
Abstract
Regulatory T cells (Tregs) are essential in the maintenance of immunity, and they are also a key to immune suppressive microenvironment in solid tumors. Many studies have revealed the biology of Tregs in various human pathologies. Here we review recent understandings of the immunophenotypes and suppressive functions of Tregs in melanoma, including Treg recruitment and expansion in a tumor. Tregs are frequently accumulated in melanoma and the ratio of CD8+ T cells versus Tregs in the melanoma is predictive for patient survival. Hence, depletion of Tregs is a promising strategy for the enhancement of anti-melanoma immunity. Many recent studies are aimed to target Tregs in melanoma. Distinguishing Tregs from other immune cells and understanding the function of different subsets of Tregs may contribute to better therapeutic efficacy. Depletion of functional Tregs from the tumor microenvironment has been tested to induce clinically relevant immune responses against melanomas. However, the lack of Treg specific therapeutic antibodies or Treg specific depleting strategies is a big hurdle that is yet to be overcome. Additional studies to fine-tune currently available therapies and more agents that specifically and selectively target tumor infiltrating Tregs in melanoma are urgently needed.
Similar content being viewed by others


Introduction
Regulatory T cells (Tregs) are well known to be involved in the immune regulatory activity [1]. Numerous mechanisms of Tregs mediated immune suppression have been investigated in autoimmune diseases, allergy, acute and chronic infection, pregnancy, and cancer [2, 3]. Depletion of Tregs in systemic immunity evokes severe human disease [4]. With the development of technology, immunophenotypes and functions of Tregs in the tumor microenvironment (TME) have been explored [5]. Tregs are identified as an immune suppressor and they have a crucial role in immune suppression in the TME [6]. The relationship of Tregs with other immune cells in the TME provides strategies to target Tregs in the tumor sites [7]. Treg depletion in tumor tissues promotes anti-tumor activity but may also induce fatal immunotherapy-related adverse events [8]. Despite rapid advances in the preclinical and clinical studies, it is still challenging to fine-tune Tregs to promote anti-tumor responses. Here, we review the biological characteristics and immunologic functions of Tregs in the context of immunotherapy. By understanding the role of Tregs in the TME, new therapies targeting Tregs in melanoma may be developed.
Phenotypes and functions of Tregs
Phenotypes of Tregs
Recent studies have established mechanisms for Tregs that mediate immune suppression towards self or non-self-antigens in immunity. In the 1970s, Gershon and colleagues made a breakthrough discovery that a subgroup of T cells dampened immune reactions, and these cells were different from helper T (Th) cells [9]. Subsequently, immunosuppressive cytokines interleukin 10 (IL-10) and transforming growth factor β (TGF-β) were identified in these suppressive T cells [10, 11]. In the 1990s, Sakaguchi et al., defined the suppressive T cells as Tregs through determining the CD25 molecule (the interleukin 2 (IL-2) receptor a-chain) on T cells [12]. CD25+ T cells constituted 5% to 50% of CD4+ T cells in the human peripheral blood and exhibited immune-suppressive activity [12, 13]. Moreover, the forkhead box P3 (FOXP3) gene was identified as a regulatory gene in CD25+CD4+ Tregs, which was crucial in autoimmune disease [14]. Therefore, Tregs can be distinguished as CD4+CD25+FOXP3+ T cells in both humans and mice. Recent studies have investigated the different phenotypes and functions of Tregs in the autoimmune diseases with the developmental marker of naive T cells, CD45RA [15]. Tregs are divided into CD45RA+FOXP3loCD25lo naive Tregs, CD45RA−FOXP3hiCD25hi effector Tregs, and CD45RA−FOXP3loCD25lo non-Tregs [16]. Naive Tregs derive from the thymus and exhibit weak suppressive activity [7]. These Tregs proliferate and differentiate into effector Treg (eTreg) cells after T cell receptor (TCR) stimulation in the lymph node, acquiring highly suppressive function [17]. The terminally differentiated effector Tregs possess highly suppressive activity but they tend to undergo apoptosis with aging, which contributes to increase reactivity to self-antigens in aging adults [4]. The heterogeneous population of CD45RA−FOXP3loCD25lo non-Tregs exhibits no suppressive activity for conventional T (Tconv) cells [18]. Other molecular markers have been suggested to classify Tregs, such as CD127 (IL-7 receptor α), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), glucocorticoid-induced TNF receptor (GITR), and lymphocyte activation gene-3 (LAG-3). In humans, CD127 is introduced as a new marker for Tregs [19, 20]. It has been suggested that CD4+CD25+T cells can differentiate into CD127low Tregs and CD127high conventional Th cells [19, 20]. However, researchers have demonstrated that CD127 expression can be downregulated after activation [21, 22]. Therefore, CD127 is identified as a variable marker in Tregs. Several reports have demonstrated that CD4+CD25high T cells express CTLA-4, which is correlated with a highly suppressive effect in humans [23, 24]. Similarly, different tissues and interindividual variations seem to determine the expression of CTLA-4 on Tregs [25]. Tumor infiltrating Tregs express much higher CTLA-4 than other T cells. Tanaka and colleagues have classified the human peripheral FOXP3+CD4+ T cells into three subsets by the expression of various molecules including CTLA-4 (Naive Treg: FOXP3+CD25+CD45RA+CTLA4+, Effector Treg: FOXP3++CD25++CD45RA−CTLA4++, Non Treg: FOXP3+CD25+CD45RA−CTLA4+) as summarized in Table 1. The effector Tregs are highly activated and proliferative with high CTLA-4 expression (FOXP3++CD25++CD45RA−CTLA4+++) in melanoma [26, 27]. Similarly, other markers such as GITR and LAG-3 are also used as Treg markers [28, 29]. Higher GITR expression is a marker for Tregs. However, the activity of GITR in the Tregs is controversial. GITR expression not only stimulates Treg expansion but also inhibits Treg suppression [30, 31]. LAG-3 is crucial for the regulation of CD4+CD25+FOXP3+ Tregs which interact with major histocompatibility complex class II (MHC II) molecules on dendritic cells (DCs) [32]. LAG-3 is a phenotypic marker of IL-10-producing Tregs both in vitro and in vivo [33].
Differentiation and proliferation of Tregs
It is well known that thymus is essential for Treg development. In newborn animals, multiple types of Tregs in lymphoid organs are derived from and developed in the thymus are called thymic Treg (tTreg) cells [4]. A proportion of Tregs is called peripheral Treg (pTreg) cells that develop in the peripheral organs such as intestine after naive T cells exposure to non-self-molecules antigens [34]. However, no reliable markers and methods can completely distinguish the development routes of Tregs. With the development and proliferation, Tregs migrate to lymphoid organs and other tissues via blood and lymphatic ducts with various functions. In physiological conditions, Tregs colonize in lymphoid organs, becoming central Treg (cTreg) cells and non-lymphoid organs such as bone marrow resident effector Treg cells [2]. In response to inflammatory stimulation, for example, acute tissue injury, Tregs expand and recruit to the local injured tissues exhibiting immune suppression [35]. In pathological conditions, cTreg cells convert into effector Tregs, and pTreg cells expand and develop into effector Tregs locally. These Tregs recover the damage tissues by releasing tissue-repairing molecules [36]. However, when the damage is incapable of being repaired completely, for instance, cancer, the Treg pool continues to expand and more Tregs are recruited into the tumor tissue, promoting tumor growth. In cutaneous melanoma, specific chemokine receptors expressed on melanoma cells attract Tregs migrating into tumor sites [37]. Tregs are recruited into tumor tissue by their expressing chemokine receptors such as CC chemokine receptor (CCR) 4, CCR7, CXC-chemokine receptor (CXCR) 4, sphingosin-1-phosphate receptor-1 (S1PR1), and others, which exacerbate the immunosuppressive TME [38,39,40,41]. Several cytokines are involved in the differentiation and proliferation of Tregs [42]. Both pTreg and tTreg are characterized by high expression of IL-2α, supporting that IL-2 plays a vital role in the Tregs differentiation [43]. Other common γ-chain cytokines such as interleukin 7 (IL-7) and interleukin 15 (IL-15) may replace the function of IL-2 in the development of tTreg cells [44]. Both IL-2 and IL-15 promote the differentiation of Tregs from tTreg cells [45]. TGF-β is known as a vital growth factor in the peripheral Treg cell differentiation [42].
