


Abstract
Epithelial ovarian cancer (EOC) is the fifth most common cancer in women and is characterized by a low 5-year survival rate. One strategy that can potentially improve the overall survival rate in ovarian cancer is the use of antitumor agents such as ABT-510. ABT-510 is a small mimetic peptide of the naturally occurring antiangiogenic compound thrombospondin-1 and has been shown to significantly reduce tumor growth and burden in preclinical mouse models and in naturally occurring tumors in dogs. This is the first evaluation of ABT-510 in a preclinical model of human EOC. Tumorigenic mouse surface epithelial cells were injected into the bursa of C57BL/6 mice that were treated with either 100 mg/kg ABT-510 or an equivalent amount of PBS. ABT-510 caused a significant reduction in tumor size, ascites fluid volume, and secondary lesion dissemination when compared with PBS controls. Analysis of the vasculature of ABT-510-treated mice revealed vascular remodeling with smaller diameter vessels and lower overall area, increased number of mature vessels, and decreased tissue hypoxia. Tumors of ABT-510-treated mice had a significantly higher proportion of apoptotic tumor cells compared with the PBS-treated controls. Immunoblot analysis of cell lysates revealed a reduction in vascular endothelial growth factor, vascular endothelial growth factor receptor-2, and proliferating cell nuclear antigen protein expression as well as expression of members of the phosphatidylinositol 3-kinase and mitogen-activated protein kinase survival pathways. In vitro, ABT-510 induced tumor cell apoptosis in mouse and human ovarian cancer cells. This study shows ABT-510 as a promising candidate for inhibiting tumor growth and ascites formation in human EOC. [Mol Cancer Ther 2009;8(1):64–74]
Introduction
Epithelial ovarian cancer (EOC) is the most common gynecologic malignancy and is the fifth leading cause of death by cancer in women (1). Despite an increased understanding of the pathogenesis of EOC, mortality rates have not changed significantly in the past three decades. EOC involves the transformation of the normal ovarian surface epithelial cells to a tumorigenic phenotype, and epithelial cells derived from ovarian tumors have a significant alteration in gene expression compared with normal ovarian surface epithelium (2). EOC is associated with the growth of large primary tumors, the formation of secondary lesions throughout the peritoneum, and the accumulation of malignant ascites. There is a paucity of early clinical signs associated with ovarian cancer, and abdominal swelling due to ascites fluid accumulation is often the first sign of EOC (3). Due to the lack of adequate screening agents, early symptoms, and predictive indicators, EOC is often not detected until the later stages of disease at which time there is often resistance to chemotherapy and high frequency of reoccurrence, so that women diagnosed with EOC have 5-year survival rates of <20% (4, 5).
During ovarian tumor formation, tumor cells undergo an angiogenic switch where they increase the expression of proangiogenic factors to facilitate the formation of new vasculature to support tumor growth (6). The degree of angiogenesis has been linked to disease progression and the length of survival in EOC (7). One of the most potent proangiogenic growth factors, vascular endothelial growth factor (VEGF), is significantly increased in advanced EOC compared with early-stage disease or benign cystadenomas (8, 9). One strategy to inhibit tumor formation is to prevent this neovascularization through antiangiogenic approaches. However, a clinical trial in ovarian cancer using a direct VEGF antagonist, Avastin, gave a high rate of intestinal perforation (10). Several endogenously occurring angiogenesis inhibitors have been identified. Thrombospondin-1 (TSP-1) has been shown to be an important negative regulator of tumor angiogenesis (11). TSP-1 is a large extracellular matrix glycoprotein with potent antiangiogenic functions (12). Reduced expression of TSP-1 is an important component of the angiogenic switch and its decrease facilitates growth of several tumor types including bladder, breast, and ovarian cancer, fibrosarcoma, and glioblastoma (13–15). TSP-1 inhibits angiogenesis by stimulating endothelial cell apoptosis and inhibiting endothelial cell migration (16) and by binding and sequestering proangiogenic growth factors such as VEGF (17). TSP-1 binds CD36, a receptor expressed on the surface of endothelial and steroidogenic cells in the ovary (18, 19) as well as numerous other cell surface receptors such as integrin-associated protein/CD47, the low-density lipoprotein receptor-related protein-1, integrins, and various heparan sulfate proteoglycans (20). Through binding to this diverse array of receptors, TSP-1 has a multitude of functions in addition to inhibiting angiogenesis. Although the native 450 kDa TSP-1 has been shown to have potent antiangiogenic and antitumorigenic effects, its large size and complex functions limit the possibility of using it as a therapeutic antitumor strategy in humans. To address these issues, smaller TSP-1 fragments have been generated and one, ABT-510, has been derived from the second properdin type I repeat from the NH2-terminal third of TSP-1 (21). ABT-510 contains a l-isoleucine-3 to d-allo-l-isoleucine substitution, and replacement of the internal arginine by norvaline in the GVITRIR sequence of the second properdin repeat of TSP-1. Nanomolar concentrations of ABT-510 are active against endothelial cells in vitro, having a 1,000-fold increase in antiangiogenic activity (21) compared with the natural 7-amino acid peptide. ABT-510, like TSP-1, causes endothelial cell apoptosis via activation of Fas/Fas ligand (FasL) and the Src-related kinase p59 Fyn pathway ultimately leading to cell death (16, 22). ABT-510 has been shown to inhibit tumor growth in xenograft and syngeneic animal models of human cancers and in companion dogs with naturally occurring cancers (23), and the peptide is currently in veterinary trials in canine cancer. In one human clinical phase I trial of ABT-510, 6 of 39 patients enrolled in the study had stable disease lasting >6 months (24). It has also been in phase II clinical trials with adults with advanced cancer (25, 26).
