





Abstract
Both the number and the activity of osteoclasts are critical for maintaining normal bone turnover. The number is determined by rates of cell differentiation and death. Fas-mediated apoptosis is a dominant mechanism for apoptosis. Here, we show the presence of the Fas receptor on mouse, human, avian, and cultured RAW264.7 (murine) derived osteoclasts and the up-regulation of its expression during mouse osteoclast differentiation. Additionally, Fas is a fully functional death receptor in osteoclasts, and its signaling pathway is consistent with classical Fas signaling in other cell systems, involving mitochondrial release of cytochrome c and activation of caspases 3 and 9. This demonstration of Fas-mediated apoptosis in mature osteoclasts provides a new and potent mechanism for the regulation of osteoclast life span. The in vivo significance of Fas-mediated apoptosis in bone (osteoclasts) was demonstrated in aged Lpr and Gld mice, which have a dysfunctional immune system. Lpr mice, which have a defect in the Fas gene, have decreased bone mineral density, bone volume, trabecular thickness, and increased osteoclast number. Gld mice, which have a Fas ligand mutation, have a slight yet insignificant decrease in bone mineral density, but a highly significant increase in osteoclast number. Taken together, these data demonstrate that the Fas/Fas ligand system is important in the regulation of bone turnover and may represent a critical link between the immune system and bone remodeling in development and in various diseases.
BONE IS THE major reservoir for calcium in the body; thus, maintaining proper metabolic balance in bone is important for maintenance of normal Ca2+ homeostasis. The balance between bone formation and resorption is determined by the number and activity of osteoblasts and osteoclasts (1). Most prevalent metabolic bone diseases, such as osteoporosis, are due to an imbalance in bone remodeling with excessive bone resorption compared with bone formation. Bone resorption has been a target for modern pharmaceutical therapy, such as bisphosphonates and calcitonin.
The rate of bone resorption is determined by both the number and activity of osteoclasts. The number of osteoclasts is dependent upon relative rates of cell differentiation and death. Osteoclast differentiation in vitro is dependent on at least two extracellular factors, macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL) (2–6). Other factors, such as IL-1, IL-6, TNF-α, and TGF-β, also affect osteoclast differentiation. It is now feasible to use in vitro models to modulate osteoclast differentiation and predict effects on bone resorption in vivo.
Another major strategy to decrease bone resorption, besides decreasing osteoclast formation, is to increase or accelerate the death rate of osteoclasts. Apoptosis, an evolutionarily conserved form of programmed cell death, plays an important role in the development and function of many tissues and cells. Agents such as estrogens, tamoxifen, and some bisphosphonates (7–10) have been reported to induce apoptosis in osteoclast-like cells; however, the cellular mechanisms by which this induction occurs are largely unknown (reviewed in Ref. 11).
The Fas/FasL system provides an important apoptotic mechanism for many cell types (12, 13), especially those of the immune system. Fas, a transmembrane death receptor of the TNF receptor family, is activated by binding of Fas ligand (FasL) or Fas activating antibody. Trimerized Fas receptors recruit FADD (Fas Associated Death Domain) and initiate a downstream caspase activation cascade, which amplifies the death signal to downstream targets. The mitochondrial component of the apoptotic process is mediated by truncated BID translocation to the mitochondria from the cytosol and subsequent cytochrome c release (reviewed in Ref. 14). It has been reported that Fas appears to be up-regulated during the differentiation of hemopoietic cells, suggesting that the Fas system participates in regulation of hemopoietic differentiation (15). Moreover, Wang et al. (16) implicated the Fas gene in alendronate-induced osteoclast apoptosis.
Defects in the Fas/FasL system cause severe immune system dysfunction, as seen in two well-characterized mouse mutations: the lymphoproliferation (Lpr) and generalized lymphoproliferative disease (Gld) mice (17–20). Aberrant transcription caused by the insertion of an early transposable element in the Fas gene in Lpr mice leads to premature termination of transcription of the Fas gene and aberrant splicing of the transcript (21, 22). Gld mice have inactive FasL due to a point mutation. These mice have normal FasL biosynthesis, surface expression, and oligomerization, but structural alterations in the Fas binding region lead to FasL dysfunction and associated phenotypic changes (23, 24). Mice bearing these two mutations develop lymphoproliferation and autoimmunity.
In this study, we have focused on cells of the monocyte/macrophage lineage, specifically examining Fas expression and function during osteoclast differentiation in vitro. Lpr and Gld mice have been used to investigate the importance of the Fas/FasL system in regulating bone remodeling in vivo. The Fas system in osteoclasts may provide an important link between the immune system and bone turnover and suggest new avenues for therapy of metabolic bone diseases.
Materials and Methods
Experimental animals
The procedures involving the animals and their care were approved by the Institutional Animal Care and Use Committee and conducted in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Osteoclast generation
Mouse osteoclasts were generated by flushing bone marrow cells from the long bones of 7- to 8-wk-old C57BL/6J mice (Harlan Industries, Indianapolis, IN, and Charles River Laboratories, Wilmington, MA) and plating overnight in α-MEM (Sigma, St. Louis, MO) containing 10% heat-inactivated fetal bovine serum (HIFBS) (Hyclone Laboratory, Inc., Logan, UT), 2 mml-glutamine (Life Technologies, Inc., Grand Island, NY), 100 U/ml penicillin (Mediatech, Inc., Herndon, VA), 100 μg/ml streptomycin (Mediatech, Inc.), and 10 ng/ml M-CSF (R&D Systems, Minneapolis, MN). After 16 h, nonadherent cells were collected and layered onto a Ficoll-Hypaque gradient (Sigma), then centrifuged at 340 × g for 15 min at room temperature. Bone marrow monocyte/macrophage lineage precursors were collected from the interface, washed, and cultured for 5–7 d in α-MEM, supplemented with 10% HIFBS, 2 mml-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 ng/ml murine M-CSF. RANKL (25) was added as indicated in the figure legends. Medium was replenished every 2 d. More than 95% of both mononuclear and multinucleated cells in these cultures stained positively for TRAP (tartrate resistant acid phosphatase) using a TRAP staining kit (Sigma).
Avian osteoclasts were isolated from the medullary bone of calcium-deprived white leghorn laying hens, Gallus domesticus, as described (26, 27). Osteoclasts, scraped from medullary bone, were enriched to 80–90% of total cells by sedimentation through 70% serum. Osteoclasts were cultured at 37 C in humidified air with 5% CO2 in DMEM (Life Technologies, Inc.), supplemented with 5% heat-inactivated calf serum and 5% heat-inactivated chicken serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Medium was changed at 1 and 3 d.
RAW 264.7 (American Type Culture Collection, Manassas, VA) cells were maintained in DMEM supplemented with 10% HIFBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Osteoclasts were differentiated from RAW264.7 cells in α-MEM supplemented with 10% HIFBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml RANKL. Medium was changed every 2 d.