Functions of Tregs
Tregs prevent autoimmunity and tissue damage via their immunosuppressive function. Tregs and effector T (Teff) cells mediate self-tolerance and deleterious immune responses against specific self and foreign antigens through mutual regulation [6]. The antigen-specific Teff cells can clear infectious agents, while uncontrolled or inappropriate Teff response results in inflammatory diseases or autoimmune diseases [46, 47]. In contrast, Tregs can prevent the pro-inflammatory and autoimmune response of Teff cells [48, 49]. The balance of tumor-antigen-specific Tregs and Teff cells is crucial for anti-tumor immunity [50, 51]. Reduced Teff: Treg ratios and enhanced Treg suppression have been observed in many cancers. Vaccines containing both CD4 and CD8 epitopes promote Teff: Treg ratios, increase tumor infiltrating lymphocytes and inhibit tumor growth [52]. Tregs also secrete a wide range of soluble anti-inflammatory mediators, such as IL-10, TGF-β, and interleukin 35 (IL-35), which contribute to suppress the proliferation of Tconv cells [53]. Studies have shown that granzyme B derived from Tregs induces effector T cells apoptosis [54, 55]. Loebbermann and co-workers have demonstrated that Tregs can control lung inflammation by expressing granzyme B during acute viral infection [56]. Moreover, Tregs express high CD25 (IL-2 receptor) that forms high affinity with IL-2 and inhibits the proliferation of effector T cells [57]. Thus, Tregs can inhibit effector T cell activation via consumption IL-2. In return, IL-2 produced by Tconv cells stimulates Treg cell expansion and enhances immune suppression [58]. CTLA-4 is a crucial molecule for Tregs in immune suppression [59]. Tregs express high level of CTLA-4 protein, which binds to CD80/CD86 on DCs, weakening their affinity of co-activation of effector T cells [60]. Furthermore, Tregs increase expression of indoleamine 2,3-dioxygenase (IDO) in DCs, which degrades tryptophan into kynurenine [7]. As a result, effector T cells exhibit a stagnation of proliferation, reducing the autoimmunity. In addition, Tregs also use other inhibitory molecules, such as CD39 and CD73 [61], which inhibit the expansion of effector T cells and antigen-presenting cells (APCs) through facilitating extracellular adenosine triphosphate (ATP) conversion into adenosine [2]. Thus, Tregs are involved in various mechanisms of immunosuppression. However, tumor infiltrating Tregs prevent anti-tumor immune response, which differs from their role in suppression of autoimmunity.
Tregs in the melanoma tumor microenvironment
Recruitment of Tregs to the tumor microenvironment
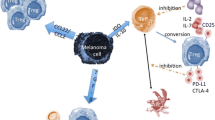
Naive Tregs (nTregs) homing to the TME is a crucial step for tumor progression [62]. Many chemokines produced by tumor and immune cells drive nTregs into the TME [63]. Studies of tumor infiltrating Tregs of cancer mouse models have demonstrated that specific chemokines and their receptors regulate the preferential homeostasis [64, 65]. A number of chemokines and chemokine receptors are involved in Treg-mediated homeostasis in melanoma, including chemokine (C-C motif) ligand (CCL)27-CCR10, CXCL12-CXCR4, CCL20-CCR6, CCL19/CCL21-CCR7, CCL9/10/11-CXCR3, CCL17/22-CCR4 and S1P-S1PR1 (Fig. 1) [39, 40, 66,67,68,69]. CCL17/22-CCR4 are involved in tumor-infiltrating Treg homeostasis and expansion in gastric cancer [64]. Mechanisms of Treg homing to the TME are underexplored. S1P is a bioactive mediator involving in tumor associated Treg expansion, tumor progression, and metastasis [70, 71]. The specific processes include endothelial adhesion, angiogenesis, and cell-cell contact [72]. Effects of S1P in tumor immunity are regulated by its binding to G-protein-coupled receptors S1PR1–5 [73]. Chemokine CXCL9 activates the expression of S1PR1 and S1PR4 on T cells, which induces T cells to migrate from the blood into tissues [74]. Moreover, S1PR1 is known to regulate the proliferation and function of Tregs through mTOR pathway [75]. Furthermore, S1PR1 signaling can activate Tregs and promote Tregs accumulation via signal transducer and activator of transcription 3 (STAT3) pathways and inhibit CD8+ T cells migration [41]. Thus, the S1P-S1PR1 axis plays an essential role in the recruitment of Tregs in melanoma. Effects of other chemokines and chemokine receptors such as CCL27-CCR10 have also been studied in the recruitment of Tregs in melanoma [76, 77].

Recruitment of Tregs to the TME. Tregs are recruited to the TME through specific chemokines and their receptors. A number of chemokines are involved in Treg-trafficking to melanoma, including CCL27-CCR10, CXCL12-CXCR4, CCL20-CCR6, CCL19/CCL21-CCR7, CCL9/10/11-CXCR3, CCL17/22-CCR4, and S1P-S1PR1
Tumor and Treg cell contact mechanisms
Various immunophenotypes and functions of Tregs in the TME are associated with the tumor burden [78]. Tumor cells produce soluble factors, extracellular vesicles, and biological-competent proteins, which activate and reprogram Tregs continuously in the TME (Fig. 2) [79,80,81]. During cancer progression, tumor cells active the TGF-β pathway to mediate tumor growth, invasion, and metastasis [82]. TGF-β is a crucial mediator for FOXP3 in tumor infiltrating Tregs. Ectopic FOXP3 is identified to confer Treg cell suppressive activity; the molecules mediating FOXP3 expression may well regulate the phenotype and function of Tregs [5]. Tumor cells secrete high levels of TGF-β binding to the TGF-β receptor on the Tregs, promoting Treg differentiation and maintenance [83]. Anti-CTLA-4-TGF-βRII fused antibodies significantly reduce the numbers of Tregs and increase CD8+ T cells in a melanoma mouse model [84]. Tumor-associated TGF-β enhances the expansion of Tregs and their immune suppressive function in the TME. IL-10 in the TME is derived from several components including tumor cells. IL-10 mRNA transcripts can be isolated from tumor tissues including ovarian, breast, renal, lung, and skin cancer [85]. Interestingly, IL-10 was originally demonstrated as an important factor in T-cell growth and differentiation [86]. Subsequently, Hsu and colleagues demonstrated that IL- 10 promotes human Treg proliferation through STAT3 and Foxo1 [53]. Moreover, tumor cells-derived exosomes are essential in the intercellular connecting system in the TME. These exosomes express high levels of immunoreceptors and ligands [87]. Exosomes deliver signals from tumor cells to immune cells including Tregs as a mimic profile of tumor cells [88]. Tumor-derived exosomes have been confirmed promoting expansion and immunosuppression of Tregs in the TME [89]. Tregs in the TME are particularly sensitive to exosomes, while CD8+ T cells are mostly inhibited by these exosomes in vitro co-incubation experiments [61]. When tumor-delivered exosomes are in touch with immune cells, they carry IL-10 and TGF-β that enhance Treg expansion and increase expression of CTLA-4 on Tregs to promote immune suppression in melanoma [90]. Melanoma cell-derived exosomes express CD39 and CD73, which significantly enhance Tregs excretion of immunosuppressive adenosine [91]. Tumor-derived exosomes regulate the expression of immune checkpoint proteins that promote the immune suppression of Tregs [92]. Melanoma cells induce Tregs to overexpress various inhibitory checkpoint receptors, including programmed cell death protein 1 (PD-1), CTLA-4, T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), LAG-3, and T cell immunoglobulin and ITIM domain (TIGIT) [93,94,95,96]. Galectin-9 expressed in melanoma cells binds to TIM-3 on Tregs, promoting tumor progression in the mouse or human melanoma tissues [97, 98]. PD-1/programmed death-ligand 1(PD-L1)-programmed death-ligand 2 (PD-L2) axis plays a crucial role in the induction and maintenance of Tregs in the TME [99]. PD-L1 and PD-L2 are highly expressed by tumor cells in the TME and they impair the infiltrating T cell function when binding with PD-1 receptor [100, 101]. It has been shown that there is a profound defect in conversion of naive CD4+ T cells into FOXP3+ iTreg cells in the absence of PD-L1 [100]. In addition, PD-L1 converts naive Tregs into effector Tregs by reducing signaling of the AKT–mTOR pathway in naive T cells [100]. Thus, the PD-1/PD-L1-PD-L2 axis synergizes with TGF-β to promote Treg differentiation and maintenance [100].

Immune suppressive functions of Tregs in the TME. Tumor cells can produce soluble factors (IL-10 and TGF-β), exosomes, and biological-competent proteins (Galectin 9 and PD-L1), which activate Tregs in the TME. Tregs release anti-inflammatory cytokines (IL-10, IL-35, and TGF-β) that directly inhibit the proliferation of effector T cells and neutrophils. They also produce perforin and granzymes to damage cell membrane and induce T cell apoptosis. In addition, high expression of CD39 and CD73 on Tregs facilitates the conversion of extracellular ATP to adenosine reducing the expansion of effector T cells. Moreover, Tregs consume IL-2 by expressing high level of CD25 (IL-2 receptor) and inhibit the proliferation of effector T cells. Tregs have been suggested to directly inhibit B cell, M2 macrophage, and effector T cells via the PD-1/PD-L1 pathway. Furthermore, Tregs contact with DCs through CTLA-4 and LAG-3. Blocking CTLA-4 can decrease the CD86/80 expression leading to upregulation of IDO. Tregs decrease the proliferation and effector functions of NK cells through IL-2 starvation and TGF-β dependent manner
Treg and other immune cell contact mechanisms
Tregs exhibit their immune-suppressive effect by multiple mechanisms through production of suppressive soluble cytokines (IL-10, TGF-β, and IL-35), upregulation checkpoint inhibitory receptors (CTLA-4, PD-1, LAG-3, TIM-3, and TIGIT), secretion of granzymes and perforin, and depletion of ATP in the TME [27, 102,103,104,105]. Tregs exert different immunosuppression by direct and indirect cell-cell contact mechanisms (Fig. 2) [106]. Tregs produce immunosuppressive cytokines to inhibit effector T cell expansion and release cell membrane soluble mediators, granzymes and perforin, to induce effector T cell apoptosis [107,108,109]. Tregs also consume IL-2 by expressing high level of CD25 (IL-2 receptor) to inhibit the proliferation of effector T cells [110].
