

Abstract
Background/Aim: Resveratrol (RSV) may have therapeutic potential for various diseases. Here we investigated the effect of RSV on oxidised low-density lipoprotein- (ox-LDL) induced apoptosis in RAW264.7 macrophages. Methods: Apoptosis of macrophages following incubation with ox-LDL (with or without RSV pre-treatment) was detected by flow cytometry. Western blotting was used to assess the protein expression of Bax, Bcl-2, and caspase-3 as well as ox-LDL receptor 1 (LOX-1) and p38 mitogen-activated protein kinase (MAPK) phosphorylation. Reactive oxygen species (ROS) generation was evaluated by 2', 7'-dichlorofluorescein diacetate, and JC-1 probe was used to determine the mitochondrial transmembrane potential of cells. Results: Ox-LDL significantly reduced viability and increased the rate of apoptosis (P < 0.05) in RAW264.7 cells. However, pre-treatment with RSV resulted in a remarkable decrease in this apoptotic effect. Moreover, ox-LDL caused the up-regulation of Bax and the down-regulation of Bcl-2, as well as the activation of caspase-3. Expression of LOX-1, phosphorylation of p38 MAPK, and intracellular ROS production also increased after ox-LDL stimulation. Strikingly, these effects were abolished by pre-treatment of cells with RSV. Conclusion: RSV suppresses ox-LDL-induced macrophage apoptosis. These beneficial effects might be exerted through inhibition of ROS generation, LOX-1, and the p38 MAPK signalling pathway.
Introduction
Atherosclerosis is the most common type of cardiovascular disease and is currently the leading cause of mortality in both men and women worldwide. Current therapy consists of pharmacologic optimisation as well as limited revascularisation, and reconstructive or replacement options that suffer from issues of durability, restenosis, and atherosclerotic progression [1]. Despite advances in interventional and pharmacological therapy of atherosclerotic disease, it is still the leading cause of death in the developed world [2,3].
In the pathogenesis of atherosclerosis, macrophages and oxidised low-density lipoproteins (ox-LDLs) are important for intracellular lipid accumulation and foam cell formation [4]. The induction of foam cell apoptosis is involved in subsequent plaque formation, and some foam cells escape from these lesions into peripheral blood. In addition, macrophages play multifaceted roles in inducing plaque rupture, blood coagulation, and fibrinolysis via the production of various enzymes, activators, inhibitors, and bioactive mediators. During the development of atherosclerosis, macrophages interact with vascular endothelial cells, medial smooth muscle cells, and infiltrated inflammatory cells [5]. Therefore, protection against macrophage apoptosis is considered a novel therapeutic strategy for atherosclerosis [6].
Recent studies have shown that sirtuin 1 (SIRT1), a molecule associated with metabolism, is likely involved in the modulation of the atherosclerotic process [7,8,9]. Stein et al. reported that partial deletion of SIRT1 down-regulated the expression of the lectin-like scavenger receptor, ox-LDL receptor 1 (LOX-1), resulting in reduced foam cell formation and atherosclerosis [10]. Resveratrol (trans-3,5,4'-trihydroxystilbene; RSV), a natural phytophenol and potent SIRT1 activator, has received great attention during the past few years due to its beneficial roles in longevity and glucose homeostasis [11]. It has been suggested that RSV could stimulate the endothelial production of nitric oxide, reduce oxidative stress, inhibit vascular inflammation and prevent platelet aggregation in vivo and in vitro [12]. Despite extensive analysis of the role of RSV in the prevention and treatment of cardiovascular diseases, few studies have focussed on the effect of RSV on ox-LDL-induced macrophage apoptosis. In the present study, we hypothesised that RSV can attenuate ox-LDL-induced macrophage apoptosis and attempted to explore the molecular mechanisms underlying this potential protective effect.
Materials and Methods
The experimental protocol was approved by the Ethics Review Committee of the Laboratory Center of Shanghai Tenth People's Hospital.
Reagents
The murine macrophage cell line RAW264.7 was purchased from the Institute of Biochemistry and Cell Biology (Shanghai Institute for Biological Science, the Chinese Academy of Sciences, Shanghai, China). Unless otherwise stated, all chemical reagents in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
RAW264.7 cells were maintained in Dulbecco's modified Eagle medium (Gibco, Life Technologies, Carlsbad, CA, USA) containing 10% penicillin-streptomycin-amphotericin B and 10% foetal bovine serum (Gibco, Life Technologies) in a humidified atmosphere of 5% CO2 and 95% air. The culture medium was replaced every 2-3 days. RSV was dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 104 M and stored at -20 °C. Further dilutions were made in cell culture medium. The final working concentration of DMSO in cell culture experiments did not exceed 0.125%. All controls for in vitro experiments contained 0.125% DMSO as a vehicle control.