Human osteoclasts were differentiated from peripheral blood mononuclear cells obtained from apheresis, enriched on a Fico/Lite-Monocytes (Atlanta Biologicals, Norcross, GA) gradient, and cultured in serum-free medium (Life Technologies, Inc.) containing human M-CSF (10 ng/ml) and mouse RANKL (100–150 ng/ml). Medium was changed every 2–3 d.
Apoptosis assay
Apoptosis was measured by Hoechst 33258 staining of condensed chromatin (28). Cells, differentiated on cover slips in the presence of 10 ng/ml of M-CSF and 25 ng/ml RANKL, were treated with various doses of the Fas activating antibody, Jo-2 (BD PharMingen, San Diego, CA) for various times, as indicated, and stained with Hoechst 33258 fluorescent dye (25 μg/ml) (Sigma) for 2 min at room temperature. The percentage of apoptotic cells was determined as apoptotic multinucleated cells/total number of multinucleated cells by fluorescence microscopy. Only cells containing more than three nuclei were included in the count.
Western blot analysis
Cells were washed twice with ice-cold PBS and scraped into a lysis buffer consisting of 150 mm NaCl, 50 mm Tris (pH 7.5), 1% Nonidet P-40, 0.25% Na-deoxycholate, 1 mm phenylmethylsulfonyl fluoride, 1 mm NaVO4, 2 mm EGTA, and 50 μg/ml leupeptin. Lysates were vortexed and incubated on ice for 15 min, and the insoluble material was cleared by centrifugation at 10,000 × g for 5 min at 4 C. Protein concentrations in the cell lysates were measured by the Lowry assay. Equal amounts of lysates were subjected to 10% SDS-PAGE and subsequently electrotransferred onto a polyvinylidene difluoride membrane (Millipore Co., Bedford, MA). After blocking with 5% nonfat milk, the membrane was incubated with purified anti-Fas (4 μg/ml) or anti-caspase-3 (4 μg/ml; catalog no. 7148, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or anti-caspase-9 antibodies (5 μg/ml; Cell Signaling Tech, Beverly, MA), followed by peroxidase-conjugated antirabbit IgG (1:2000 dilution). Immunoreactive proteins were visualized with ECL Western blotting detection reagents (Amersham, Piscataway, NJ), following the manufacturer’s instructions. As an additional control for equal loading of gels, after transfer, gels were stained with Coomassie blue R-250, destained, and examined for any abnormal protein band densities.
Caspase activity measurement
Caspase 3 activity in cells was measured using the ApoAlert Caspase Fluorescent Assay Kit (CLONTECH Laboratories, Inc., Palo Alto, CA), as directed in the manufacturers’ protocol. Briefly, cells treated with or without the Fas activating antibody were lysed in 50 μl cell lysis buffer and incubated on ice for 10 min. Cell lysates were centrifuged, and the supernatants were collected. The Reaction Buffer/DTT Mix and 1 mm caspase-3 substrate were added into each reaction and incubated for 1 h at 37 C. To confirm the correlation between protease activity and signal detection, an induced sample cell lysate was incubated with the caspase 3 inhibitor, DEVD-CHO, before adding substrate. A Fusion Universal Microplate Analyzer (PerkinElmer Life Sciences, Inc., Boston, MA) was used to measure fluorescence (400 nm excitation filter and 505 nm emission filter).
Immunohistochemistry
Cells were differentiated on cover slips and fixed in 3% formaldehyde at room temperature for 30–45 min. Cells were permeablized in 100% methanol at −20 C for 7–30 min, blocked in 1% BSA at room temperature for 15 min, and incubated with purified primary antibody at a 1:500 dilution (Fas antibody and cytochrome c antibody, Santa Cruz Biotechnology, Inc.) overnight at 4 C. Cells were reblocked in 1% BSA and incubated with fluorescence-conjugated secondary antibody (Alexa546 fluorescent-conjugated antirabbit IgG, or Alexa 488 fluorescent-conjugated antirabbit IgG, Molecular Probes, Eugene, OR) at a 1:1000 dilution at room temperature in the dark for 1 h. Cells were then stained with Hoechst 33258 Stain (25 μg/ml) for 2 min at room temperature and visualized by fluorescence microscopy.
Mitochondrial staining
After differentiation on cover slips, the medium was replaced with prewarmed (37 C) medium containing 100 nm MitoTracker Red CMXRos (Molecular Probes) for 30 min at 37 C. After incubation, the medium was replaced, and the cells were fixed. Staining was visualized by confocal fluorescence microscopy.
RT-PCR
Total RNA was extracted from cells using an RNA extraction kit (Promega, Madison, WI). cDNA was prepared using a First Strand cDNA synthesis kit (Life Technologies Inc.). The sequences of the synthesized primers used for PCR were based on the mouse sequence for actin: β-actin forward, (accgtgaaaagatgacccag); β-actin reverse, (tctcagctgtggtggtgaag); mouse primers—Fas3 forward, (ggaggcccattttgctgtcaacca); Fas4 reverse, (gtccttctggaccatgtcctg); avian primers—Fas forward, (cctgctcctcatcattgtgt); Fas reverse, (tgatccatgtactcctctcc) (Kong, F.-K., and C. H. Chen, unpublished observations; and Ref. 29). Amplification was conducted for 35–40 cycles at 94 C for 1 min, 55 C for 1 min, and 72 C for 1 min, in a 50-μl reaction mixture containing 1× reaction buffer, 1 μl of each cDNA, 20 pmol of each primer, 0.25 mm deoxynucleoside triphosphate, and 1 U of Taq DNA polymerase (Promega). After amplification, 10 μl of each reaction mixture was analyzed by 1% agarose gel electrophoresis, and the bands were visualized by ethidium bromide staining.
Flow cytometric determination of Fas
Monocyte/macrophage precursor cells were cultured in petri dishes with M-CSF (10 ng/ml) and RANKL (concentrations as indicated in figure legends). After the number of days indicated, cells were trypsinized, washed in cold PBS and FACS buffer (1% HIFBS, 0.1% NaN3 in PBS), and stained with R-phycoerythrin conjugated hamster antimouse Fas monoclonal antibody (BD PharMingen) at a 1:50 dilution at room temperature for 30 min. Cells were washed with FACS buffer one to two times, fixed with 300 μl 1% paraformaldhyde, and analyzed without gating.
Dual-energy x-ray absorptiometry (DXA) analysis
Five each of B6.mrl-tnfrsf6lpr, B6smn-C3H-tnfsf6gld, and C57BL/6J (the background control strain for both Lpr and Gld, recommended by Jackson Laboratories, Bar Harbor, ME) mice, obtained from Jackson Laboratories and from the Frederick Cancer Research Institute (Frederick, MD) were raised until 6 months of age. At 4 and 6 months of age, in vivo DXA measurements were performed with a GE-LUNAR PIXImus densitometer (GE-Lunar, Madison, WI) using software version 1.4. To measure whole body bone mineral density (BMD), mice were anesthetized with isofluorane and placed in sternal recumbency on the scanner as previously described and validated (30). Because of the small image area of the DXA, a portion of the animal’s head was outside the image. Therefore, it was necessary to exclude the remaining portion of the skull from the region of interest (ROI) during the analysis as previously described (30). All DXA scans were conducted by the same technician.