Moreover, high expression of CD39 and CD73 on Tregs facilitates the conversion of extracellular ATP to adenosine, which reduces the expansion of effector T cells and inhibits dendritic and myeloid cells [111]. In melanoma patients, upregulation of CD73 expression was found as a cause for anti-PD-1 therapy resistance, which correlates with poor prognosis [112]. B-cells express high levels of PD-1 and PD-L1 in an antigen-specific manner. Thus, Treg may use PD-1 ligands to directly inhibited B cell activation, suppressed their proliferation via PD-1/PD-L1 axis [113]. Furthermore, Tregs can decrease the proliferation and effector functions of NK cells by IL- 2 starvation in a TGF-β dependent manner [57]. CTLA-4 on Tregs binds to CD80/CD86, and LAG-3 binds to MHC-II, reducing the proliferation of APCs and inducing upregulation of IDO [57], which directly reduces effector T cells and DCs expansion and promotes the differentiation of naive Tregs into effector Tregs [114]. In melanoma patients, Tregs upregulate TIGIT expression and reduce the expression of CD226, which results in a decreased proliferation of effector T cells and DCs [96, 115]. Recently, Tregs was found that they can skew monocyte differentiation into M2 macrophages by reduction of sterol regulatory element-binding protein 1 (SREBP1) in a melanoma mouse model [116]. Other mechanisms of how Tregs affect other immune cells, such as gamma delta T (γδT) cells in melanoma are being investigated.
Immunotherapy targeting Tregs in melanoma
Blocking recruitment of Tregs to the TME
As discussed above, many chemokines and chemokine receptors are involved in recruiting Tregs into tumor tissues. Therapies are being explored to prevent Treg recruitment to tumor sites (Fig. 3). CCR4 is expressed on effector Tregs that significantly reduce the immune response in the TME [117]. CCR4 plays a crucial role in Tregs migration and infiltration into tumor site [6]. Moreover, CCR4 is overexpressed in melanoma tissues from brain metastasis compared with primary melanoma [38]. The recruitment by CCR4 can be abrogated by an anti-CCL17 antibody, which blocks CCL17 binding to CCR4 in the TME [118]. A CCR4 antagonist, FLX475, has shown to effectively inhibit Treg migration into the TME and deplete effector Tregs in many tumor models [119]. Clinical trials of combination of FLX475 with pembrolizumab are ongoing for advanced cancers. In melanoma patients, the anti-CCR4 antibody, KM2160, effectively depletes effector Tregs and promotes the immune response of CD8+ T cells in vivo [120]. Only a minor population of patients who received anti-CCR4 monoclonal antibodies (mAbs) experienced severe immune-related adverse events [121]. In addition, animal models demonstrated that it requires a much longer period and profounder degree for autoimmunity than effective antitumor immunity when drugs are applied in Treg depletion [122, 123]. In addition, Sugiyama and colleagues demonstrated that the residual CCR4− eTreg cells and naive Tregs are sufficient to prevent deleterious autoimmunity when using anti-CCR4 mAb to decrease eTreg cells in the immune system [120]. In melanoma patients, high CCR10 expression is associated with a short survival [40]. In addition, overexpression of CCR10 in the B16 melanoma mouse model resulted in increased tumor size and lymph node metastases [124]. CCR10 antagonist (brintonamide D) demonstrates a potential anti-tumor effect in breast cancer [125]. Melanoma patients with higher CXCR4 expression show poorer overall survival [126]. An oral CXCR4 antagonist, X4P-001, improves the efficacy of checkpoint inhibitor therapy and modulates tumor infiltrating immune cells by disrupting the CXCL12-CXCR4 axis [127]. The combination of X4P-001 with anti-PD-1 therapy is being evaluated in clinical trials in advanced melanoma patients. Furthermore, S1PRs antagonist, FTY720, is an immunomodulatory prodrug [128, 129]. FTY720 decreases the recruitment of CD4+ T cells directly and prevents the differentiation of Th1 T cells into Tregs via targeting S1PR1 [130]. In the B16F10 mouse melanoma model, FTY720 induces immunomodulatory effect by inhibiting the recruitment of Tregs into tumor tissues [131].

Chemokine and small molecule inhibitors targeting Tregs in melanoma. Anti-CCR4 antibody, CXCR4 antagonist, CCR10 antagonist, and S1PRs antagonist may inhibit recruitment of Tregs to the TME. Other small molecule inhibitors such as STAT3 inhibitors and TLR8 agonists inhibit proliferation of Tregs and enhance TCR activation
Small molecule antagonists for Treg depletion
STAT3 plays an essential role in tumor progression and tumor immunity [132]. Phosphorylation of STAT3 mediates immune escape and is associated with poor survival in melanoma patients [133]. STAT3 knockout mice have reduced tumor infiltrating Tregs and pronounced anti-tumor response [134]. Therefore, STAT3 is a potential target for melanoma immunotherapy (Fig. 3). WP1066, a novel small molecule inhibitor of STAT3, exhibits significant antitumor effect in advanced melanoma patients by inhibiting the proliferation of Tregs and enhancing the TCR activation on ZAP-70 [135]. Similarly, another small molecule STAT3 inhibitor, Stattic, decreases the immune suppressive function of Tregs in vitro [93]. Another small molecule STAT3 inhibitors, OPB-31121, inhibited cancer cell lines by targeting the STAT3-SH2 domain. However, the clinical trials for these drugs were abrogated due to low anti-tumor activity, high toxicity events, and poor pharmacokinetics [136]. In addition, toll like receptor 8 (TLR8) activation on Tregs can prevent their suppression on effector T cells and DCs [137]. TLR8 reverses immunosuppression by suppressing the glucose uptake and the process of glycolysis in Tregs [138]. These findings need to be confirmed in human studies in the future. Nevertheless, depletion of tumor infiltrating Tregs by signaling molecules is a potential promising therapy for melanoma.
Immune checkpoint blockade therapies targeting Tregs
The anti-PD-1 antibody is one of the recent breakthroughs in cancer immunotherapy, which has excellent results in many cancers [139, 140]. Nevertheless, only a minority of patients who receive anti-PD-1 therapy exhibits significant responses [141, 142]. Resistance to anti-PD-1 therapy in melanoma correlates with lymphatic vessel density in the tumor tissues and lymph nodes [143]. Blockade of PD-1/PD-L1 axis induces the recruitment of exhausted T cells and Tregs in anti-PD-1 therapy-resistant melanoma patients [144]. The PD-1 expression on Tregs function similarly to that on effector T cells; and the anti-PD-1 mAbs may activate the immunosuppressive function of Tregs in the TME (Fig. 4) [145]. While another study found that anti-PD-1 mAbs , pembrolizumab, did not affect the phenotype or function of Tregs through PD-1/PD-L1 axis [146]. Thus, carefully designed studies to elucidate the effects of anti-PD-1 therapy on Tregs are needed. A few recent studies suggest that anti-CTLA-4 mAbs play a major role in regulating the function of tumor infiltrating Tregs. As discussed above, CTLA-4 is highly expressed on activated Tregs and also upregulated in activated CD4+ and CD8+ T cells in melanoma tissues compared to other tumors [7, 27]. Anti-CTLA-4 mAbs were initially thought to suppress the inhibition on activated CD4+ and CD8+ T cells and augment the anti-tumor immune response in the TME [147]. However, more recent studies discover that anti-CTLA-4 mAbs predominantly deplete Tregs in the TME to promote the anti-tumor immune response [148]. Treg depletion by anti-CTLA-4 mAbs decreases the immunosuppression in the TME, but it also results in severe cancer immunotherapy-related adverse events [59]. Thus, maintenance of balanced Tregs in the immune system and the TME is crucial for preventing cancer immunotherapy-related adverse events while promoting anti-tumor response.

