 4.5 | 7.3 | 5.7 | 4.7 | 5.9 | 1.5 | 1.9 | 2.2–5.8 |
| pH | 7.42 | 7.36 | 7.38 | 7.39 | 7.36 | N/A | 7.36 | 7.37 | 7.38–7.41 |
| Bicarbonate (mmol/L) | 22.4 | 22.5 | 23 | 24.2 | 26.2 | N/A | 24 | 25 | 22–26 |
| Intact PTH (pg/mL) | Undetectable | 19 | Undetectable | 12 | 22 | 24 | 45 | 29 | 8–54 |
| 25(OH)D3 (ng/mL) | N/A | 31 | 69 | 35.7 | 31,9 | 46.1 | 12.4 | 8.3 | >30 |
| 1,25 (OH)2D3 (pg/mL) | N/A | N/A | 213 | N/A | N/A | N/A | 21, previously elevated | 12, previously elevated | 20–71 |
| FGF23 sequencing | N/A | N/A | normal | N/A | N/A | N/A | N/A | N/A | N/A |
| GH tests (max peak-ng/mL) | N/A | N/A | N/A | 10.1 | N/A | N/A | N/A | N/A | >6 |
| Urine tests | |||||||||
| Glucose (mg/dL) | N/A | N/A | N/A | 90 (units) | N/A | N/A | 100 | 300 | N/A |
| TRP (%) | N/A | 88 | 90 | 91.6 | N/A | N/A | 49 | 66 | 85–95 |
| Calcium: creatinine ratio | 0.55 | 0.21 | 2.2 | 0.4 | N/A | N/A | 0.19 | 0.1 | <0.2 |
| Protein (g/24 h) | N/A | 0.09 | N/A | 0.04 | N/A | N/A | 1.8 | 0.6 | <0.2 |
| Citrate | N/A | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | N/A | N/A | 375 mg/24 h (Nl > 320) | 420 mg/24 h (Nl > 320) | N/A |
| This Report | Homozygous Duplication, Magen D et al (8) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 (male) | Patient 2 (female) | Mother | Father | ||||||
| Admission | Last Visit | Admission | Last Visit | Patient 1 (male) | Patient 2 (female) | Reference Values | |||
| Age (y) | 1.5 | 10.3 | 0.24 | 3.2 | N/A | N/A | 39 | 43 | N/A |
| Height (Z-score) | −1.2 | −1.8 | −3.1 | −3.4 | −2.0 | −1.5 | −4.1 | −2.8 | N/A |
| Dysmorphisms | None | None | Narrow forehead, thin lips, low-set ears | Narrow forehead, thin lips, low-set ears | No | No | No | No | N/A |
| Hypercalciuria | Yes | Yes | Yes | Yes | No | No | In adolescence but not adulthood | In adolescence but not adulthood | N/A |
| Nephrocalcinosis | Yes | Yes | Yes | Yes | No | N/A | No | No | N/A |
| Skeletal deformities | No | No | Rhizhomelia, short neck | Rhizhomelia, short neck | No | No | Bone pain, leg deformities, osteomalacia | N/A | N/A |
| Proximal tubulopathy | No | No | No | No | No | N/A | Yes | Yes | N/A |
| BMD (L1–L4) gm/cm2 | N/A | 0.864 | N/A | 0.651 | N/A | N/A | N/A | N/A | N/A |
| Blood tests | |||||||||
| Calcium (mg/dL) | 11 | 9.7 | Between 11.5 and 14.7 | 9.8 | 9.8 | 9.3 | 9.9, previously elevated | 9.1, previously elevated | 8.5–10.8 |
| Phosphate (mg/dL) | 4.3 (Nl 4.3–5.4) | 3.9 (Nl 3.7–5.4) | 3.7 (Nl 4.3–5.4) | 4.7 (Nl 4.3–5.4) | 3.5 (Nl 2.5–4.5) | 3.2 | 2 (Nl 2.5–4.5) | 2.2 (Nl 2.5–4.5) | Varies according to age and gender |
| Magnesium (mg/dL) | 1.9 | 1.8 | 2 | 1.9 | 1.9 | 2 | N/A | N/A | 1.7–2.3 |
| Alkaline phosphatase (UI/L) | 906 | 287 | 791 | 339 | 79 | 151 | 113 | 102 | 10–270 |
| Creatinine (mg/dL) | 0.38 | 0.7 | 0.3 | 0.3 | 0.78 | 0.89 | 1.5 | 0.9 | 0.5–1.3 |
| Uric acid (mg/dL) | 2.8 | 4.5 | 7.3 | 5.7 | 4.7 | 5.9 | 1.5 | 1.9 | 2.2–5.8 |
| pH | 7.42 | 7.36 | 7.38 | 7.39 | 7.36 | N/A | 7.36 | 7.37 | 7.38–7.41 |
| Bicarbonate (mmol/L) | 22.4 | 22.5 | 23 | 24.2 | 26.2 | N/A | 24 | 25 | 22–26 |
| Intact PTH (pg/mL) | Undetectable | 19 | Undetectable | 12 | 22 | 24 | 45 | 29 | 8–54 |
| 25(OH)D3 (ng/mL) | N/A | 31 | 69 | 35.7 | 31,9 | 46.1 | 12.4 | 8.3 | >30 |
| 1,25 (OH)2D3 (pg/mL) | N/A | N/A | 213 | N/A | N/A | N/A | 21, previously elevated | 12, previously elevated | 20–71 |
| FGF23 sequencing | N/A | N/A | normal | N/A | N/A | N/A | N/A | N/A | N/A |