We show that administration of ABT-510 to an orthotopic, syngeneic model for EOC is effective at inhibiting primary tumor growth, secondary lesion formation, and ascites accumulation. ABT-510 decreased morbidity associated with ovarian tumor growth and increased survival time.
Materials and Methods
Animals
All animals were housed and treated in accordance with the Canadian Council on Animal Care. Tumor induction was done as described previously (27). Briefly, 106 spontaneously transformed mouse epithelial cells (ID8; generously provided by Drs. Kathy Roby and Paul Terranova, Kansas State University) were suspended in 5 μL PBS and orthotopically injected under the ovarian bursa of anesthetized syngeneic C57BL/6 mice using a Hamilton syringe (Fisher) and a 30-gauge needle. The contralateral ovary received an injection of 5 μL PBS under the ovarian bursa. Mice were then divided into two groups for treatment starting 3 weeks after the above surgical inoculation: one group received daily i.p. injections of 100 mg/kg ABT-510 suspended in 200 μL PBS (ABT-510 group), whereas the other group received daily i.p. injections of 200 μL PBS alone (PBS group). Mice were treated for 30, 60, or 90 days post-tumor induction at which time they were euthanized by CO2 asphyxiation and had their abdomens opened with a midline incision. Tumor induction was also done in TSP-1-null mice. TSP-1−/− mice (on C57BL/6 background) were injected with ID8 cells and divided into three groups: TSP-1−/− treated with ABT-510, TSP-1−/− treated with PBS, and wild-type littermate controls. The same protocol as above was followed. At the time of euthanasia for all mice, both ID8- and PBS-injected ovaries were removed and weighed. Some ovaries were fixed overnight in 10% neutral buffered formalin for immunohistochemistry experiments. For immunoblot experiments, the isolated ovaries were homogenized in lysis buffer (10 mmol/L Tris-HCl, 5 mmol/L EDTA, 50 mmol/L NaCl, 30 mmol/L Na4P2O7, 1% Triton X-100, 50 mmol/L NaF, 200 μmol/L Na3VO4, 1 mmol/L phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin, 1 μg/mL pepstatin A, and 2 μg/mL leupeptin) and total protein content was measured using a modified Bio-Rad assay (DC Bio-Rad Assay; Bio-Rad). Other ovaries were embedded in cryomedia and frozen in liquid nitrogen for evaluation of blood vessel density using cryosections.
Collection of Ascites Fluid and Scoring of Secondary Peritoneal Lesions
At euthanasia, mice were positioned in dorsal recumbency and a 25-gauge needle attached to a 10 mL syringe was introduced into the peritoneal space, and the ascites fluid was aspirated. After initial aspiration, a vertical incision was made in the abdomen and any residual ascites fluid not recovered at the initial aspiration was collected. Ascites fluid was transferred to either 10 or 20 mL graduated cylinder for measurement of volume. Tumor dissemination was scored by the number of visible metastasis present within the abdominal cavity. Abdomens with no visible secondary tumors were scored a 0. Presence of one or two secondary lesions scored a 1. Three to 10 secondary lesions were scored 2. Presence of >10 lesions throughout the abdomen received a score of 3.
Immunohistochemistry
Ovaries were removed from PBS-treated and ABT-510-treated mice at 30, 60, or 90 days post-tumor induction and fixed in 10% neutral buffered formalin overnight at 4°C. Tissues were embedded in paraffin wax and then cut into 5 μm sections using a rotary microtome. Following deparaffinization and rehydration, endogenous peroxide activity was inhibited with a 10 min incubation in 3% (v/v) hydrogen peroxide and then blocked in 5% bovine serum albumin (containing 0.02% sodium azide) for 10 min. Tissues were then exposed to the following primary antibodies: anti-VEGF (1:600 dilution; Santa Cruz Biotechnology), anti-Ki-67 (1:1,000 dilution; Abcam), anti-VEGF receptor (VEGFR)-2 (1:500 dilution; Santa Cruz Biotechnology), and anti-smooth muscle actin (1:600 dilution; RDI) overnight at 4°C. After rinsing in PBS, biotinylated secondary antibodies from the appropriate species were applied for 2 h at room temperature (all at 1:100 dilution; Sigma) followed by 1 h incubation at room temperature with horseradish peroxidase (Extravidin, 1:50 dilution; Sigma). Staining localization was visualized by brief incubation with diaminobenzidine tetrahydrochloride (Sigma) and tissues were counterstained for 1 min in Carazzi's hematoxylin followed by dehydration and mounting with Permount (Sigma).