Immune checkpoint therapies targeting Tregs in melanoma. In the TME, anti-PD-1 mAbs block the PD-1 function on the Tregs. Anti-CTLA-4 mAbs induce Treg cell depletion by activating the Fc receptors. However, current CTLA-4 mAbs often result in severe immune-therapy related adverse events. A new version of anti-CTLA-4 (pH-sensitive CTLA-4) mAbs do not degrade in the lysosomal and recirculate to the cell surface of Tregs, reducing the adverse immune events. Anti-TIM-3 mAbs exhibit dual functions in depleting Tregs and stimulating CD8+ T cells. Anti-LAG-3 mAbs prevent Treg recruitment and promote DCs function in melanoma therapy. Other antibodies targeting OX-40 and ICOS inhibit proliferation of Tregs in the TME. The anti-GITR mAbs reduce circulating and intratumor Tregs in advanced melanoma patients
TIM-3 is expressed on Tregs with enhanced regulatory function [104]. However, TIM-3 is rarely found on Tregs in the peripheral immune system, the majority of tumor-associated Tregs express TIM-3 comprising a particular subset of tissue Treg [149]. Thus, it is possible to target TIM-3 in the TME in cancer immunotherapy (Fig. 4). In the preclinical mouse melanoma model, blockade TIM-3 exhibits anti-tumor response by stimulating CD8+ T cells [150]. Moreover, TIM-3 signaling appears to be a crucial mediator in both innate and adaptive immune responses. Therefore, targeting TIM-3 combined with other checkpoints such as anti-PD-1 mAbs is currently being tested as a new cancer immunotherapy strategy. Recently, a combination of TIM-3 inhibitor TSR-022 and PD-1 inhibitor dostarlimab is conducted at the University of Pittsburgh in a clinical trial for Stage III or IV melanoma patients. Similar to TIM-3, LAG-3 expressed on a variety of T cells, including CD4+ T cells, CD8+ T cells, and Tregs. LAG-3 expression is essential for Treg cell function and is also associated with treatment resistance [151]. Recent studies have demonstrated that LAG-3 blockade prevents Treg recruitment and promotes DCs function in melanoma treatment [152]. Nevertheless, anti-LAG-3 therapy is still in its infancy, further studies are needed to explore the therapeutic efficacy in various tumors. Furthermore, other molecules are explored to target Tregs either deliberately or inadvertently in cancer immunotherapy such as TIGIT, V-domain Ig suppressor of T cell activation (VISTA), and CD73.
In addition to checkpoint inhibitors, other molecules can also inhibit immune suppressive functions of Tregs. GITR is a member of the tumor necrosis factor receptor family. It is expressed on Tregs and serves as a mediator in Treg regulated immunosuppression [153]. Anti-GITR mAbs abrogate tumor infiltrating Tregs, decrease the suppressive function of Tregs, and promote the effector function of Tconv cells in a preclinical melanoma model [154]. The anti-GITR mAbs (TRX518) reduce circulating and intratumoral Tregs and demonstrate significant clinical efficacy in advanced melanoma patients [155]. In addition, combination of anti-PD-1 mAbs with TRX518 reverses the resistance to anti-GITR therapy in a mouse tumor model [155]. The efficacy of the combination therapy should be explored in patients with advanced melanoma.
OX40 (CD134) is a co-stimulatory molecule on Tregs [156]. Earlier studies on tumor inhibition using anti-OX40 antibody showed that anti-OX40 mAbs augmented anti-tumor immunity with the depletion of tumor infiltrating Tregs in several types of cancer animal models [157, 158]. Moreover, OX40 antibodies, such as MOXR0916, demonstrated excellent therapeutic effect in some patients with low adverse events in a phase I trial [157]. Inducible T-cell costimulator (ICOS) that binds to ICOS ligands on APC has also been identified on the Tregs [159]. Recent studies have shown that increased number of ICOS+ Tregs is found in various cancers, including melanoma [160] and breast cancers [161]. Increased proliferation of ICOS+ Tregs is also found in melanoma patients after IL-2 therapy [162]. Thus, targeting ICOS on Tregs and interrupt the interaction between ICOS and ICOSL may be an effective measure for anti-tumor immunity. In preclinical studies of an ICOS agonistic mAbs alone showed a promising effect and but resulted in severe side effect [163]. Another ICOS agonistic mAbs (JTX-2011) is currently being tested in a clinical trial alone and in combination with anti-PD-1 mAbs.
Vaccine immunotherapy for Treg depletion
A potential application of anti-cancer vaccine may be used to target Tregs [164]. Vaccination targeting FOXP3 provides an excellent measure for depleting Tregs in anti-tumor immunity [165]. A study has been performed by using Fox-Fc DNA vaccine/recombinant FOXP3-Fc fusion protein, which demonstrates an increased cytotoxic T lymphocytes (CTL) response against FOXP3 Tregs [166]. Moreover, in the B16 melanoma mouse model, the DC vaccine exhibits a significant antitumor effect by enhancing the CTL response and decreasing the percentages of FOXP3+ Tregs [165]. Tumor cell vaccine plus FOXP3 gene silencing inhibits tumor growth and enhances the efficacy of vaccination immunotherapy [167]. Similarly, therapy that combined dendritic cell-based tumor vaccine with toll-like receptor 7 (TLR7) agonist showed excellent anti-tumor response that resulted in a decrease of tumor infiltrating Tregs [168]. However, more studies are needed for vaccine-based therapies against Tregs.
Challenges of targeting Tregs in melanoma immunotherapy
Tregs: a friend or foe for melanoma
Based on the immune suppressive role of Tregs, their presence in the TME is expected to be associated with tumor progression and short survival in cancer patients. However, tumor infiltrating Tregs seem to correlate with a favorable overcome in cancers with characteristics of chronic inflammation, such as colorectal cancer [169]. In colorectal cancers, abundant tumor infiltrating naive FOXP3low Tregs exhibit better survival than those patients with FOXP3hi Tregs in the tumors. The differentiation and proliferation of inflammatory FOXP3low naive Tregs are dependent on the production of IL-2 and TGF-β by tissues [170]. Strategies that deplete the FOXP3hi Tregs and increase the FOXP3low naive Tregs in the tumor tissue might show high anti-tumor therapeutic efficacy [170]. Remarkably, genetic depletion of caspase recruitment domain-containing membrane-associated guanylate kinase protein-1 (CARMA1) which is a critical component mediated by TCR engagement in FOXP3hi Tregs produces an anti-tumor effect without affecting systemic autoimmunity [171]. Moreover, combination of anti-PD-1 mAbs with CARMA1 deletion therapy reverses resistance to PD-1 blockade therapy in cancer [171]. By analyzing PBMC from melanoma patients, flow data classifies FOXP3+ cells into FOXP3hi Tregs, FOXP3low naive Tregs, and FOXP3low non-Tregs [172]. With tumor progression, both FOXP3hi Tregs and FOXP3low Tregs increase in melanoma patients [172]. Despite methods to suppress different Treg subpopulations in melanoma have not been discovered, targeting subpopulation of Tregs in the TME may be proven as an effective cancer therapy without concomitant significant adverse immunological reactions.
Balance of autoimmunity and Treg-targeting cancer immunotherapy
As described above, CTLA-4-targeting immunotherapy significantly depletes tumor infiltrating Tregs but may also induce fatal immunotherapy-related adverse events in some patients. Anti-CTLA-4 mAbs suppress the binding of CTLA-4 to CD80 and CD86 [173]. These actions promote the tumor-activated T cells migrating into the tumor site, and enhance the anti-tumor therapeutic effect. The therapeutic effect of CTLA-4 mAbs is determined by their ability to engage Fc receptors for antibody-dependent cell mediated cytotoxicity (ADCC) on host cells [59]. The interaction between CTLA-4 mAbs and the activating Fc receptors is critical for selective depletion of Tregs in the tumor. In addition, the anti-CTLA-4 mAbs selectively reduce Tregs in the tumor sites by activating the Fc receptors, increasing the anti-tumor activity in cancer immunotherapy [148]. To reduce anti-CTLA-4 mAbs associated adverse immune reaction, it is urgent to discover new versions of anti-CTLA-4 mAbs. A pH-sensitive CTLA-4 antibody has been shown to reduce cancer immunotherapy-related adverse events with increased anti-cancer activity in the tumor site [174]. pH-insensitive CTLA-4 mAbs degraded by lysosomal may cause autoimmune events [148]. However, pH-sensitive CTLA-4 mAbs maintain their bioavailability that is not degraded by the lysosomal, recirculate to the Treg surface, reduce adverse immune events, and exert high anti-tumor efficacy in the TME [175]. One of the pH-sensitive CTLA-4 mAbs, ONC-392, has entered in the clinical trials of metastatic melanoma patients. It may be important for patients to perform human leukocyte antigen (HLA) haplotypes analysis to determine susceptibility to autoimmunity in Treg depletion treatment. Intratumor immunotherapy may also resolve the issues of immune-related adverse events caused by the immune checkpoint blockade [176]. Intratumor immunotherapy could selectively abrogate the tumor infiltrating Tregs by not affecting naive Tregs elsewhere. Sato and colleagues demonstrated that tumor-associated Tregs were depleted by near-infrared photoimmunotherapy in a preclinical study [177]. Studies have shown that combination of anti-CTLA-4 with anti-PD-1 mAbs provides an excellent immune response but also causes severe immune-related adverse events [178]. Intratumoral administration of smaller dose of Treg depleting antibodies or immune checkpoint antibodies is a potential promising therapy. However, additional clinical trials are needed to test this approach.