Lipoprotein preparation
LDL was separated from the plasma of normal healthy volunteers by discontinuous ultracentrifugation as previously described [13], diluted in PBS to a concentration of 1 mg· mL-1 and subsequently dialysed against PBS three times to remove residual EDTA. Ox-LDL was prepared by incubating LDL with freshly prepared 0.5 µM CuSO4 in PBS for 20 h at 37 °C in a shaking water bath. The degree of oxidation of ox-LDL was assessed on the basis of increased mobility in an agarose gel (compared with native LDL) and an increased level of thiobarbituric acid-reactive substances [14]. Protein concentrations were determined using the BCA protein assay kit (Pierce Chemical Company, Rockford, IL, USA). The prepared Ox-LDL was endotoxin free.
Cell viability assay
The viability of RAW264.7 cells was evaluated using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto, Japan), according to the manufacturer's instructions. In this assay, RAW264.7 cells were rested in 96-well plates (100 μl/well) at a density of 1-2 × 104 cells· mL-1 for 24 h, followed by incubation with RSV for 24 h. Cells were subsequently treated with different concentrations of ox-LDL in complete medium for 24 h, prior to incubation with the Cell Counting Kit-8 reagent for 2 h. Absorbance at 450 nm was then measured using the Synergy HT multi-mode plate reader (Bio-Tek, Winooski, VT, USA).
Apoptosis assay
Apoptosis was assessed in three independent experiments using an annexin V apoptosis detection kit (Roche, Indianapolis, IN, USA). Macrophages (1 × 105 cells per well) were pre-treated with various concentrations of RSV for 24 h, and then incubated with ox-LDL (150 mg· L-1) for 48 h. After RSV and ox-LDL treatment, cells were harvested and resuspended in binding buffer. Cells were double stained with fluorescein isothiocyanate-conjugated annexin V and propidium iodide for 15 min at 20 °C in Ca2+-enriched binding buffer, and fluorescence was then detected by flow cytometry (FCM). The EPICS XL flow cytometer (Beckman Coulter, Fullerton, CA, USA) was used to analyse ≥10 000 cells in each of the three independent experiments.
Protein extraction and western blot analysis
The total proteins of RAW264.7 were analysed using western blotting. The mitochondrial proteins were extracted from cells using the Minute Mitochondria Membrane Protein Isolation Kit (Invent Biotechnologies Inc., MN, USA). To perform western blot experiments, macrophages were removed from plates after treatment with lysis solution (4% SDS, 2 mM EDTA, 50 mM Tris-HCl, pH 6.8). The homogenates were centrifuged at 15 000 × g for 15 min at 4 °C and then supernatants were collected and removed for protein quantification using the Bradford method. Equal amounts of protein (50 µg) were separated by 8% or 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. These membranes were then washed with Tris-buffered saline, blocked with 5% non-fat dry milk (except in the analysis of phosphorylated myosin phosphatase targeting protein, where 5% BSA was used for blocking) in Tris-buffered saline containing Tween-20 for 1 h, and incubated with the appropriate primary antibody at dilutions recommended by the supplier. Each membrane was then washed before incubation with a secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. The blots were then developed with SuperSignal enhanced chemiluminescent substrate solution (Pierce Chemical Company, Rockford, IL, USA). Anti-β-actin, anti-caspase-3, anti-Bcl-2, anti-Bax, and anti-LOX-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p38 mitogen-activated protein kinase (MAPK) and anti-phosphorylated p38 MAPK were purchased from Cell Signaling Technology (Danvers, MA, USA).
JC-1 staining and mitochondrial membrane potential test
Loss of mitochondrial membrane potential within RAW264.7 cells was assessed using the Leica DMI6000 fluorescence microscope (Leica Microsystems, Wetzlar, Germany) following cell staining with the JC-1 dye (Beyotime Institute of Biotechnology, Jiangsu, China). Mitochondrial membrane potentials were monitored by determining the relative amounts of dual emissions from mitochondrial JC-1 monomers or polymers using the fluorescent microscope. Mitochondrial depolarisation was detected by an increase in the red/green fluorescence intensity ratio. Red emission of the dye is indicative of the formation of JC-1 polymers after potential-dependent aggregation in the mitochondria, reflecting normal mitochondrial membrane potential. Green fluorescence, which indicates the presence of the monomeric form of JC-1, appeared in the cytosol after depolarisation of the mitochondrial membrane. Fluorescence was detected using the Infinite 200 microplate reader (Tecan, Männedorf, Switzerland). The excitation and emission wavelengths used to detect the monomeric form of JC-1 were 514 nm and 529 nm, whereas 585 nm and 590 nm were used to detect JC-1 polymers. The ratio of JC-1 monomer to polymer demonstrated the condition of the membrane potential of RAW264.7 mitochondria.