Bone histomorphometry
At 6 months of age, the animals were euthanized by exposure to carbon dioxide. All procedures were approved by the Institutional Animal Care and Use Committee. The left rear limbs were dissected, fixed in 10% phosphate-buffered formalin, decalcified with EDTA, embedded in paraffin, sectioned, and stained with hematoxylin and eosin and TRAP. Bone histomorphometry was performed by the methods of Parfitt et al. (31) using Bioquant Image Analysis software (R&M Biometrics, Nashville, TN). Specifically bone area, tissue area, bone area/tissue area (BV/TV), trabecular thickness, trabecular number, and trabecular space were measured. A ROI was selected that was exactly 1 mm distal to the growth plate and extending 2 mm downward through the metaphysis of the long bones. For TRAP staining measurements, the ROI was centered at the midpoint of the epiphysial growth plate of the tibia, containing the provisional calcification layer and the primary spongiosa, excluding cortical bone. Five circles, having radii of 100 μm, were placed adjacent to each other and following the curvature of the growth plate to form the ROI. The proximal side of the circles contained the primary spongiosa. The tangents of the circles were connected to form the 1.85-mm2 ROI. The total tissue area was quantified by measuring the cumulative pixel area within the ROI. The pixels from the interior of the ROI that visually contained the positive red stain color, indicating TRAP activity, were selected. The TRAP-positive area was measured by a video count array in which the area of the red-colored pixels, relative to the magnification, was reported as square microns. The total percentage of TRAP activity was reported as the percentage of TRAP-positive area divided by the total tissue area. This technique correlates extremely well with standard osteoclast counting with a correlation coefficient (r2) of 0.903 (n = 50; Sawyer, A. A., and J. M. McDonald, submitted for publication).
Statistical analysis
Differences between data were analyzed with the Student’s unpaired one-tailed t test.
Results
Fas is present in mouse and avian osteoclasts and osteoclasts derived from the RAW 264.7 cell line
We used a number of different methods to detect Fas in osteoclasts from several species. By RT-PCR, Fas was detected at the mRNA level in osteoclasts differentiated from mouse primary bone marrow cells, RAW264.7 cells, and mature avian osteoclasts (Fig. 1A). A-20 cells, mouse B cell line, were analyzed as a positive control. Because PCR products of murine Fas and avian Fas were different sizes, they migrated differently in two experiments compared with the actin control. By Western blotting, using an antibody against mouse Fas, we detected Fas protein in cell lysates from mouse osteoclasts differentiated from primary bone marrow cells (Fig. 1B, lane 1) and from RAW264.7 cells (Fig. 1B, lane 3). In both preparations, a band at 45–49 kDa was detected, which is consistent with the reported molecular mass of Fas (32). The specificity of antibody binding was determined by preabsorbing the antibody with the peptide that the polyclonal antibody was raised against (Fig. 1B, lane 2). Fas protein was also detected in osteoclasts derived from human precursors and in mature osteoclasts from chickens (Fig. 1B, lanes 4 and 5). The weak signal for avian Fas is probably due to the fact that antihuman Fas antibody was used, which has poor cross-reactivity with avian Fas protein, no specific antibody for avian Fas being available. To demonstrate Fas expression on the surface of osteoclasts, we analyzed cell surface staining with another Fas antibody (R-PE conjugated hamster antimouse Fas monoclonal antibody, Jo-2) by flow cytometry, using osteoclasts differentiated from mouse primary bone marrow cells and RAW264.7 cells (Fig. 1C). The top panel shows Fas staining in osteoclasts derived from mouse primary bone marrow cells. The bottom panel of Fig. 1C demonstrates the Fas staining in mouse osteoclasts differentiated from RAW264.7 cells. Osteoclasts from primary bone marrow cells have more Fas expression at the cell surface compared with osteoclasts derived from RAW264.7 cells (Fig. 1C).
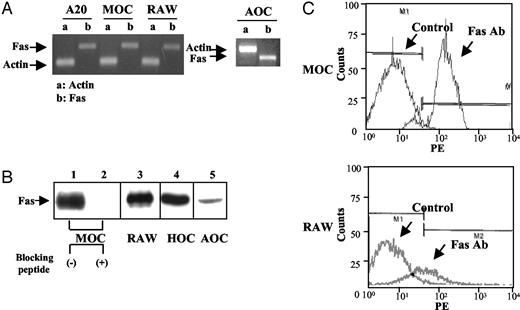
Expression of Fas in osteoclasts. A, Detection of Fas mRNA by RT-PCR. Total RNA was extracted from mouse osteoclasts differentiated from mouse primary bone marrow cells (MOC); RAW264.7 cell-derived mouse osteoclasts (RAW); and purified avian osteoclasts (AOC). A-20 cells (mouse B cell line) were used as positive control. Fas and actin mRNA were detected by RT-PCR, which was performed as described in Materials and Methods. B, Detection of Fas protein in osteoclasts by Western blotting. Osteoclasts were generated as described in Materials and Methods. Lysates from osteoclasts generated from mouse primary bone marrow cells (MOC, 45 μg, lane 1) and RAW264.7 cell-derived osteoclasts (RAW, 30 μg, lane 3) were blotted with purified antimouse Fas antibody. Blocking peptide (20 μg/ml) was added to the blot-containing lysate from osteoclasts differentiated from mouse primary bone marrow cells (MOC; 45 μg, lane 2). Cell lysates from human peripheral blood cell-derived osteoclasts (HOC, 30 μg) and mature avian osteoclasts (AOC, 50 μg) were blotted with antihuman Fas antibody (lanes 4 and 5). Bands at 45–49 kDa were detected. C, Detection of Fas by flow cytometry. Osteoclasts derived from both mouse bone marrow and RAW264.7 cells were cultured in a petri dish and detached by incubation with trypsin-EDTA. Cells were fixed, stained with anti-Fas-PE (1:50) at room temperature for 30 min, and flow cytometry was performed. Top, Fas expression in osteoclasts derived from mouse primary bone marrow cells. Bottom, Fas expression in RAW264.7-derived osteoclasts. Left peaks show control staining. Right peaks show Fas expression in the cell surface.
Fas is present on the surface of both TRAP-positive mononuclear cells and multinucleated mature osteoclasts
Mouse osteoclasts differentiated in vitro from monocyte/macrophage precursors yield a mixed cell population that includes both TRAP-positive mononuclear and mature multinucleated cells. To observe Fas expression on individual cells, we used immunohistochemistry. Immunostaining was strongly localized around the periphery of multinucleated osteoclasts (double arrows) with little or no cytoplasmic staining, and was present over the cytoplasm in mononuclear cells (single arrow) (Fig. 2B). The specificity of Fas immunostaining was demonstrated using controls lacking primary antibody (Fig. 2A) and by addition of a blocking peptide (Fig. 2C).