Conclusions and prospects
Tregs play a crucial role in the melanoma progression. Tumor infiltrating Tregs migrate into the TME via specific chemokines and chemokine receptors to promote tumor growth by enhancing immune suppression. The immune suppressive functions of Tregs in the TME are multifaceted through production of suppressive soluble cytokines (IL-10, TGF-β, and IL-35), upregulation of checkpoint inhibitory receptors (CTLA-4, PD-1, LAG-3, TIM-3, and TIGIT), secretion of granzymes and perforin, consumption of IL-2 and depletion of ATP in the TME. Subpopulations Tregs in the TME may have different functions. By understanding the mechanisms of Treg differentiation, recruitment, expansion, and immune suppression, therapeutic strategies of depleting subpopulation of Tregs can be developed to increase anti-tumor response without causing severe adverse immune response. Targeting tumor infiltrating Tregs may be achieved using antibodies to TIM-3, LAG-3, TIGIT, VISTA, and CD73. CTLA-4-targeting immunotherapy significantly depletes tumor infiltrating Tregs, but it also may induce severe immunotherapy-related adverse events. Cancer immunotherapy that aims at depletion of tumor infiltrating Tregs needs the balance of anti-tumor response and autoimmunity.
Availability of data and materials
Not applicable.
Abbreviations
- Treg:
-
Regulatory T
- IL-10:
-
Interleukin 10
- TGF-β:
-
Transforming growth factor β
- IL-2:
-
Interleukin 2
- FOXP3:
-
Forkhead box P3
- Teff:
-
Effector T
- Tconv:
-
Conventional T
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- GITR:
-
Glucocorticoid-induced tumor necrosis factor receptor family-related gene
- LAG-3:
-
Lymphocyte activation gene-3
- Tfh:
-
T follicular helper cells
- Th:
-
T helper cells
- DCs:
-
Dendritic cells
- NK:
-
Natural killer
- MHC-II:
-
Major histocompatibility complex class II
- tTreg:
-
Thymic Treg
- pTreg:
-
Peripheral Treg
- cTreg:
-
Central Treg
- eTreg:
-
Effector Treg
- CCR:
-
CC chemokine receptor
- CXCR:
-
CXC-chemokine receptor
- CCL:
-
Chemokine C-C motif ligand
- S1PR1:
-
Sphingosin-1-phosphate receptor-1
- IL-7:
-
Interleukin 7
- IL-15:
-
Interleukin 15
- IL-35:
-
Interleukin 35
- nTreg:
-
Naive Treg
- IDO:
-
Indoleamine 2,3-dioxygenase
- APCs:
-
Antigen-presenting cells
- TME:
-
Tumor microenvironment
- STAT3:
-
Signal transducer and activator of transcription 3
- PD-1:
-
Programmed cell death protein 1
- TIM-3:
-
T cell immunoglobulin and mucin domain-containing protein 3
- TIGIT:
-
T cell immunoglobulin and ITIM domain
- PD-L1:
-
Programmed death-ligand 1
- PD-L2:
-
Programmed death-ligand 2
- ATP:
-
Adenosine triphosphate
- SREBP1:
-
Sterol regulatory element-binding protein 1
- γδT:
-
Gamma delta T
- VISTA:
-
V-domain Ig suppressor of T cell activation
- ICOS:
-
Inducible T-cell costimulator
- mAbs:
-
Monoclonal antibodies
- CTL:
-
Cytotoxic T lymphocytes
- OVA:
-
Ovalbumin
- TLR7:
-
Toll like receptor 7
- TLR8:
-
Toll like receptor 8
- CARMA1:
-
Caspase recruitment domain-containing membrane-associated guanylate kinase protein-1
- TCR:
-
T cell receptor
- ADCC:
-
Antibody-dependent cell mediated cytotoxicity
- HLA:
-
Human leukocyte antigen
References
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–87. https://doi.org/10.1016/j.cell.2008.05.009.
Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. 2016;34(1):609–33. https://doi.org/10.1146/annurev-immunol-032712-095948.
Wolf D, Sopper S, Pircher A, Gastl G, Wolf AM. Treg(s) in cancer: friends or foe? J Cell Physiol. 2015;230(11):2598–605. https://doi.org/10.1002/jcp.25016.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30(1):531–64. https://doi.org/10.1146/annurev.immunol.25.022106.141623.
Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–18. https://doi.org/10.1038/cr.2016.151.
Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. https://doi.org/10.1016/j.coi.2013.12.005.
Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110(7):2080–9. https://doi.org/10.1111/cas.14069.
Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev. 2014;259(1):173–91. https://doi.org/10.1111/imr.12173.
Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18(5):723–37.
O'Garra A, Murphy K. Role of cytokines in determining T-lymphocyte function. Curr Opin Immunol. 1994;6(3):458–66. https://doi.org/10.1016/0952-7915(94)90128-7.
Cottrez F, Hurst SD, Coffman RL, Groux H. T regulatory cells 1 inhibit a Th2-specific response in vivo. J Immunol. 2000;165(9):4848–53. https://doi.org/10.4049/jimmunol.165.9.4848.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, et al. The number of human peripheral blood CD4+CD25high regulatory T cells increases with age. Clin Exp Immunol. 2005;140(3):540–6. https://doi.org/10.1111/j.1365-2249.2005.02798.x.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. https://doi.org/10.1126/science.1079490.
Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the Foxp3 transcription factor. Immunity. 2009;30(6):899–911. https://doi.org/10.1016/j.immuni.2009.03.019.
Wing JB, Tanaka A, Sakaguchi S. Human FOXP3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. 2019;50(2):302–16. https://doi.org/10.1016/j.immuni.2019.01.020.
Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4 Tregs. J Clin Invest. 2005;115(7):1953–62. https://doi.org/10.1172/JCI23963.
Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, et al. A distinct subpopulation of CD25− T-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci U S A. 2017;114(31):E6400–E9. https://doi.org/10.1073/pnas.1705551114.
Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7):1693–700. https://doi.org/10.1084/jem.20060468.
Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7):1701–11. https://doi.org/10.1084/jem.20060772.
Del Pozo-Balado MM, Leal M, Mendez-Lagares G, Pacheco YM. CD4+CD25+/hiCD127lo phenotype does not accurately identify regulatory T cells in all populations of HIV-infected persons. J Infect Dis. 2010;201(3):331–5. https://doi.org/10.1086/649840.
Alves NL, van Leeuwen EM, Derks IA, van Lier RA. Differential regulation of human IL-7 receptor α expression by IL-7 and TCR signaling. J Immunol. 2008;180(8):5201–10. https://doi.org/10.4049/jimmunol.180.8.5201.
Levings MK, Sangregorio R, Roncarolo MG. Human CD25+CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193(11):1295–302. https://doi.org/10.1084/jem.193.11.1295.
Zheng Y, Manzotti CN, Burke F, Dussably L, Qureshi O, Walker LS, et al. Acquisition of suppressive function by activated human CD4+CD25− T cells is associated with the expression of CTLA-4 not FoxP3. J Immunol. 2008;181(3):1683–91. https://doi.org/10.4049/jimmunol.181.3.1683.
Rodriguez-Perea AL, Arcia ED, Rueda CM, Velilla PA. Phenotypical characterization of regulatory T cells in humans and rodents. Clin Exp Immunol. 2016;185(3):281–91. https://doi.org/10.1111/cei.12804.
Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. Eur J Immunol. 2019;49(8):1140–6. https://doi.org/10.1002/eji.201847659.
Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A. 2019;116(2):609–18. https://doi.org/10.1073/pnas.1812186116.
Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19(4):345–54. https://doi.org/10.1093/intimm/dxm014.
Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5):1393–405. https://doi.org/10.1084/jem.171.5.1393.
Ronchetti S, Ricci E, Petrillo MG, Cari L, Migliorati G, Nocentini G, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory T cells. J Immunol Res. 2015;2015:171520–17. https://doi.org/10.1155/2015/171520.
van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, et al. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol. 2009;182(12):7490–500. https://doi.org/10.4049/jimmunol.0802751.
Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–13. https://doi.org/10.1016/j.immuni.2004.08.010.
Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 2011;12(4):304–11. https://doi.org/10.1038/ni.2006.
Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14(4):307–8. https://doi.org/10.1038/ni.2554.
Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14(10):1007–13. https://doi.org/10.1038/ni.2683.
Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. https://doi.org/10.1126/science.1242454.
Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11(9):597–606. https://doi.org/10.1038/nri3049.
Klein A, Sagi-Assif O, Meshel T, Telerman A, Izraely S, Ben-Menachem S, et al. CCR4 is a determinant of melanoma brain metastasis. Oncotarget. 2017;8(19):31079–91. https://doi.org/10.18632/oncotarget.16076.
Monteagudo C, Ramos D, Pellin-Carcelen A, Gil R, Callaghan RC, Martin JM, et al. CCL27-CCR10 and CXCL12-CXCR4 chemokine ligand-receptor mRNA expression ratio: new predictive factors of tumor progression in cutaneous malignant melanoma. Clin Exp Metastasis. 2012;29(6):625–37. https://doi.org/10.1007/s10585-012-9476-2.