Intracellular reactive oxygen species (ROS) measurement
The level of intracellular ROS was measured with the fluorescent probe 2', 7'-dichlorofluorescein diacetate (DCFH-DA; Beyotime Institute of Biotechnology). RAW264.7 macrophages were seeded in a 24-well plate at 1-2 × 105 cells per well and rested for 24 h. Next, cells were treated with various concentrations of RSV for 24 h, or left untreated, and then incubated with 150 mg· L-1 ox-LDL for 2 h. Cells were then incubated with serum-free medium containing 10 μM DCFH-DA for 30 min at 37 °C in the dark, washed twice with PBS, trypsinised, resuspended, and then immediately analysed by FCM. Negative controls were simultaneously created using the same protocol, but with DCFH-DA staining omitted. A 530 nm band-pass filter was used to detect DCFH-DA fluorescence, and each determination was based on the mean fluorescence intensity of 10 000 cells.
Statistical analysis
All data were analysed with GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA) and are expressed as mean ± SD. Statistical analyses were performed using one-way ANOVA, followed by Tukey's post-hoc test. Differences with P< 0.05 were considered statistically significant.
Results
The effect of RSV on cell viability with and without exposure to ox-LDL
Cell viability was measured to evaluate whether RSV could protect RAW264.7 cells from injuries induced by ox-LDL. As shown in Fig. 1A, RSV exerted no cytotoxicity on RAW264.7 cells at 1 µM (P > 0.05), 10 µM (P > 0.05) or 100µM (P > 0.05), whereas cytotoxicity was caused by the treatment of cells with 200 µM (P < 0.05) and 500 µM (P < 0.01) RSV. The cytotoxicity of RSV increased in a concentration-dependent manner.

The viability of RAW264.7 macrophages incubated with different concentrations of RSV. (A) RAW264.7 macrophages were incubated for 24 h in medium containing 1-500 µM RSV, and then viability was assessed using the Cell Counting Kit-8 according to the manufacturer's protocol. (B) The cells were incubated with various concentrations of ox-LCL for 24 h after pre-treatment with RSV, and then viability was assessed. The cytotoxic effects of ox-LDL on RAW264.7 cells were reduced by RSV at 10 and 100 µM. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the control group. # P < 0.05 indicates significant differences from the ox-LDL 150 mg· L-1 group.
The viability of RAW264.7 macrophages incubated with different concentrations of RSV. (A) RAW264.7 macrophages were incubated for 24 h in medium containing 1-500 µM RSV, and then viability was assessed using the Cell Counting Kit-8 according to the manufacturer's protocol. (B) The cells were incubated with various concentrations of ox-LCL for 24 h after pre-treatment with RSV, and then viability was assessed. The cytotoxic effects of ox-LDL on RAW264.7 cells were reduced by RSV at 10 and 100 µM. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the control group. # P < 0.05 indicates significant differences from the ox-LDL 150 mg· L-1 group.
Next, we investigated whether RSV exerted a dose-dependent protective effect on RAW264.7 macrophages exposed to ox-LDL. As shown in Fig. 1B, the exposure of cells to ox-LDL at 150 mg· L-1 for 48 h significantly decreased cell viability compared with untreated control cells (P < 0.01; control cell viability 100%, ox-LDL treated cell viability 61.0 ± 3.1%). However, RSV pre-treatment significantly reduced the ox-LDL-induced loss of cell viability in a concentration-dependent manner, demonstrating the anti-cytotoxic effect of this chemical compound (Fig. 1B).
Ox-LDL induced apoptosis in macrophages and foam cell formation
We investigated whether ox-LDL could induce apoptosis in RAW264.7 cells and foam cell formation. RAW264.7 cells were incubated with 50-200 mg·L-1 ox-LDL for 48 h or 150 mg·L-1 ox-LDL for 12, 24, 48, or 72 h, and then the level of apoptosis was determined by FCM. As shown in Fig. 2A and 2B, exposure of RAW264.7 cells to ox-LDL induced a time- and dose-dependent increase in apoptosis. In addition, the exposure of RAW264.7 cells to 150 mg·L-1 ox-LDL for 24 h and 48 h increased the rate of foam cell formation (Fig. 2C).