| GH tests (max peak-ng/mL) | N/A | N/A | N/A | 10.1 | N/A | N/A | N/A | N/A | >6 |
| Urine tests | |||||||||
| Glucose (mg/dL) | N/A | N/A | N/A | 90 (units) | N/A | N/A | 100 | 300 | N/A |
| TRP (%) | N/A | 88 | 90 | 91.6 | N/A | N/A | 49 | 66 | 85–95 |
| Calcium: creatinine ratio | 0.55 | 0.21 | 2.2 | 0.4 | N/A | N/A | 0.19 | 0.1 | <0.2 |
| Protein (g/24 h) | N/A | 0.09 | N/A | 0.04 | N/A | N/A | 1.8 | 0.6 | <0.2 |
| Citrate | N/A | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | 2 mg/kg/24 h (Nl ≥ 2) | N/A | N/A | 375 mg/24 h (Nl > 320) | 420 mg/24 h (Nl > 320) | N/A |
Abbreviation: N/A, not applicable.
A novel homozygous point mutation in SLC34A1 is identified in the siblings
Whole exome analysis revealed a single G→A transition that resulted in the substitution of histidine for arginine at position 495 of the sodium-phosphate transporter, SLC34A1 (Figure 3, A and D). The G→A substitution was further confirmed by Sanger sequencing (Figure 3B). The high degree of conservation of the arginine residue across species (Figure 3C) and rarity of this variant in the population suggest that the mutation we identified might play a causative role in defective phosphate metabolism in this patient. A different variant at the same residue (rs199565633, p.R495C) was only observed once, and in the heterozygous state, among 6503 individuals sequenced for this region in the Exome Variant Server release version v.0.0.22. (October 17, 2013).
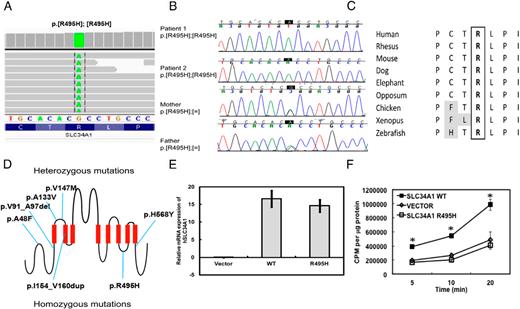
Mutation in SLC34A1 results in decreased phosphate uptake in 93 cells.
A, Results from whole exome analysis. B, Sanger sequencing of the patients and parents. C, Conservation of the arginine residue among vertebrates. D, Model of heterozygous and homozygous mutations found in SLC34A1. E, Real time PCR performed in HEK 293 cells overexpressing either the empty vector, wild type or mutant hSLC34A1 constructs. Student's t-test was performed and the asterisk indicates significance at P < .01. F, Phosphate uptake assay performed in 293 cells overexpressing either the empty vector (diamonds), wild type (squares), or mutant SLC34A1 (triangles) constructs. Student's t-test was performed and the asterisk indicates significance at P < .05, compared to the vector alone. The uptake assay was performed three separate times and each assay was done in duplicate.
Phosphate uptake is markedly reduced in cells overexpressing mutant SLC34A1
To test whether the p.Arg495His mutation resulted in dysregulated phosphate transport, we performed phosphate uptake assays using radioactive 32P in HEK 293 cells transfected with wild-type or mutant SLC34A1 construct. Both wild type and mutant hSLC34A1 constructs are expressed at similar levels and this expression was significantly higher than the control (Figure 3e). Immunofluorescence assays showed that both the wild-type and mutant proteins colocalize with wheat germ agglutinin to the cell surface (Supplemental Figure 1). Forty-eight hours post-transfection, cells were incubated with 1 μCi/mL 32P-labeled potassium phosphate for 5, 10, or 20 min and phosphate uptake assay was performed as previously described (10). Cells overexpressing the wild type SLC34A1 showed an increased phosphate uptake at all three time points as compared to cells expressing an empty vector (3F). Overexpression of mutant SLC34A1 did not result in an increased phosphate uptake and levels of radioactive phosphate remained similar to that of an empty vector control (Figure 3F).