Kuhnelt-Leddihn L, Muller H, Eisendle K, Zelger B, Weinlich G. Overexpression of the chemokine receptors CXCR4, CCR7, CCR9, and CCR10 in human primary cutaneous melanoma: a potential prognostic value for CCR7 and CCR10? Arch Dermatol Res. 2012;304(3):185–93. https://doi.org/10.1007/s00403-012-1222-8.
Priceman SJ, Shen S, Wang L, Deng J, Yue C, Kujawski M, et al. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell Rep. 2014;6(6):992–9. https://doi.org/10.1016/j.celrep.2014.02.016.
Richards DM, Delacher M, Goldfarb Y, Kagebein D, Hofer AC, Abramson J, et al. Treg cell differentiation: from thymus to peripheral tissue. Prog Mol Biol Transl Sci. 2015;136:175–205. https://doi.org/10.1016/bs.pmbts.2015.07.014.
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–51. https://doi.org/10.1038/ni1263.
Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, −7, and −15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181(5):3285–90. https://doi.org/10.4049/jimmunol.181.5.3285.
Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28(1):100–11. https://doi.org/10.1016/j.immuni.2007.11.021.
Hirahara K, Nakayama T. CD4+ T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int Immunol. 2016;28(4):163–71. https://doi.org/10.1093/intimm/dxw006.
Dornmair K, Goebels N, Weltzien HU, Wekerle H, Hohlfeld R. T-cell-mediated autoimmunity: novel techniques to characterize autoreactive T-cell receptors. Am J Pathol. 2003;163(4):1215–26. https://doi.org/10.1016/S0002-9440(10)63481-5.
Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation. 2017;14(1):117. https://doi.org/10.1186/s12974-017-0892-8.
Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13(4):423–31. https://doi.org/10.1038/nm1564.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8. https://doi.org/10.1038/ni1102-991.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22(1):329–60. https://doi.org/10.1146/annurev.immunol.22.012703.104803.
Bui JD, Uppaluri R, Hsieh CS, Schreiber RD. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res. 2006;66(14):7301–9. https://doi.org/10.1158/0008-5472.CAN-06-0556.
Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol. 2015;195(8):3665–74. https://doi.org/10.4049/jimmunol.1402898.
Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med. 2004;199(11):1479–89. https://doi.org/10.1084/jem.20040179.
Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174(4):1783–6. https://doi.org/10.4049/jimmunol.174.4.1783.
Loebbermann J, Thornton H, Durant L, Sparwasser T, Webster KE, Sprent J, et al. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. 2012;5(2):161–72. https://doi.org/10.1038/mi.2011.62.
Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun. 2018;95:1–14. https://doi.org/10.1016/j.jaut.2018.10.017.
Jeffery HC, Jeffery LE, Lutz P, Corrigan M, Webb GJ, Hirschfield GM, et al. Low-dose interleukin-2 promotes STAT-5 phosphorylation, Treg survival and CTLA-4-dependent function in autoimmune liver diseases. Clin Exp Immunol. 2017;188(3):394–411. https://doi.org/10.1111/cei.12940.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5. https://doi.org/10.1126/science.1160062.
Corthay A. How do regulatory T cells work? Scand J Immunol. 2009;70(4):326–36. https://doi.org/10.1111/j.1365-3083.2009.02308.x.
Muller L, Mitsuhashi M, Simms P, Gooding WE, Whiteside TL. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci Rep. 2016;6(1):20254. https://doi.org/10.1038/srep20254.
Hagar A, Wang Z, Koyama S, Serrano JA, Melo L, Vargas S, et al. Endurance training slows breast tumor growth in mice by suppressing Treg cells recruitment to tumors. BMC Cancer. 2019;19(1):536. https://doi.org/10.1186/s12885-019-5745-7.
Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–55. https://doi.org/10.4049/jimmunol.182.3.1746.
Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122(10):2286–93. https://doi.org/10.1002/ijc.23392.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. https://doi.org/10.1038/nm1093.
Martin-Garcia D, Silva-Vilches C, Will R, Enk AH, Lonsdorf AS. Tumor-derived CCL20 affects B16 melanoma growth in mice. J Dermatol Sci. 2020;97(1):57–65. https://doi.org/10.1016/j.jdermsci.2019.12.005.
Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328(5979):749–52. https://doi.org/10.1126/science.1185837.
Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22(1):50–60. https://doi.org/10.1016/j.tcb.2011.09.003.
Redmer T. Deciphering mechanisms of brain metastasis in melanoma-the gist of the matter. Mol Cancer. 2018;17(1):106. https://doi.org/10.1186/s12943-018-0854-5.
Liu YN, Zhang H, Zhang L, Cai TT, Huang DJ, He J, et al. Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: leading to poor survival in bladder cancer. Cell Death Dis. 2019;10(2):50. https://doi.org/10.1038/s41419-018-1298-y.
Colie S, Van Veldhoven PP, Kedjouar B, Bedia C, Albinet V, Sorli SC, et al. Disruption of sphingosine 1-phosphate lyase confers resistance to chemotherapy and promotes oncogenesis through Bcl-2/Bcl-xL upregulation. Cancer Res. 2009;69(24):9346–53. https://doi.org/10.1158/0008-5472.CAN-09-2198.
Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY, et al. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc Natl Acad Sci U S A. 2006;103(46):17384–9. https://doi.org/10.1073/pnas.0600050103.
Bolli MH, Abele S, Binkert C, Bravo R, Buchmann S, Bur D, et al. 2-imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J Med Chem. 2010;53(10):4198–211. https://doi.org/10.1021/jm100181s.
Chimen M, McGettrick HM, Apta B, Kuravi SJ, Yates CM, Kennedy A, et al. Homeostatic regulation of T cell trafficking by a B cell-derived peptide is impaired in autoimmune and chronic inflammatory disease. Nat Med. 2015;21(5):467–75. https://doi.org/10.1038/nm.3842.
Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T (reg) cells. Nat Immunol. 2010;11(11):1047–56. https://doi.org/10.1038/ni.1939.
Martinez-Rodriguez M, Thompson AK, Monteagudo C. High CCL27 immunoreactivity in 'supratumoral' epidermis correlates with better prognosis in patients with cutaneous malignant melanoma. J Clin Pathol. 2017;70(1):15–9. https://doi.org/10.1136/jclinpath-2015-203537.
Simonetti O, Goteri G, Lucarini G, Filosa A, Pieramici T, Rubini C, et al. Potential role of CCL27 and CCR10 expression in melanoma progression and immune escape. Eur J Cancer. 2006;42(8):1181–7. https://doi.org/10.1016/j.ejca.2006.01.043.
Whiteside TL. FOXP3+ Treg as a therapeutic target for promoting anti-tumor immunity. Expert Opin Ther Targets. 2018;22(4):353–63. https://doi.org/10.1080/14728222.2018.1451514.
Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-β pathway for cancer therapy. Clin Cancer Res. 2012;18(17):4514–21. https://doi.org/10.1158/1078-0432.CCR-11-3224.
Whiteside TL. Targeting adenosine in cancer immunotherapy: a review of recent progress. Expert Rev Anticancer Ther. 2017;17(6):527–35. https://doi.org/10.1080/14737140.2017.1316197.
Li P, Liu C, Yu Z, Wu M. New insights into regulatory T cells: exosome- and non-coding RNA-mediated regulation of homeostasis and resident Treg cells. Front Immunol. 2016;7:574. https://doi.org/10.3389/fimmu.2016.00574.
Fan P, Li Z, Zuo C, Fang M. Promotion effects of mono-2-ethyhexyl phthalate (MEHP) on migration and invasion of human melanoma cells via activation of TGF-β signals. Cell Biochem Funct. 2020;38(1):38–46. https://doi.org/10.1002/cbf.3447.
Oh E, Hong J, Yun CO. Regulatory T cells induce metastasis by activating TGF-β and enhancing the epithelial-mesenchymal transition. Cells. 2019;8(11). https://doi.org/10.3390/cells8111387.
Oliva M, Rullan AJ, Piulats JM. Uveal melanoma as a target for immune-therapy. Ann Transl Med. 2016;4(9):172. https://doi.org/10.21037/atm.2016.05.04.
Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. 2011;51(2–3):170–82. https://doi.org/10.1007/s12026-011-8262-6.
MacNeil IA, Suda T, Moore KW, Mosmann TR, Zlotnik A. IL-10, a novel growth cofactor for mature and immature T cells. J Immunol. 1990;145(12):4167–73.
Whiteside TL. Exosomes carrying immunoinhibitory proteins and their role in cancer. Clin Exp Immunol. 2017;189(3):259–67. https://doi.org/10.1111/cei.12974.
Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol. 2016;36(3):301–12. https://doi.org/10.1007/s10571-016-0366-z.
Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One. 2010;5(7):e11469. https://doi.org/10.1371/journal.pone.0011469.
Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol. 2009;183(6):3720–30. https://doi.org/10.4049/jimmunol.0900970.
Sharma P, Diergaarde B, Ferrone S, Kirkwood JM, Whiteside TL. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci Rep. 2020;10(1):92. https://doi.org/10.1038/s41598-019-56542-4.
Tucci M, Mannavola F, Passarelli A, Stucci LS, Cives M, Silvestris F. Exosomes in melanoma: a role in tumor progression, metastasis and impaired immune system activity. Oncotarget. 2018;9(29):20826–37. https://doi.org/10.18632/oncotarget.24846.