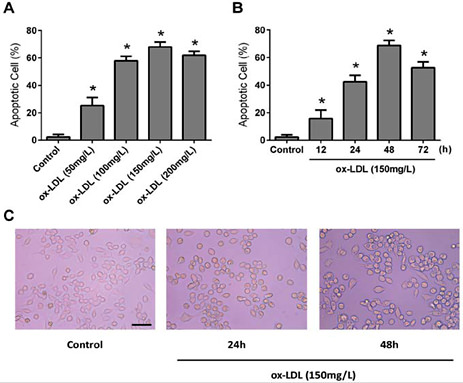
Ox-LDL- induced apoptosis of cultured RAW264.7 macrophages. Apoptosis was determined by annexin V and propidium iodide dual staining followed by analysis on a flow cytometer. (A) Cells were treated with 50, 100, 150 and 200 mg· L-1 of ox-LDL for 48 h prior to analysis of apoptosis. (B) Cells were incubated with ox-LDL (150 mg· L--1) for 12, 24, 48 and 72 h. Values are expressed as means ± SD from triplicate experiments. (C) RAW264.7 cells were exposed to ox-LDL at 150 mg· L-1 for 24 h and 48 h, prior to staining and flow cytometric analysis. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the control group.
Ox-LDL- induced apoptosis of cultured RAW264.7 macrophages. Apoptosis was determined by annexin V and propidium iodide dual staining followed by analysis on a flow cytometer. (A) Cells were treated with 50, 100, 150 and 200 mg· L-1 of ox-LDL for 48 h prior to analysis of apoptosis. (B) Cells were incubated with ox-LDL (150 mg· L--1) for 12, 24, 48 and 72 h. Values are expressed as means ± SD from triplicate experiments. (C) RAW264.7 cells were exposed to ox-LDL at 150 mg· L-1 for 24 h and 48 h, prior to staining and flow cytometric analysis. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the control group.
RSV inhibits the ox-LDL induced apoptosis of RAW264.7 macrophages
To further confirm the inhibitory effect of RSV on ox-LDL induced apoptosis, annexin V/propidium iodide dual staining and FCM analysis were performed. Representative images from FCM assays and summarised data are presented in Fig. 3A and 3B. These analyses revealed that, compared with the control, ox-LDL increased the late apoptosis rate and early apoptosis rate, from 0.8 ± 0.3% to 2.2 ± 1.2% and 1.4 ± 0.2% to 72.5 ± 2.5%, respectively (P < 0.01). Pre-treatment with RSV (1, 10 and 100 µM) significantly reduced apoptosis in a concentration-dependent manner (Fig. 3A and 3B, P < 0.05).

The inhibitory effect of RSV on ox-LDL-induced macrophage apoptosis. (A) Flow cytometry dot plots show necrotic and apoptotic cell populations, based on annexin V and propidium iodide (PI) staining. In each figure, the upper left quadrant corresponds to necrotic cells (annexin V- PI+), the upper right quadrant contains the late apoptotic cells (annexin V+ PI+), the lower left quadrant shows viable cells (annexin V- PI-), and the lower right quadrant represents the early apoptotic cells (annexin V+ PI-). (B) The rate of apoptosis as quantified by flow cytometry. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the control group. # P < 0.01 indicates significant differences from the control group.
The inhibitory effect of RSV on ox-LDL-induced macrophage apoptosis. (A) Flow cytometry dot plots show necrotic and apoptotic cell populations, based on annexin V and propidium iodide (PI) staining. In each figure, the upper left quadrant corresponds to necrotic cells (annexin V- PI+), the upper right quadrant contains the late apoptotic cells (annexin V+ PI+), the lower left quadrant shows viable cells (annexin V- PI-), and the lower right quadrant represents the early apoptotic cells (annexin V+ PI-). (B) The rate of apoptosis as quantified by flow cytometry. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the control group. # P < 0.01 indicates significant differences from the control group.
The effect of RSV on ox-LDL-induced expression of Bcl-2 and Bax
Cell survival is dependent on the ratio of Bcl-2 (antiapoptotic) to Bax (proapoptotic) and in this study, the levels of these apoptotic regulators were determined by western blotting. The treatment of RAW264.7 cells with 150 mg·L-1 ox-LDL for 48 h caused a significant decrease in the level of Bcl-2 (Fig. 4A and 4B, P < 0.05), but enhanced Bax expression (Fig. 4A and 4C, P < 0.05), resulting in a decrease in the Bcl-2/Bax ratio (Fig. 4D, P < 0.05). In contrast, various concentrations of RSV increased the Bcl-2/Bax ratio (Fig. 4D, P < 0.05) by increasing Bcl-2 and simultaneously decreasing Bax expression (Fig. 4A-C, all P < 0.05).