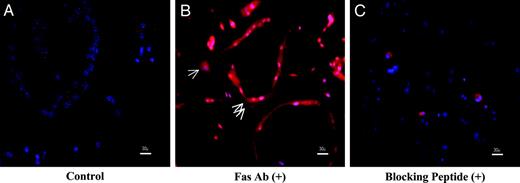
Immunohistochemistry of Fas in osteoclasts. Differentiated mouse osteoclasts were stained with Fas antibody and subsequently with Alexa546 fluorescent-conjugated antirabbit IgG. A, Control, without Fas antibody. B, Stained with Fas antibody. The red color represents specific Fas staining. Single arrow, mononuclear cells; double arrows, multinuclear cells. C, Stained with Fas antibody in the presence of blocking peptide (20 μg/ml). Scale bar, 30 μm.
Expression of Fas during osteoclast differentiation
During osteoclast differentiation, Fas expression on the cell surface of osteoclast precursors increases. Only a small portion of monocyte/macrophage precursors isolated from bone marrow had Fas expressed on the surface, as determined by flow cytometry. Fas expression was highly up-regulated after 6 d incubation with M-CSF and RANKL (Fig. 3A). Consistent with the flow cytometry results, Fas expression, detected by Western blotting, was highly up-regulated after 6 d incubation with M-CSF and RANKL (Fig. 3B). The relative abundance of Fas protein by Western blotting was evaluated by densitometric scanning of four separate experiments (Fig. 3C).
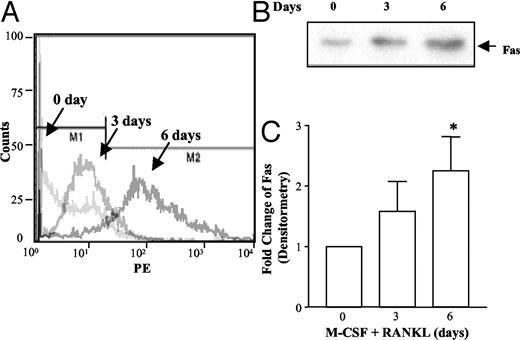
Expression of Fas during differentiation. Mouse osteoclast precursors were differentiated by the addition of M-CSF (10 ng/ml) and RANKL (50 ng/ml), as indicated for various times: 0, 3, and 6 d. On d 0, precursors were collected after Ficoll-Hypaque gradient centrifugation; on d 3 and 6, cells were collected after incubation with M-CSF (10 ng/nl) and RANKL (50 ng/ml). Differential expression of Fas was demonstrated by flow cytometry and Western blotting. A, Cells were collected after treatment as indicated, fixed, stained with Fas-PE antibody, and analyzed by FACS. B, Cells were lysed after being treated as indicated, and identical amounts of protein were separated by SDS-PAGE and immunoblotted with anti-Fas antibody. C, Densitometric scanning of Fas bands from four independent Western blot experiments was performed and is presented as mean ± sem. *, P < 0.05 vs. d 0.
Fas activating antibody induces apoptosis in multinucleated osteoclast-like cells
Stimulation of apoptosis by Fas activating antibody is both time- and dose-dependent (Fig. 4, A and B). Both binding of FasL to the Fas receptor and cross-linking of Fas activating antibody induce apoptosis in Fas-bearing cells (33–35). Mouse osteoclast-like cells treated with increasing concentrations of Fas activating antibody, ranging from 0–1 μg/ml for 24 h at 37 C, showed a concentration-dependent increase in apoptosis. The maximal number of apoptotic cells was seen at 0.5 μg/ml (Fig. 4A). Apoptosis induced by Fas activating antibody was time-dependent, with maximum apoptosis at 20 h (Fig. 4B). These data were obtained using Hoechst 33258 staining to detect characteristic apoptotic changes in the nuclei (Fig. 4C). The nuclei of apoptotic cells are condensed and fragmented (Fig. 4C, right panel, arrows). In nonapoptotic cells, nuclei were round and smooth (Fig. 4C, left panel). Only those multinucleated cells that had condensed chromatin were counted as apoptotic.
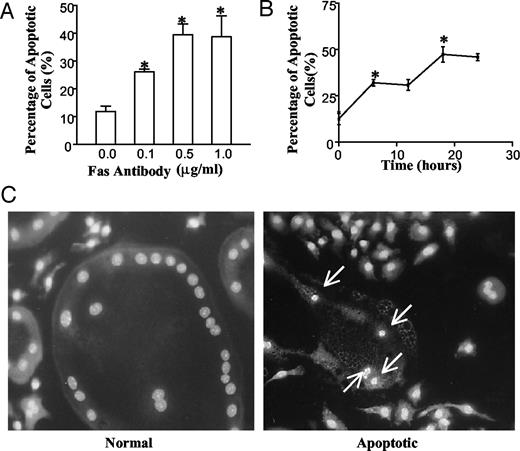
Fas-mediated apoptosis in multinucleated osteoclasts. Osteoclasts were generated as described in Materials and Methods. A, Dose-dependent induction of apoptosis. Cells were treated with anti-Fas activating antibody at concentrations from 0 to 1 μg/ml for 24 h. Results are presented as the mean percentage of apoptotic cells ± sem (n = 3). *, P < 0.05 vs. control. B, Time-dependent induction of apoptosis. Osteoclast-like cells were treated with 0.5 μg/ml anti-Fas activating antibody for different time periods as indicated. Results are presented as the mean percentage of apoptotic cells ± sem (*, n = 3), each point P < 0.05 vs. control. C, Demonstration of nuclear staining. Hoechst 33258 staining of normal nuclei (left) and apoptotic nuclei (right, arrows). In panels A and B, apoptosis was quantified by counting multinucleated cells with condensed nuclei, stained with Hoechst.
Signaling molecules involved in Fas-mediated apoptosis
Caspase activation is involved in Fas-mediated apoptosis in osteoclasts. Cleavage of procaspases into their enzymatically active forms is a critical step in most forms of apoptosis, whereas caspase-3 is the common effector caspase for most pathways. Disappearance of the noncleaved form of caspase-3, as detected by Western blotting, was used to demonstrate its activation (Fig. 5A). As can be seen, the amount of procaspase 3 was decreased due to cleavage by treatment with increasing concentrations of Fas antibody (Fig. 5A). The relative abundance of procaspase 3 protein by Western blotting was evaluated by densitometric scanning of five separate experiments (Fig. 5B). A fluorescent assay was also used to confirm the activation of caspase 3 induced by Fas activating antibody (Fig. 6). Caspase 3 activation induced by Fas activating antibody in A-20 cells was used as a positive control. The caspase 3 inhibitor DEVD-CHO was able to inhibit caspase 3 activity induced by the antibody (Fig. 6). Caspase 9 is also activated by Fas antibody (Fig. 7). Shown in Fig. 7 is the increase in the cleaved forms of caspase 9 with increasing concentrations of Fas antibody. Band density was quantitated by densitometry, and fold changes are indicated beneath the blot (Fig. 7).