Woods DM, Ramakrishnan R, Laino AS, Berglund A, Walton K, Betts BC, et al. Decreased suppression and increased phosphorylated STAT3 in regulatory T cells are associated with benefit from adjuvant PD-1 blockade in resected metastatic melanoma. Clin Cancer Res. 2018;24(24):6236–47. https://doi.org/10.1158/1078-0432.CCR-18-1100.
Sharma N, Vacher J, Allison JP. TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc Natl Acad Sci U S A. 2019;116(21):10453–62. https://doi.org/10.1073/pnas.1819004116.
Wang JJ, Burger P, Taube J, Soni A, Chaichana K, Sheu M, et al. PD-L1, PD-1, LAG-3, and TIM-3 in melanoma: expression in brain metastases compared to corresponding extracranial tumors. Cureus. 2019;11(12):e6352. https://doi.org/10.7759/cureus.6352.
Fourcade J, Sun Z, Chauvin JM, Ka M, Davar D, Pagliano O, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. 2018;3(14). https://doi.org/10.1172/jci.insight.121157.
Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97–111. https://doi.org/10.1111/imr.12520.
Kageshita T, Kashio Y, Yamauchi A, Seki M, Abedin MJ, Nishi N, et al. Possible role of galectin-9 in cell aggregation and apoptosis of human melanoma cell lines and its clinical significance. Int J Cancer. 2002;99(6):809–16. https://doi.org/10.1002/ijc.10436.
Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol. 2009;39(11):3147–59. https://doi.org/10.1002/eji.200839103.
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–29. https://doi.org/10.1084/jem.20090847.
Cai J, Wang D, Zhang G, Guo X. The role of PD-1/PD-L1 axis in Treg development and function: implications for cancer immunotherapy. Onco Targets Ther. 2019;12:8437–45. https://doi.org/10.2147/OTT.S221340.
Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 limits anti-tumor immunity. Immunity. 2016;44(2):316–29. https://doi.org/10.1016/j.immuni.2016.01.013.
Liu B, Zhang H, Li J, Lu C, Chen G, Zhang G, et al. Triptolide downregulates Treg cells and the level of IL-10, TGF-β, and VEGF in melanoma-bearing mice. Planta Med. 2013;79(15):1401–7. https://doi.org/10.1055/s-0033-1350708.
Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. 2013;190(9):4899–909. https://doi.org/10.4049/jimmunol.1300271.
Leclerc M, Voilin E, Gros G, Corgnac S, de Montpreville V, Validire P, et al. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat Commun. 2019;10(1):3345. https://doi.org/10.1038/s41467-019-11280-z.
Leignadier J, Favre S, Luther SA, Luescher IF. CD8 engineered cytotoxic T cells reprogram melanoma tumor environment. Oncoimmunology. 2016;5(3):e1086861. https://doi.org/10.1080/2162402X.2015.1086861.
Budhu S, Schaer DA, Li Y, Toledo-Crow R, Panageas K, Yang X, et al. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci Signal. 2017;10(494). https://doi.org/10.1126/scisignal.aak9702.
Liu Z, Liu JQ, Shi Y, Zhu X, Liu Z, Li MS, et al. Epstein-Barr virus-induced gene 3-deficiency leads to impaired antitumor T-cell responses and accelerated tumor growth. Oncoimmunology. 2015;4(7):e989137. https://doi.org/10.4161/2162402X.2014.989137.
Yan H, Zhang P, Kong X, Hou X, Zhao L, Li T, et al. Primary Tr1 cells from metastatic melanoma eliminate tumor-promoting macrophages through granzyme B- and perforin-dependent mechanisms. Tumour Biol. 2017;39(4):1010428317697554. https://doi.org/10.1177/1010428317697554.
Diller ML, Kudchadkar RR, Delman KA, Lawson DH, Ford ML. Exogenous IL-2 induces FoxP3+ Th17 cells in vivo in melanoma patients. J Immunother. 2016;39(9):355–66. https://doi.org/10.1097/CJI.0000000000000139.
Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother. 2018;67(8):1271–84. https://doi.org/10.1007/s00262-018-2186-0.
Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011;71(8):2892–900. https://doi.org/10.1158/0008-5472.CAN-10-4246.
Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, et al. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res. 2015;3(4):345–55. https://doi.org/10.1158/2326-6066.CIR-14-0196.
Chu CL, Lee YP, Pang CY, Lin HR, Chen CS, You RI. Tyrosine kinase inhibitors modulate dendritic cell activity via confining c-kit signaling and tryptophan metabolism. Int Immunopharmacol. 2020;82:106357. https://doi.org/10.1016/j.intimp.2020.106357.
Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–58. https://doi.org/10.1172/JCI80445.
Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8+ T cell-derived interferon-γ. Immunity. 2019;51(2):381–97e6. https://doi.org/10.1016/j.immuni.2019.06.017.
Wong SQ, Behren A, Mar VJ, Woods K, Li J, Martin C, et al. Whole exome sequencing identifies a recurrent RQCD1 P131L mutation in cutaneous melanoma. Oncotarget. 2015;6(2):1115–27. https://doi.org/10.18632/oncotarget.2747.
Jacquelot N, Duong CPM, Belz GT, Zitvogel L. Targeting chemokines and chemokine receptors in melanoma and other cancers. Front Immunol. 2018;9:2480. https://doi.org/10.3389/fimmu.2018.02480.
Ketcham JM, Marshall LA, Talay O. CCR4 antagonists inhibit Treg trafficking into the tumor microenvironment. ACS Med Chem Lett. 2018;9(10):953–5. https://doi.org/10.1021/acsmedchemlett.8b00351.
Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A. 2013;110(44):17945–50. https://doi.org/10.1073/pnas.1316796110.
Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30(8):837–42. https://doi.org/10.1200/JCO.2011.37.3472.
Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163(10):5211–8.
Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202(7):885–91. https://doi.org/10.1084/jem.20050940.
Murakami T, Cardones AR, Finkelstein SE, Restifo NP, Klaunberg BA, Nestle FO, et al. Immune evasion by murine melanoma mediated through CC chemokine receptor-10. J Exp Med. 2003;198(9):1337–47. https://doi.org/10.1084/jem.20030593.
Al-Awadhi FH, Gao B, Rezaei MA, Kwan JC, Li C, Ye T, et al. Discovery, synthesis, pharmacological profiling, and biological characterization of brintonamides A-E, novel dual protease and GPCR modulators from a marine cyanobacterium. J Med Chem. 2018;61(14):6364–78. https://doi.org/10.1021/acs.jmedchem.8b00885.
Longo-Imedio MI, Longo N, Trevino I, Lazaro P, Sanchez-Mateos P. Clinical significance of CXCR3 and CXCR4 expression in primary melanoma. Int J Cancer. 2005;117(5):861–5. https://doi.org/10.1002/ijc.21269.
D'Alterio C, Buoncervello M, Ierano C, Napolitano M, Portella L, Rea G, et al. Targeting CXCR4 potentiates anti-PD-1 efficacy modifying the tumor microenvironment and inhibiting neoplastic PD-1. J Exp Clin Cancer Res. 2019;38(1):432. https://doi.org/10.1186/s13046-019-1420-8.
Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest. 2015;125(4):1379–87. https://doi.org/10.1172/JCI76369.
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. https://doi.org/10.1097/WNF.0b013e3181cbf825.
Muls N, Dang HA, Sindic CJ, van Pesch V. Fingolimod increases CD39-expressing regulatory T cells in multiple sclerosis patients. PLoS One. 2014;9(11):e113025. https://doi.org/10.1371/journal.pone.0113025.
Pereira FV, Arruda DC, Figueiredo CR, Massaoka MH, Matsuo AL, Bueno V, et al. FTY720 induces apoptosis in B16F10-NEX2 murine melanoma cells, limits metastatic development in vivo, and modulates the immune system. Clinics (Sao Paulo). 2013;68(7):1018–27. https://doi.org/10.6061/clinics/2013(07)21.
Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24(2):315–27. https://doi.org/10.1007/s10555-005-1580-1.
Rietschel P, Chapman PB. Immunotherapy of melanoma. Hematol Oncol Clin North Am. 2006;20(3):751–66. https://doi.org/10.1016/j.hoc.2006.02.005.
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–21. https://doi.org/10.1038/nm1325.
Kong LY, Wei J, Sharma AK, Barr J, Abou-Ghazal MK, Fokt I, et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol Immunother. 2009;58(7):1023–32. https://doi.org/10.1007/s00262-008-0618-y.
Leung KH, Liu LJ, Lin S, Lu L, Zhong HJ, Susanti D, et al. Discovery of a small-molecule inhibitor of STAT3 by ligand-based pharmacophore screening. Methods. 2015;71:38–43. https://doi.org/10.1016/j.ymeth.2014.07.010.
Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA, et al. Tumor-derived γδ regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol. 2013;190(5):2403–14. https://doi.org/10.4049/jimmunol.1202369.
Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. TLR8-mediated metabolic control of human Treg function: a mechanistic target for cancer immunotherapy. Cell Metab. 2019;29(1):103–23e5. https://doi.org/10.1016/j.cmet.2018.09.020.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–87. https://doi.org/10.1038/nrc.2016.36.
Yang Y, Xu W, Peng D, Wang H, Zhang X, Wang H, et al. An oncolytic adenovirus targeting transforming growth factor β inhibits protumorigenic signals and produces immune activation: a novel approach to enhance anti-PD-1 and anti-CTLA-4 therapy. Hum Gene Ther. 2019;30(9):1117–32. https://doi.org/10.1089/hum.2019.059.
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30. https://doi.org/10.1056/NEJMoa1412082.
Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. 2015;21(4):712–20. https://doi.org/10.1158/1078-0432.CCR-14-2468.
Bordry N, Broggi MAS, de Jonge K, Schaeuble K, Gannon PO, Foukas PG, et al. Lymphatic vessel density is associated with CD8+ T cell infiltration and immunosuppressive factors in human melanoma. Oncoimmunology. 2018;7(8):e1462878. https://doi.org/10.1080/2162402X.2018.1462878.
Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, et al. Sustained type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29(10):846–61. https://doi.org/10.1038/s41422-019-0224-x.
Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci U S A. 2016;113(30):8490–5. https://doi.org/10.1073/pnas.1608873113.
Toor SM, Syed Khaja AS, Alkurd I, Elkord E. In-vitro effect of pembrolizumab on different T regulatory cell subsets. Clin Exp Immunol. 2018;191(2):189–97. https://doi.org/10.1111/cei.13060.
Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210(9):1685–93. https://doi.org/10.1084/jem.20130573.
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42. https://doi.org/10.1158/2326-6066.CIR-13-0013.
Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol. 2014;44(9):2703–11. https://doi.org/10.1002/eji.201344392.
Liu Y, Cai P, Wang N, Zhang Q, Chen F, Shi L, et al. Combined blockade of Tim-3 and MEK inhibitor enhances the efficacy against melanoma. Biochem Biophys Res Commun. 2017;484(2):378–84. https://doi.org/10.1016/j.bbrc.2017.01.128.
Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173–83. https://doi.org/10.4049/jimmunol.1002050.
Khair DO, Bax HJ, Mele S, Crescioli S, Pellizzari G, Khiabany A, et al. Combining immune checkpoint inhibitors: established and emerging targets and strategies to improve outcomes in melanoma. Front Immunol. 2019;10:453. https://doi.org/10.3389/fimmu.2019.00453.
Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3(2):135–42. https://doi.org/10.1038/ni759.
Avogadri F, Zappasodi R, Yang A, Budhu S, Malandro N, Hirschhorn-Cymerman D, et al. Combination of αvirus replicon particle-based vaccination with immunomodulatory antibodies: therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer Immunol Res. 2014;2(5):448–58. https://doi.org/10.1158/2326-6066.CIR-13-0220.
Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med. 2019;25(5):759–66. https://doi.org/10.1038/s41591-019-0420-8.
Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol. 2010;28(1):57–78. https://doi.org/10.1146/annurev-immunol-030409-101243.
Jeffrey RIA, Michael JP, Laura QMC, Grant AM, Todd MB, Stephen VL, et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J Clin Oncol. 2016;34(15):1. https://doi.org/10.1200/JCO.2016.34.15_suppl.101.
Omid HJ, Adi D, Willeke R, Ferry E, Candy B, Cyril K, et al. First in human (FIH) study of an OX40 agonist monoclonal antibody (mAb) PF-04518600 (PF-8600) in adult patients (pts) with select advanced solid tumors- preliminary safety and pharmacokinetic (PK):pharmacodynamic results. J Clin Oncol. 2016;34(15):1.
Wikenheiser DJ, Stumhofer JS. ICOS co-stimulation: friend or foe? Front Immunol. 2016;7:304. https://doi.org/10.3389/fimmu.2016.00304.
Strauss L, Bergmann C, Szczepanski MJ, Lang S, Kirkwood JM, Whiteside TL. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: implications and impact on tumor-mediated immune suppression. J Immunol. 2008;180(5):2967–80. https://doi.org/10.4049/jimmunol.180.5.2967.
Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res. 2012;72(23):6130–41. https://doi.org/10.1158/0008-5472.CAN-12-2409.
Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2014;124(1):99–110. https://doi.org/10.1172/JCI46266.
Howard ABM, Anthony WT, Shivaani K, Gerald SF, Russell KP, Scott ST, et al. Phase 1 safety of ICOS agonist antibody JTX-2011 alone and with nivolumab (nivo) in advanced solid tumors; predicted vs observed pharmacokinetics (PK) in ICONIC. J Clin Oncol. 2017;35(15):1. https://doi.org/10.1200/JCO.2017.35.15_suppl.3033.
Mousavi-Niri N, Naseroleslami M, Hadjati J. Anti-regulatory T cell vaccines in immunotherapy: focusing on FoxP3 as target. Hum Vaccin Immunother. 2019;15(3):620–4. https://doi.org/10.1080/21645515.2018.1545625.
Nair S, Boczkowski D, Fassnacht M, Pisetsky D, Gilboa E. Vaccination against the forkhead family transcription factor Foxp3 enhances tumor immunity. Cancer Res. 2007;67(1):371–80. https://doi.org/10.1158/0008-5472.CAN-06-2903.
Mousavi Niri N, Memarnejadian A, Pilehvar-Soltanahmadi Y, Agha Sadeghi M, Mahdavi M, Kheshtchin N, et al. Improved anti-Treg vaccination targeting Foxp3 efficiently decreases regulatory T cells in mice. J Immunother. 2016;39(7):269–75. https://doi.org/10.1097/CJI.0000000000000133.
Miguel A, Sendra L, Noe V, Ciudad CJ, Dasi F, Hervas D, et al. Silencing of Foxp3 enhances the antitumor efficacy of GM-CSF genetically modified tumor cell vaccine against B16 melanoma. Onco Targets Ther. 2017;10:503–14. https://doi.org/10.2147/OTT.S104393.
Ren S, Wang Q, Zhang Y, Song Y, Dong X, Zhang W, et al. Imiquimod enhances the potency of an exogenous BM-DC based vaccine against mouse melanoma. Int Immunopharmacol. 2018;64:69–77. https://doi.org/10.1016/j.intimp.2018.08.026.
Droeser R, Zlobec I, Kilic E, Guth U, Heberer M, Spagnoli G, et al. Differential pattern and prognostic significance of CD4+, FOXP3+ and IL-17+ tumor infiltrating lymphocytes in ductal and lobular breast cancers. BMC Cancer. 2012;12(1):134. https://doi.org/10.1186/1471-2407-12-134.
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22(6):679–84. https://doi.org/10.1038/nm.4086.
Di Pilato M, Kim EY, Cadilha BL, Prussmann JN, Nasrallah MN, Seruggia D, et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature. 2019;570(7759):112–6. https://doi.org/10.1038/s41586-019-1215-2.
Fujii H, Josse J, Tanioka M, Miyachi Y, Husson F, Ono M. Regulatory T cells in melanoma revisited by a computational clustering of FOXP3+ T cell subpopulations. J Immunol. 2016;196(6):2885–92. https://doi.org/10.4049/jimmunol.1402695.
Blankenstein SA, van Akkooi ACJ. Adjuvant systemic therapy in high-risk melanoma. Melanoma Res. 2019;29(4):358–64. https://doi.org/10.1097/CMR.0000000000000604.
Zhang Y, Du X, Liu M, Tang F, Zhang P, Ai C, et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. 2019;29(8):609–27. https://doi.org/10.1038/s41422-019-0184-1.
Altman A, Kong KF. pH-sensitive anti-CTLA4 antibodies: yes to efficacy, no to toxicity. Cell Res. 2019;29(8):601–2. https://doi.org/10.1038/s41422-019-0198-8.
Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28(suppl_12):xii33–43. https://doi.org/10.1093/annonc/mdx683.
Sato K, Sato N, Xu B, Nakamura Y, Nagaya T, Choyke PL, et al. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci Transl Med. 2016;8(352):352ra110. https://doi.org/10.1126/scitranslmed.aaf6843.
Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(13):1270–1. https://doi.org/10.1056/NEJMc1509660.
Acknowledgements
Not applicable.
Code availability
Not applicable.
Funding
The research was funded by the NIH grants CA224070, CA217489, CA210944 and CA170340.
Author information
Authors and Affiliations
Contributions
LLH drafted the manuscript. XX reviewed and edited the manuscript. YYG and SJL conducted the literature search. HSW and LLO prepared the figures. JJZ performed manuscript review. The authors have reviewed and approved the manuscript before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Corresponding author Xiaowei Xu is a member of the Editorial Board for Molecular Biomedicine, Xiaowei Xu was not involved in the journal's review of, or decisions related to, this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, L., Guo, Y., Liu, S. et al. Targeting regulatory T cells for immunotherapy in melanoma. Mol Biomed 2, 11 (2021). https://doi.org/10.1186/s43556-021-00038-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-021-00038-z