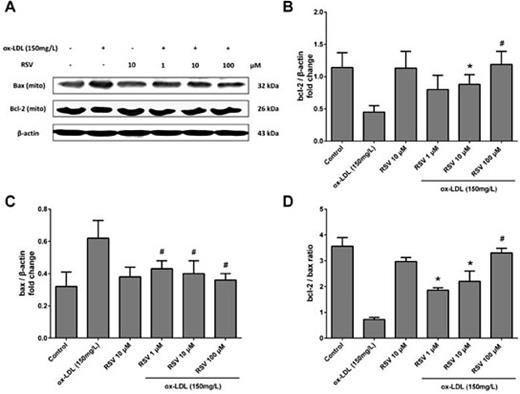
The effect of RSV on ox-LDL-induced expression of Bax and Bcl-2 in RAW264.7 cells. (A) Representative western blots show ox-LDL-induced expression of Bax and Bcl-2 in RAW264.7 cells with and without pre-treatment with RSV. (B) Densitometric analysis shows that treatment with ox-LDL decreased the expression of Bcl-2, but RSV significantly restored Bcl-2 expression. (C) Densitometric analysis of RSV's effect on Bax expression. Treatment with ox-LDL at 150·mg· L-1 significantly increased Bax expression. (D) Densitometric analysis shows that treatment with ox-LDL reduced the Bcl-2/Bax ratio, and that this effect could be inhibited by RSV pre-treatment. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the ox-LDL 150·mg· L-1 group. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
The effect of RSV on ox-LDL-induced expression of Bax and Bcl-2 in RAW264.7 cells. (A) Representative western blots show ox-LDL-induced expression of Bax and Bcl-2 in RAW264.7 cells with and without pre-treatment with RSV. (B) Densitometric analysis shows that treatment with ox-LDL decreased the expression of Bcl-2, but RSV significantly restored Bcl-2 expression. (C) Densitometric analysis of RSV's effect on Bax expression. Treatment with ox-LDL at 150·mg· L-1 significantly increased Bax expression. (D) Densitometric analysis shows that treatment with ox-LDL reduced the Bcl-2/Bax ratio, and that this effect could be inhibited by RSV pre-treatment. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. * P < 0.05 indicates significant differences from the ox-LDL 150·mg· L-1 group. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
The effect of RSV on ox-LDL-induced caspase-3 activation
We used western blotting to determine the effect of RSV on the activated caspase-3 induced by ox-LDL. As seen in Fig. 5A-C, treatment of RAW264.7 macrophages with ox-LDL for 48 h significantly increased caspase-3 cleavage and cleaved caspase-3/ total caspase-3 ratio. This caspase-3 activation was reduced by pre-treatment of cells with RSV at 1, 10 and 100 µM (Fig. 5A and 5B, all P < 0.05).

The effect of RSV on ox-LDL-induced caspase-3 activation. (A) Representative western blots show ox-LDL-induced expression of total caspase-3 and cleaved caspase-3 in RAW264.7 cells with and without pre-treatment with RSV. (B) Densitometric analysis shows that treatment with ox-LDL for 48 h increased the expression of cleaved caspase-3, which was reduced by RSV. (C) Quantitative analysis of the ratios of cleaved caspase-3 to total caspase-3 shows that RSV pretreatment significantly decreased the cleaved caspase-3/total caspase-3 ratio in ox-LDL treated macrophages. (D) Quantitative analysis of the ratios of JC-1 monomer to JC-1 polymer (green/red fluorescence) shows that ox-LDL increased and RSV cDNA decreased the green/red fluorescence ratio. Values are expressed as means ± SD from triplicate experiments. The ratio was significantly lower for cells treated with RSV (P < 0.01). R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
The effect of RSV on ox-LDL-induced caspase-3 activation. (A) Representative western blots show ox-LDL-induced expression of total caspase-3 and cleaved caspase-3 in RAW264.7 cells with and without pre-treatment with RSV. (B) Densitometric analysis shows that treatment with ox-LDL for 48 h increased the expression of cleaved caspase-3, which was reduced by RSV. (C) Quantitative analysis of the ratios of cleaved caspase-3 to total caspase-3 shows that RSV pretreatment significantly decreased the cleaved caspase-3/total caspase-3 ratio in ox-LDL treated macrophages. (D) Quantitative analysis of the ratios of JC-1 monomer to JC-1 polymer (green/red fluorescence) shows that ox-LDL increased and RSV cDNA decreased the green/red fluorescence ratio. Values are expressed as means ± SD from triplicate experiments. The ratio was significantly lower for cells treated with RSV (P < 0.01). R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
RSV restores ox-LDL-induced loss of mitochondrial membrane potential
To determine whether RSV protects ox-LDL-induced apoptosis of RAW264.7 cells through a mitochondrial pathway, we investigated the effect of RSV on mitochondrial membrane potential. The JC-1 dye was used to assess mitochondrial membrane potential, with a decrease in potential indicated by an increase in green fluorescence, and an increase in potential indicated by an increase in red fluorescence. Membrane potential was therefore determined by the ratio of green/red fluorescence intensity. When RAW264.7 cells were treated with ox-LDL, this ratio was increased relative to untreated control cells. As shown in Fig. 5D, the green/red fluorescence ratio was restored by the incubation of cells with RSV at 1, 10 and 100 µM prior to ox-LDL exposure (Fig. 5D, all P < 0.01).