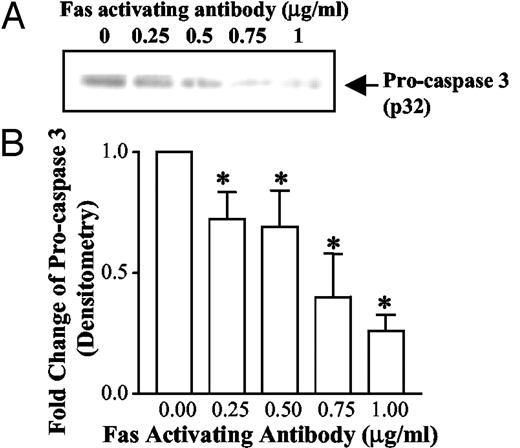
Caspase 3 is activated by Fas activating antibody in osteoclasts. Osteoclasts were differentiated in the presence of M-CSF and RANKL for 6–9 d. Differentiated osteoclasts were treated with Fas-activating antibody at concentrations from 0–1 μg/ml for 24 h. A, Analysis of caspase-3 activation by Western blotting. Cell lysates were immunoblotted with an anti-caspase-3 antibody that recognizes the proform (molecular mass = 32 kDa). B, Densitometric scanning of results from five Western blotting experiments was performed, and results are presented as mean ± sem. *, P < 0.05, vs. no Fas activating antibody.
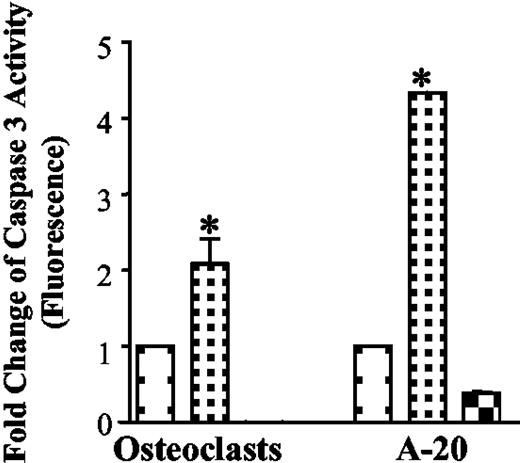
Caspase 3 activity is increased by Fas activating antibody. Differentiated osteoclasts were treated with or without Fas activating antibody (1 μg/ml) for 24 h. Caspase 3 activity was measured as in the manufacturer’s instructions. Six experiments were combined, and the results are presented as mean ± sem. Caspase 3 activity induced by Fas activating antibody (1 μg/ml) for 4 h in A-20 cells was used as positive control (n = 2). Caspase 3 inhibitor was added to A-20 cell lysate to confirm the correlation between protease activity and signal detection (n = 2). □, Treatment with Fas activating antibody, 0 μg/ml; □, treatment with Fas activating antibody, 1 μg/ml; and □, treatment with Fas activating antibody (1 μg/ml) plus DEVD-CHO. *, P < 0.05, vs. Fas activating antibody (0 μg/ml).

Caspase 9 is activated by Fas activating antibody in osteoclasts. Differentiated osteoclasts were treated with Fas activating antibody at concentrations from 0 to 1 μg/ml for 24 h. Cell lysates were immunoblotted with a caspase-9 antibody that recognizes the cleaved forms (molecular mass = 39 and 37 kDa). Band densities were measured (p39), and fold changes were indicated beneath the blot.
The osteoclast is a very metabolically active cell requiring high ATP use to drive the H+-ATPase of the ruffled membrane. For this reason, these cells are rich in mitochondria, like mitochondria-rich, proton-secreting epithelial cells (36, 37). Mitochondria are also important in mediating apoptosis (38). Apoptotic stimuli, such as the Fas/FasL interaction, trigger the release of cytochrome c from the mitochondria into the cytosol (39, 40). This cytochrome c release can be observed directly by immunostaining for cytochrome c and simultaneously staining the mitochondria with MitoTracker and observation by confocal microscopy (Fig. 8). The top panels of Fig. 8 show cytochrome c staining (left, green color), mitochondrial staining (middle, red color), and the overlay for both (right, yellow and orange color) in a normal, multinucleated mouse osteoclast. In normal cells, cytochrome c is colocalized almost entirely with the mitochondria. The bottom panels show cytochrome c release from osteoclast mitochondria after Fas antibody treatment. In the apoptotic cell, the pattern of cytochrome c staining (green color) was altered to a diffuse pattern seen in the cytosol (indicated by arrows in the overlay), only partially colocalizing with mitochondrial staining (yellow color). These data demonstrate that mitochondrial release of cytochrome c is involved in Fas-mediated apoptosis in mouse osteoclasts.
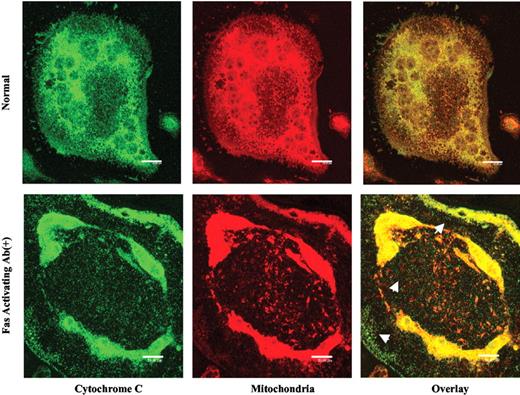
Localization of cytochrome c and mitochondria. Osteoclasts were differentiated on coverslips in the presence of M-CSF and RANKL for 6 d. Cells were stained with a cytochrome c antibody (left panels) and MitoTracker Red CMXRos (middle panels), and subsequently with Alexa488-conjugated antirabbit IgG. The right panels show the overlay of the left and middle panels. Staining was visualized by confocal fluorescence microscopy. The upper panels show localization of cytochrome c and mitochondria in normal multinucleated osteoclasts. The lower panels show localization of cytochrome c and mitochondria in an apoptotic cell induced by Fas activating antibody. Cytochrome c (green) released from the mitochondria into the cytosol is indicated by arrows. Scale bar, 20 μm.