Discussion
We have identified a homozygous single nucleotide variation in SLC34A1 in siblings who presented with hypercalcemia, hypercalciuria, hypophosphatemia, and nephrocalcinosis. The causative role of the SLC34A1 R495H mutation in the pathogenesis of the disease in our patients is supported by the following results: (a) the mutation segregates with the disease phenotype (b) the single nucleotide variant we report is novel and a different variant at the same residue is very rare (minor allele frequency of 0.00008) (c) the arginine residue is highly conserved across vertebrates and lies in the region that is thought to be involved in PTH-mediated regulation of SLC34A1 and (d) in vitro phosphate uptake experiments showed a reduction in the amount of phosphate transported by the NaPi-IIa R495H mutant.
The role of SLC34A1 or NaPi-IIa in human renal phosphate homeostasis is not well understood (8, 11). Recently, a homozygous mutation was identified in SLC34A1 in two siblings from a consanguineous family. While patients studied in the Magen et al study have renal Fanconi syndrome and hypophosphatemic rickets, we did not detect these in our patients (8). Phenotypic heterogeneity is observed both in our patients, as evidenced by the few phenotypic signs in patient 1 as compared to patient 2, his sister, and also by the difference with the other reported family though with a different mutation (8). We observe hypercalciuria but not persistent hypercalcemia in our patients; this is consistent with results from previous studies showing that a low serum phosphate concentration could stimulate 1,25 dihydroxy vitamin D synthesis, which, in turn could lead to hypercalciuria (12). Hypercalcemia has not been documented previously, but it could be due to differences in the age at which the patients were examined since in our patients, hypercalcemia was transient and spontaneously resolved at a young age (13). A study done by Chau et al showed that the expression on NaPi-IIa is key for the development of nephrocalcinosis in the NaPi-IIa null mouse model (14). They showed that Npt2a mRNA and protein are expressed in the prenatal period and the onset of nephrocalcinosis coincides with the absence of Npt2a expression (14). Additionally, hypercalciuria secondary to the increase in serum 1,25 dihydroxy vitamin D, is a risk factor for nephrocalcinosis in both rodent models and humans (12).
The mutation we identified in SLC34A1, which resulted in the substitution of histidine for arginine at position 495, resides in a domain thought to be essential for PTH-mediated endocytosis of SLC34A1 (15). Parathyroid hormone functions as a phosphaturic hormone by inhibiting sodium-phosphate co-transport, inducing transporter internalization and targeting the internalized transporter for lysosomal degradation (16–20).
In conclusion, we report a novel homozygous single nucleotide mutation in SLC34A1 that plays a causative role in the altered calcium phosphate handling in this family. The phenotypic difference from the previously reported family highlights the clinical variability of this condition. Exome sequencing, by providing detailed sequence information on most coding regions, is a powerful tool for research and the clinic. However, there are some limitations in the data that it can provide: some regions (such as GC rich regions) remain difficult to sequence, genes with other homologous sequences (eg, paralogs, pseudogenes) are difficult to map, the sensitivity for whole exon deletions is variable, and by definition it does not cover noncoding regions which could affect gene expression (promoters and enhancers, deep-intronic splicing regulators). Nevertheless, in the field of endocrinology and metabolism, it has the potential to identify new disease genes, to provide a rapid genetic diagnosis for known diseases, and to expand the phenotypic variability of previously described disorders.
Acknowledgments
We thank Shalini N Jhangiani for exome sequencing coordination, Yuqing Chen and Terry Bertin for technical support, and Erich Fradinger (IDIM) for 1,25 dihydroxy vitamin D determinations.
This work was supported by the Baylor College of Medicine (BCM) Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the BCM Advanced Technology Cores with funding from the National Institutes of Health (NIH) (AI036211, CA125123, and RR024574), the Rolanette and Berdon Lawrence Bone Disease Program of Texas, the BCM Center for Skeletal Medicine and Biology, National Insitute of Diabetes and Digestive and Kidney Diseases training Grant No. 5T32DK060445-10 (A.R.) and NIH Grant No. HD070394 (B.L.).
The ethics committee approved our study protocol and written informed consent was obtained from the parents.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
References

{kind=link}
{kind=link}
{kind=link}