The inhibition of ROS production by RSV
The level of ROS within RAW264.7 macrophages treated with RSV and/or ox-LDL was determined by staining cells with DCFH-DA and analysing their fluorescence by FCM. Representative FCM images are presented in Fig. 6A-E, and the summarised data are shown in Fig. 6F The treatment of RAW264.7 macrophages with 150 mg·L-1 ox-LDL for 2 h induced substantial production of ROS (Fig. 6B and 6F), while pre-treatment with RSV for 24 h significantly inhibited ROS production in a dose-dependent manner (Fig. 6C-F, all P < 0.05).
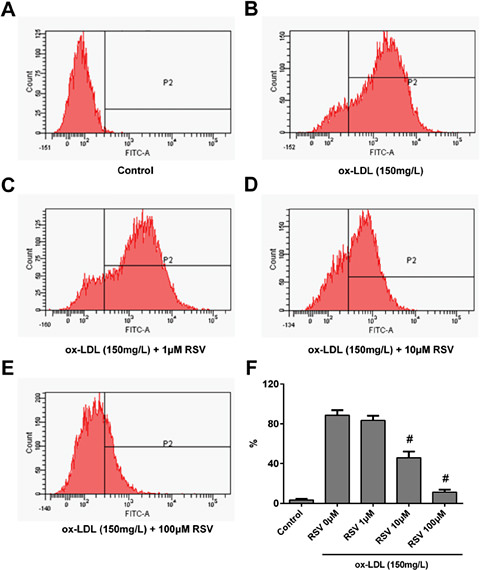
The inhibitory effect of RSV on ox-LDL-induced reactive oxygen species (ROS) over-production. Cells were pre-treated with RSV for 24 h prior to ox-LDL exposure for 2 h. ROS were detected using a 2',7'-dichlorofluorescin diacetate (DCFH-DA) probe. (A-E) representative flow cytometric images of cells in different groups: (A) Control; (B) Ox-LDL 150 mg·L-1; (C-E) cells treated with different concentrations of RSV (1, 10, 100 mM) prior to 150·mg· L-1 ox-LDL exposure. (F) Quantitative analysis of the ratio of DCFH-DA probe positive cells. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein * P < 0.05 indicates significant differences from the ox-LDL 150 mg·L-1 group. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
The inhibitory effect of RSV on ox-LDL-induced reactive oxygen species (ROS) over-production. Cells were pre-treated with RSV for 24 h prior to ox-LDL exposure for 2 h. ROS were detected using a 2',7'-dichlorofluorescin diacetate (DCFH-DA) probe. (A-E) representative flow cytometric images of cells in different groups: (A) Control; (B) Ox-LDL 150 mg·L-1; (C-E) cells treated with different concentrations of RSV (1, 10, 100 mM) prior to 150·mg· L-1 ox-LDL exposure. (F) Quantitative analysis of the ratio of DCFH-DA probe positive cells. Values are expressed as means ± SD from triplicate experiments. R: resveratrol (RSV); ox-LDL: oxidised low-density lipoprotein * P < 0.05 indicates significant differences from the ox-LDL 150 mg·L-1 group. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
RSV down-regulates LOX-1 protein up-regulation and p38 MAPK phosphorylation by ox-LDL
In this study, we investigated the effect of RSV on ox-LDL-induced LOX-1 up-regulation by undertaking western blot analysis of RAW264.7 cell proteins. We found that RSV (100 µM) did not alter LOX-1 expression in cells treated without ox-LDL, and ox-LDL (150 mg·L-1) markedly up-regulated the expression of LOX-1 compared with an untreated control (Fig. 7A and 7B). However, pre-treatment of the cells with RSV (100 µM) resulted in suppression of this ox-LDL-mediated increase in LOX-1 expression at 6, 24 and 48 h (Fig. 7B, P < 0.05).