Abnormal BMD and bone histomorphometry in Lpr and Gld mice
Lpr and Gld mice have spontaneous mutations in Fas and FasL, respectively. Defective Fas/FasL signaling in these two strains results in autoimmunity and lymphoproliferation. Figure 9A shows that control, Lpr, and Gld mice had similar body weights (at 6 months of age: control, 31.2 ± 2.5 g, n = 4; Lpr, 30.1 ± 5.4 g, n = 5; and Gld, 33.6 ± 2.2 g, n = 5; P > 0.05 vs. control, and no difference was also seen at 4 months of age). At 4 months of age, Lpr mice had a small, but significant decrease in whole body BMD compared with control mice. In contrast, Gld mice had a slight, yet nonsignificant decrease in BMD compared with the control group (Fig. 9B). Repeated scans were performed on the same groups of animals two months later, and similar results were obtained (Fig. 9C). We investigated whether decreased BMD was due to increased osteoclast number in mice with defective Fas signaling. At 6 months of age, both Lpr and Gld mice had stronger staining for TRAP activity than control mice (see Fig. 10B for two examples of each). Quantitatively, in Lpr and Gld mice, TRAP activity was increased 61.5 ± 6.25% (mean ± sem) and 76.6 ± 13.9% (mean ± sem), respectively (Fig. 10A). Consistent with this, we observed that osteoclast surface/bone perimeter is increased 42 and 53% in Lpr and Gld mice, respectively, compared with control mice (data not shown). Assessment of other bone histomorphometric parameters showed that Lpr mice had significantly decreased bone volume and trabecular bone volume fraction (BV/TV), compared with those in the control group. Lpr mice also had a significant decrease in trabecular thickness, compared with control mice (Table 1). However, trabecular number and trabecular space did not change significantly. Consistent with the BMD results, Gld mice had no change in the various bone histomorphometric parameters.
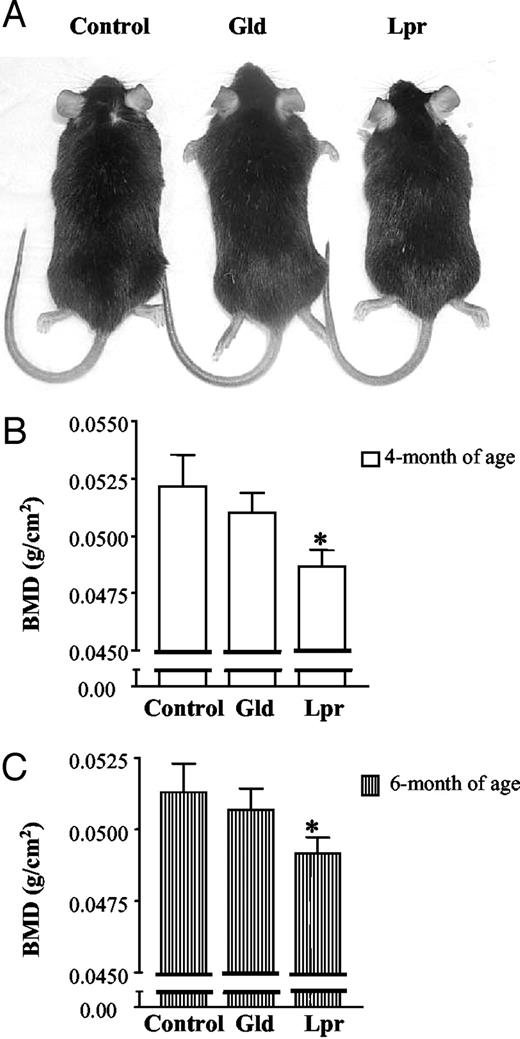
BMD change in Lpr and Gld mice. A, Sizes of control, Gld, and Lpr mice at 6 months of age. B, BMD at 4 months of age. At 4 months of age, four control mice and five Lpr and Gld mice were placed on a PIXImus densitometer, and whole body BMD was measured. C, BMD at 6 months of age for the same groups of mice. Results are presented as mean ± sem. *, P < 0.05 vs. control.
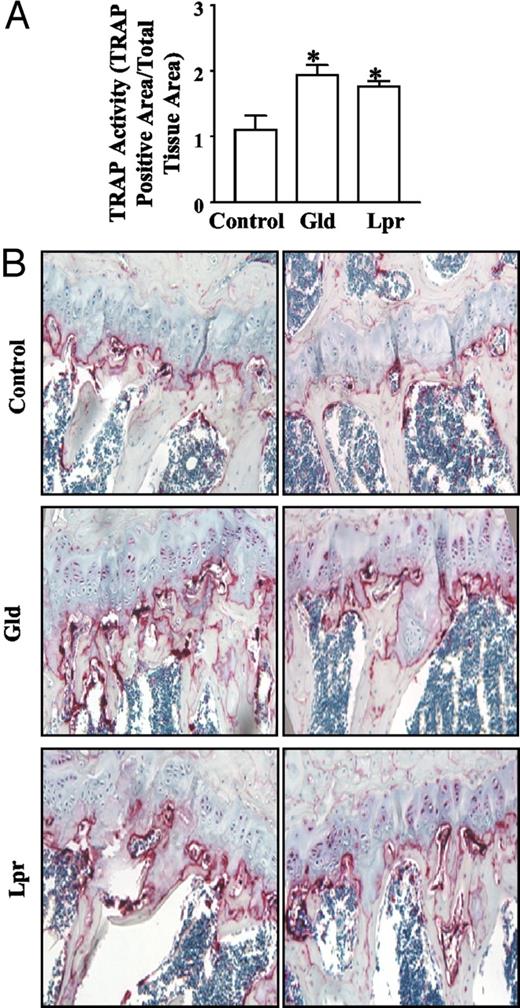
TRAP activity changes in Lpr and Gld mice. At 6 months of age, mice were euthanized, and long bones were isolated and processed for histomorphometric analysis. A, Three bone sections from two control mice, four bone sections from three Gld mice, and four bone sections from three Lpr mice were processed from paraffin-embedded left rear limbs and stained for TRAP activity. Bioquant Digital software was used to analyze the data. TRAP activity was presented as TRAP-positive stained area/total tissue area. Results are presented as mean ± sem. *, P < 0.05 vs. control. B, Examples of TRAP-stained images. Two representative images from the bones of control, Gld, and Lpr mice are shown. TRAP staining is red.
Bone histomorphometric parameters (in 6-month-old mice)
| Control (n = 2) | Gld (n = 3) | Lpr (n = 3) | |
|---|---|---|---|
| BV (mmb) | 0.20 ± 0.03 | 0.15 ± 0.03 | 0.08 ± 0.00a |
| TV (mmb) | 1.55 ± 0.12 | 1.37 ± 0.06 | 1.28 ± 0.02 |
| BV/TV | 13.0 ± 0.8 | 10.9 ± 2.6 | 6.1 ± 0.2b |
| Trabecular thickness (μm) | 55.4 ± 4.8 | 46.6 ± 9.8 | 27.4 ± 1.6a |
| Trabecular number (mm−1) | 2.4 ± 0.2 | 2.36 ± 0.39 | 2.3 ± 0.1 |
| Trabecular space (μm) | 369.2 ± 25.2 | 400.8 ± 67.8 | 419.0 ± 19.0 |
| Control (n = 2) | Gld (n = 3) | Lpr (n = 3) | |
|---|---|---|---|
| BV (mmb) | 0.20 ± 0.03 | 0.15 ± 0.03 | 0.08 ± 0.00a |
| TV (mmb) | 1.55 ± 0.12 | 1.37 ± 0.06 | 1.28 ± 0.02 |
| BV/TV | 13.0 ± 0.8 | 10.9 ± 2.6 | 6.1 ± 0.2b |
| Trabecular thickness (μm) | 55.4 ± 4.8 | 46.6 ± 9.8 | 27.4 ± 1.6a |
| Trabecular number (mm−1) | 2.4 ± 0.2 | 2.36 ± 0.39 | 2.3 ± 0.1 |
| Trabecular space (μm) | 369.2 ± 25.2 | 400.8 ± 67.8 | 419.0 ± 19.0 |
Data represents mean ± sem.