The effect of RSV on LOX-1 expression and p38 MAPK phosphorylation in ox-LDL-exposed cells. Western blot data showed the effect of 100 µM RSV on (A and B) LOX-1 expression and (C and D) phosphorylation of p38 MAPK in 150 mg·L-1 ox-LDL-exposed RAW264.7 cells. (A and C) RSV exerts no significant effect on LOX-1 and p38 MAPK expression at 6, 12, 24 and 48 h. Total cell protein extracts were blotted with primary antibodies against LOX-1 and phosphorylated p38 MAPK. The β-actin protein was used as an internal control. Values are expressed as means ± SD from three independent experiments. * P < 0.05 indicates significant differences from the ox-LDL 150 mg·L-1 group. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
The effect of RSV on LOX-1 expression and p38 MAPK phosphorylation in ox-LDL-exposed cells. Western blot data showed the effect of 100 µM RSV on (A and B) LOX-1 expression and (C and D) phosphorylation of p38 MAPK in 150 mg·L-1 ox-LDL-exposed RAW264.7 cells. (A and C) RSV exerts no significant effect on LOX-1 and p38 MAPK expression at 6, 12, 24 and 48 h. Total cell protein extracts were blotted with primary antibodies against LOX-1 and phosphorylated p38 MAPK. The β-actin protein was used as an internal control. Values are expressed as means ± SD from three independent experiments. * P < 0.05 indicates significant differences from the ox-LDL 150 mg·L-1 group. # P < 0.01 indicates significant differences from the ox-LDL 150 mg·L-1 group.
Discussion
The main aim of this study was to investigate whether RSV, a polyphenol compound found in red wine and grapes, could protect RAW264.7 macrophages against ox-LDL-induced apoptosis. In addition, the potential mechanisms of this anti-apoptotic effect were explored. These data complement findings from our previous study [15], which suggested that the SIRT1 activator RSV may improve efferocytosis of apoptotic RAW264.7 cells by up-regulation of SIRT1-mediated autophagy.
It has been shown that LDLs can be modified by oxidation and/or enzymatic modification, for example by phospholipases [16]. LDL is normally present in tissues such as the intima of arteries, where it can bind to the proteoglycan matrix, especially after modification. This binding is thought to be an early event in atherogenesis according to the “response to retention” hypothesis [17]. Indeed, human and animal studies have revealed that elevated levels of plasma lipoproteins are associated with a high risk of atherosclerosis, and lipoproteins with a high affinity for arterial proteoglycans are strongly atherogenic [18].
The internalisation of native LDLs by macrophages and the subsequent down-regulation of the LDL receptor account for only a small portion of foam cell formation [19]. However, oxidation of LDL increases its uptake by macrophages via scavenger receptors such as scavenger receptor A, CD36 and LOX-1 [20]. Interstitial free radicals and excess ox-LDL particles can injure and kill foam cells, resulting in the formation of the necrotic extracellular lipid core, which is a key transitional step in lesion progression [21]. Therefore, further understanding macrophage apoptosis may enable the development of more efficient therapies for the prevention and treatment of atherothrombosis [22].
RSV has been the subject of intense scientific and public interest in recent years, mainly due to its widely reported ability to delay ageing and prevent age-related diseases [23,24,25]. The beneficial effects of RSV were originally thought to derive from its antioxidant properties [26]. Moreover, RSV has been shown to possess anti-platelet, anti-cancer and lifespan-extending effects in various experimental models [27,28,29,30]. It has been reported that SIRT1 regulates both inflammatory processes and cholesterol metabolism in macrophages by reducing the uptake of ox-LDL and preventing the formation of macrophage foam cells [31]. In the present study, we found the SIRT1 activator RSV could also significantly inhibit foam cell formation and ox-LDL induced macrophage apoptosis (Fig. 2 and 3). In published animal studies, RSV was shown to ameliorate metabolic disorders in mice on a high-fat diet [32,33] and improved hyperglycaemia in diabetic animal models [34].
Oxidative stress is a common mediator in the pathogenicity of established cardiovascular risk factors and evokes many intracellular events including apoptosis. In recent years, a growing body of evidence has shown that oxidative stress can cause cellular apoptosis via both the mitochondria-dependent and mitochondria-independent pathways [35]. The mitochondria-independent apoptosis pathway involves death receptors and the Fas-mediated activation of caspase-8, whereas caspase-9 activation is central to the mitochondria-dependent pathway [36]. Caspase-3 activation is pivotal in both pathways and leads to DNA fragmentation and morphological changes. Oxidative stress also induces the release of cytochrome c from mitochondria and the activation of caspases, p53, and a range of kinases [37,38,39,40]. The regulation of mitochondrial function is a key focus of apoptosis and redox research, as this organelle is critical for the induction and inhibition of apoptosis.