P < 0.05, and
P < 0.01 compared with control, by unpaired t test.
Bone histomorphometric parameters (in 6-month-old mice)
| Control (n = 2) | Gld (n = 3) | Lpr (n = 3) | |
|---|---|---|---|
| BV (mmb) | 0.20 ± 0.03 | 0.15 ± 0.03 | 0.08 ± 0.00a |
| TV (mmb) | 1.55 ± 0.12 | 1.37 ± 0.06 | 1.28 ± 0.02 |
| BV/TV | 13.0 ± 0.8 | 10.9 ± 2.6 | 6.1 ± 0.2b |
| Trabecular thickness (μm) | 55.4 ± 4.8 | 46.6 ± 9.8 | 27.4 ± 1.6a |
| Trabecular number (mm−1) | 2.4 ± 0.2 | 2.36 ± 0.39 | 2.3 ± 0.1 |
| Trabecular space (μm) | 369.2 ± 25.2 | 400.8 ± 67.8 | 419.0 ± 19.0 |
| Control (n = 2) | Gld (n = 3) | Lpr (n = 3) | |
|---|---|---|---|
| BV (mmb) | 0.20 ± 0.03 | 0.15 ± 0.03 | 0.08 ± 0.00a |
| TV (mmb) | 1.55 ± 0.12 | 1.37 ± 0.06 | 1.28 ± 0.02 |
| BV/TV | 13.0 ± 0.8 | 10.9 ± 2.6 | 6.1 ± 0.2b |
| Trabecular thickness (μm) | 55.4 ± 4.8 | 46.6 ± 9.8 | 27.4 ± 1.6a |
| Trabecular number (mm−1) | 2.4 ± 0.2 | 2.36 ± 0.39 | 2.3 ± 0.1 |
| Trabecular space (μm) | 369.2 ± 25.2 | 400.8 ± 67.8 | 419.0 ± 19.0 |
Data represents mean ± sem.
P < 0.05, and
P < 0.01 compared with control, by unpaired t test.
Discussion
Control of osteoclast apoptosis has recently been recognized as a critical regulatory factor in bone remodeling (1), and alteration in osteoclast apoptosis has been shown to contribute to the pathogenesis of postmenopausal osteoporosis (1, 41). Pharmaceutical agents such as bisphosphonates and tamoxifen inhibit osteoclastic bone resorption, at least in part, by inducing osteoclast apoptosis (7, 42, 43), further supporting osteoclast apoptosis as an important therapeutic target in the treatment of bone disease. However, key regulatory mechanisms underlying osteoclast apoptosis have not been fully identified and characterized. Elucidation of the mechanisms controlling osteoclast apoptosis will not only help improve the efficacy of available drugs, but more importantly, may also reveal novel strategies for drug development.
Fas, a pivotal member of the death receptor family, has been reported to be present in hematopoietic cells and is potentially important in the maintenance of their homeostasis (15). In human bone marrow, FasL expression has been identified in myeloid, lymphoid, and a subset of CD34 (+) cells (44). We also detected a small amount of FasL expressed on the surface of differentiated osteoclasts (data not shown). With the presence of FasL in the environment, several studies have been initiated to explore the role of Fas in osteoclast apoptosis. Wang et al. (16) also reported that alendronate, one of the bisphosphonates, induced apoptosis in osteoclast-like cells expressing the Fas gene, thus implicating Fas in osteoclast apoptosis. Furthermore, a recently published study also described the involvement of the Fas/FasL system in TNF-α/IL-12 mediated apoptosis in mouse bone marrow cell cultures (45), indicating that functional FasL is present in adherent stromal/osteoblast cells and triggers Fas-mediated apoptosis in nonadherent TRAP-positive cells. However, the use of a whole bone marrow cell preparation, i.e. a mixed population containing many cell types, did not permit these investigators to definitively elucidate either the underlying molecular mechanisms or the in vivo effects of the Fas/FasL system.
Here, we demonstrate the presence of a potent mechanism for osteoclast apoptosis in mature osteoclasts, the Fas system. We show that Fas is expressed in mature osteoclasts and is functional, inducing apoptosis after treatment with Fas activating antibody in both concentration- and time-dependent manners. Fas expression is up-regulated during osteoclast differentiation, which further suggests that Fas might critically influence osteoclast life span. In other apoptotic systems, cytochrome c released into the cytoplasm binds to Apaf-1, which interacts with pro-caspase-9 promoting its proteolytic activation. Furthermore, we observed that Fas activating antibody induced chromatin condensation in mouse osteoclasts, for which caspase 3 has been shown to be indispensable (46). Consistent with these data, we demonstrated that the Fas-mediated death signal in mouse osteoclasts involves activation of both caspase 3 and caspase 9 and release of cytochrome c from the mitochondria. These results are consistent with the general mechanism of Fas-mediated apoptosis.
More significantly, our in vitro data are supported by in vivo data obtained from studies using Lpr and Gld mice. Lpr and Gld mice have a defect in Fas gene transcription and a mutation of the FasL genes, respectively, causing defective regulation of apoptosis, e.g. lymphocyte apoptosis in vitro (47). As in mice, human autoimmune lymphoproliferative syndrome patients have been reported to have human Lpr and Gld-like mutations (48, 49). Therefore, we reasoned that Lpr and Gld mice, models for Fas/FasL system defects, are excellent animal models for studying the in vivo role of the Fas/FasL system in the regulation of bone remodeling. We showed that 6-month-old Lpr mice had more osteoclasts (increased bone TRAP staining) compared with the control mice, indicating that the defect in Fas leads to prolonged osteoclast survival. The increase in osteoclast number resulted in a significantly decreased BMD (Fig. 7A), also decreased bone volume and trabecular thickness. Similarly, Gld mice had more osteoclasts compared with control animals, further supporting the concept that the Fas/FasL system plays a pivotal role in osteoclast apoptosis in vivo. However, in contrast to the Lpr mice, the Gld mice had a small but not statistically significant decrease in BMD, bone volume, and trabecular thickness. This discrepancy may be explained by the observation that osteoblasts, the bone forming cells, also express functional Fas (Ref. 50 , and Zayzafoon, M., and J. M. McDonald, unpublished data). Therefore, the defect in the Fas/FasL system enhances osteoblast life span as well. The net effect of the Fas/FasL system on BMD results from the actions of this system on both osteoclasts and osteoblasts. Thus, bone turnover rates and osteoblast number will need to be determined to clarify the role of Fas in bone biology using this model. Moreover, our unpublished data show that RANKL, a potent inhibitor of osteoclast apoptosis, antagonizes the effect of the Fas/FasL system on osteoclast apoptosis, in part, by down-regulation of Fas expression in osteoclasts (51). The potential effect of RANKL on the Fas signal in osteoclasts might also be responsible for the difference between Lpr, the Fas-defective mice, and Gld, the FasL-defective mice. This effect on Fas must be fully characterized at the molecular level.