Although the scavenger receptor LOX-1 has low-level basal expression, higher expression can be induced by pro-inflammatory and pro-oxidative stimuli in a variety of cell types involved in atherogenesis. This includes vascular endothelial cells, smooth muscle cells, macrophages, platelets and cardiomyocytes [41]. These cell types are targets of pathophysiologic effects of ox-LDL in atherogenesis, such as endothelial cell dysfunction, smooth muscle cell growth and migration, monocyte transformation into macrophages, and platelet aggregation [42]. In line with these findings, our results demonstrate that ox-LDL can up-regulate LOX-1 expression in RAW264.7 cells, and that this effect increases with time.
It has previously been shown that LOX-1 up-regulation by ox-LDL can induce MAPK activation [43]. Therefore, we postulated that RSV would down-regulate cellular LOX-1 expression and abolish this effect. Our data revealed that RSV can indeed exert an inhibitory effect on the ox-LDL-mediated up-regulation of LOX-1 expression. This suggests that the blockage of ox-LDL-stimulated MAPK activation by RSV is mediated by the regulation of LOX-1 expression, and that other molecular mechanisms are potentially involved in this process.
The p38 MAPK signalling pathway plays an important role in the ability of cells to integrate external cues and direct the appropriate responses [44]. Four p38 MAPK family members are known, of which p38α is ubiquitously expressed—usually at high levels— and p38β is thought to be expressed at lower levels. Despite its lower expression, p38β can potentially perform overlapping roles with p38α, whereas p38γ and p38δ have more restricted expression patterns [45]. Recently, many studies have reported that the inhibition of p38 MAPK phosphorylation could either lead to or explain the beneficial effects of particular compound and proteins on cell apoptosis [46,47,48]. In the present investigation, we focussed on the relationship between RSV and p38 MAPK in the ox-LDL-mediated apoptosis of RAW264.7 macrophages.
Deby-Dupont et al. [49] reported that an increased respiratory burst linked to increased NADPH oxidase activity could be inhibited by RSV in THP1 monocytes. Furthermore, Voloshyna et al. [50] found that RSV regulates expression of proteins involved in cholesterol transport, promotes apoA-1 and HDL-mediated efflux, down-regulates ox-LDL uptake and diminishes foam cell formation. These data support our finding that RSV can exert an inhibitory effect on macrophage apoptosis.
Although we observed the anti-apoptotic function of RSV in the present study, the effects of RSV are believed to extend further. For example, RSV has been shown to inhibit proliferation and induce apoptosis of human cancer cell lines [51,52]. Indeed, little is known about the contributions of RSV or SIRT1 during the development and proliferation of macrophages. In particular, the mechanisms behind the RSV- or SIRT1-mediated inhibition of the ox-LDL-induced intrinsic pathway of macrophage apoptosis require further clarification.
Conclusion
In summary (Fig. 8), our results provide new insights into the roles of ROS, LOX-1 and p38 MAPK in the effect of RSV on cell survival after ox-LDL injury. Ox-LDL-induced RAW264.7 cell apoptosis was abolished by RSV through inhibition of intracellular ROS generation, LOX-1 down-regulation, and the blocking of the p38 MAPK pathway. Herein, our findings suggest a beneficial role for RSV in cardiovascular health.

Overview of the signal cascade involved in ox-LDL-induced macrophage apoptosis, and the effect of RSV on this system. The binding of ox-LDL to LOX-1 initiates a downstream signal cascade and the up-regulation of LOX-1 expression. It subsequently promotes ROS overproduction and p38 MAPK phosphorylation. Bursts of ROS could activate caspase-3 and the mitochondria-mediated apoptosis pathway. This pathway includes the elevation of the expression of Bax and the down-regulation of Bcl-2. The activation of p38 MAPK, caspase-3 and a decrease in Bcl-2 expression play a critical role in macrophage apoptosis. The symbol cal role in poptosis pathway Burstexp.
Overview of the signal cascade involved in ox-LDL-induced macrophage apoptosis, and the effect of RSV on this system. The binding of ox-LDL to LOX-1 initiates a downstream signal cascade and the up-regulation of LOX-1 expression. It subsequently promotes ROS overproduction and p38 MAPK phosphorylation. Bursts of ROS could activate caspase-3 and the mitochondria-mediated apoptosis pathway. This pathway includes the elevation of the expression of Bax and the down-regulation of Bcl-2. The activation of p38 MAPK, caspase-3 and a decrease in Bcl-2 expression play a critical role in macrophage apoptosis. The symbol cal role in poptosis pathway Burstexp.
Disclosure Statement
The authors declare that there is no conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (no. 81070107), and partly supported by Foundation for Distinguished Graduate Student of Shanghai Tenth People's Hospital. The authors are grateful to Dr Wenhui Peng for critical guidance during the study.