We did not detect any significant changes in BMD, bone volume, and trabecular thickness in 4- and 6-month-old male Gld mice. Katavic et al. (52) reported that increased BMD is a characteristic of 3-month-old female Gld mice. It is well known that estrogen participates in the regulation of bone turnover and favors bone formation over bone resorption. Moreover, FasL has been shown to be increased by estrogen in human monocytes (53). Katavic et al. (52) also showed that in female Gld mice, osteoclast number did not increase as dramatically as that in control mice after ovariectomy. This is consistent with estrogen causing a decrease in osteoclast number by increasing FasL in osteoclasts or other bone marrow cells. Therefore, the gender difference may account, at least in part, for the difference between our results and those of Katavic et al. (52). In addition, age is an important factor that affects bone turnover. Mice used in our histomorphometric studies were older (6 months) than those of Katavic et al. (3 months). Osteoblasts are dominant during young bone growth in young animals; however, osteoclasts dominate bone remodeling after bones reach maturity. Therefore, differences in age and gender in the two experiments make direct comparisons impossible. Further study is needed to address the question whether estrogen/androgens play a role in the regulation of the Fas/FasL system in osteoclasts. Measurements of bone formation rate and bone turnover rate will be required to interpret the difference between two studies.
Osteoclast number is determined by the net effect of osteoclastogenesis and apoptosis. Although we showed that osteoclasts undergo Fas-mediated apoptosis and Fas-defective mice have lower BMD and increased osteoclasts, we cannot exclude the possibility that the Fas system plays a role in the regulation of osteoclast formation. Osteoclast generation involves cell proliferation and differentiation. Like RANK, Fas is a member of the TNF receptor family, and in human glioma cells, Fas has been shown to stimulate cell proliferation in addition to mediating apoptosis (54). To rule out the involvement of Fas in stimulating osteoclast formation, cell proliferation rate was measured in osteoclast precursors isolated from control, Lpr, and Gld mice. There is no difference in cell proliferation rate among cells from the three types of mice (data not shown), however, osteoclast precursor cells isolated from Lpr and Gld mice, cultured for 4 d in the presence of RANKL (50 ng/ml) and M-CSF (10 ng/ml), had more TRAP activity than cells from control mice (data not shown). Because cell proliferation is not affected by Fas signaling, the increased number of osteoclasts from Fas-defective mice is mainly due to increased osteoclast survival.
There is increasing evidence linking the immune system to bone turnover. In addition to the physical proximity of the cells of these systems, they share the same progenitors; they share several cytokines and signaling molecules, which are essential for normal development and function of both systems, such as RANKL/RANK/ osteoprotegerin (reviewed in Ref. 55). The same molecules regulate the function of the two different systems within the same microenvironment, and thus their interactions may be involved in modulating bone abnormalities seen in pathological conditions such as rheumatoid arthritis, periodontal disease, metastatic cancer, chronic viral infection, and even in fracture healing, which involves immune cell infiltration. For example, rheumatoid arthritis affects skeletal tissue remodeling. Studies show that patients with early rheumatoid arthritis have a generalized reduction in bone mass and increased risk of fracture (56). An increase in bone resorption, rather than decreased bone formation, appears to be responsible for the loss of bone, which is associated with disease activity (57, 58) (reviewed in Ref. 59).
Our demonstration of fully functional Fas on mature osteoclasts provides a potentially important link between the immune and skeletal systems that may be especially significant under inflammatory circumstances. T cell infiltration and activation may contribute substantially to bone destruction. Inflammatory cytokine secretion by T cells and the cytokine network in the bone marrow microenvironment determine the response of osseous cells, thereby regulating bone remodeling. For example, the secretion of interferon (IFN)-γ by activated T cells inhibits osteoclastogenesis by increasing TRAF6 degradation and opposing the positive effect of RANKL on osteoclast formation and bone turnover (60). IFN-γ has been shown to up-regulate Fas expression in other systems (61, 62). Therefore, IFN-γ may decrease osteoclast number by sensitizing cells to Fas-mediated apoptosis. On the other hand, T-helper type II cytokines (IL-4, IL-5, and granulocyte macrophage-colony-stimulating factor) have a specific inhibitory effect on Fas mRNA (63). Also, in rheumatoid arthritis, some proinflammatory cytokines, such as TNF-α and IL-1β, can inhibit Fas antibody-induced apoptosis in synovial cells (64). Negative regulation of Fas expression and thus Fas-mediated apoptosis in osteoclasts may result in an extended life span of osteoclasts, thus deeper bone erosions. Hence, regulation of Fas in osteoclasts by cytokines, secreted by activated T cells, may be responsible for the destructive effects on bone, under inflammatory conditions.
In conclusion, both our in vitro and in vivo studies show that Fas-mediated apoptosis is a potent mechanism for apoptosis in osteoclasts. This finding not only defines an important mechanism governing osteoclast apoptosis but also raises many important questions. Given that estrogen deficiency has been implicated in causing postmenopausal osteoporosis by decreasing osteoclast apoptosis, an important question to be answered is whether estrogen promotes osteoclast apoptosis directly or indirectly, via the Fas/FasL system. A second question is whether the Fas/FasL system regulates osteoclast apoptosis using any cell-type-specific signaling pathways. If such pathways exist, they would provide foundations for the design of osteoclast-specific therapeutic interventions in bone disorders, especially given that many cell types, including osteoblasts and immune cells, use the general apoptosis-signaling system. Thus, future research will elucidate the precise Fas-mediated intracellular signaling pathways involved in osteoclast apoptosis.
Acknowledgments
We thank Dr. Steve L. Teitelbaum (Professor, Department of Pathology, Washington University School of Medicine) and Dr. Chen Lo H Chen [Research Professor, Department of Microbiology, University of Alabama at Birmingham (UAB)] for providing reagents. We also appreciate the Histomorphometry and Molecular Analysis Core Laboratory of the Center for Metabolic Bone Disease and the High Resolution Imaging Facility at UAB for their invaluable assistance.
This work is funded by National Institutes of Health Grants RO1 AR 43225 (to J.M.M.), P30 AR 46031 (to J.M.M.), and DK56336 (to T.R.N.).
Abbreviations
- BMD
Bone mineral density
- BV/TV
bone area/tissue area
- DXA
dual-energy x-ray absorptiometry
- FasL
Fas ligand
- Gld
generalized lymphoproliferative disease
- HIFBS
heat-inactivated fetal bovine serum
- IFN
interferon
- Lpr
lymphoproliferation
- M-CSF
macrophage-colony stimulating factor
- RANKL
receptor activator of nuclear factor-κB ligand
- ROI
region of interest
- TRAP
tartrate resistant acid phosphatase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}