Evaluation of Blood Vessel Density
For blood vessel measurements, ovaries were collected at euthanasia 30, 60, and 90 days post-tumor induction and flash frozen in cryomedia. Cryosections (5 μm) were cut on cryostat, placed on glass slides, and fixed with acetone on ice. Cryosections were thawed and hydrated and incubated overnight at 4°C with a fluorescently labeled anti-isolectin antibody (1:500 dilution; Invitrogen), which stains endothelial cells. Tissues were counterstained with 4′,6-diamidino-2-phenylindole to visualize nuclei and coverslips were added using Antifade (Prolong Gold; Invitrogen). To evaluate vascularity, both blood vessel area and blood vessel perimeter were measured using integrated morphometry software (Image Pro Plus). For these measurements, 6 PBS-treated and ABT-510-treated sections were used for quantification and a minimum of 5 fields of view per section were randomly selected for each tissue.
Tissue Hypoxia Detection by Hypoxyprobe
Hypoxyprobe-1 (Millipore) solution was injected 60 mg/kg i.p. 30 min before euthanasia in ABT-510-treated and PBS-treated mice. Ovaries were removed and processed for histology as described above. Sections were incubated with anti-hypoxyprobe-1MaAb1 antibody overnight at 4°C. After rinsing in PBS, biotinylated mouse secondary antibodies were applied for 2 h at room temperature (1:100 dilution; Sigma) followed by 1 h incubation at room temperature with horseradish peroxidase (Extravidin, 1:50 dilution; Sigma). The color reaction was visualized by diaminobenzidine tetrahydrochloride as described above. At least 3 fields of view per section were used for analysis from 4 animals per group. Similar areas of tissue were measured between groups.
Apoptosis Detection
Histologic sections of ABT-510 and control ovaries were processed and fixed as above. Detection of apoptotic cells was done with a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) kit (Roche Applied Sciences) according to the manufacturer's protocol. Following deparaffinization and rehydration, tissue sections were washed in PBS and incubated with the FITC-conjugated TUNEL enzyme for 60 min to detect DNA fragmentation. Incubation for 60 min with an anti-fluorescein antibody conjugated to a peroxidase to detect incorporated fluorescein. Apoptotic cells were visualized by brief incubation with diaminobenzidine tetrahydrochloride and sections were counterstained by hematoxylin and mounted with Permount. In vitro, apoptosis was evaluated in ID8 cells following RNA interference (RNAi) knockdown of CD36 (described below) and in ID8, SKOV3, OVCAR3, and CAOV3 cells in the presence or absence of 50 mmol/L ABT-510 for 24 h. SKOV3, OVCAR3, and CAOV3 cells were purchased from the American Type Culture Collection. Following treatment, cells were fixed in 10% neutral buffered formalin for 1 h at room temperature and then permeabilized in 0.1% Triton X-100 (Sigma) in PBS. The TUNEL protocol was then done as described above. Following TUNEL detection, cells were counterstained with 4′,6-diamidino-2-phenylindole and TUNEL-positive and TUNEL-negative cells were counted and expressed as percent apoptotic.
Immunoblotting
VEGF, VEGFR-2, and proliferating cell nuclear antigen (PCNA) protein expression was detected and analyzed in ovarian tumors collected from PBS-treated and ABT-510-treated animals by immunoblot analysis. At 90 days post-tumor induction, tumor tissue was removed and immediately immersed in liquid nitrogen and stored at −80°C. Tissues were homogenized in cold lysis buffer and total protein lysates (10 μg) were separated by SDS-PAGE on 12% gels. Proteins were blotted onto polyvinylidene difluoride (Millipore) membranes then blocked 1 h at room temperature in 5% (w/v) skim milk. Primary antisera [VEGF, 1:500 (Santa Cruz Biotechnology); VEGFR-2, 1:500 (Cell Signaling Technologies); PCNA, 1:600 (Sigma); α-tubulin, 1:1,000 (Santa Cruz Biotechnology); phospho-Akt, 1:1,000 (Cell Signaling Technologies); phospho-p38 mitogen-activated protein kinase (MAPK), 1:1,000 (Cell Signaling Technologies); phospho-p85 phosphatidylinositol 3-kinase, 1:600 (Cell Signaling Technologies); and FasL, 1:600 (Pharmingen)] were applied overnight at 4°C on a rocking platform. Blots were washed in TBS-1% Tween 20 and incubated with the appropriate peroxidase-conjugated secondary antibody for 1 h at room temperature. Reactive protein was detected with enhanced chemiluminescence (Boehringer Mannheim) and Konica medical X-ray film.
Small Interfering RNA and Transfection Reagents
Stealth small interfering RNA (siRNA) targeting either murine (ID8 cells) or human (SKOV3, OVCAR3, and CAOV3 cells) CD36 (Stealth select RNAi) and negative control siRNAs were purchased from Invitrogen. Negative controls included in this study were vehicle control and Stealth RNAi-negative control with medium GC content (Invitrogen). Cells were transfected with siRNAs using Lipofectamine 2000 (Invitrogen) in Opti-MEM.
Transfection of siRNAs
Mouse and human EOC cell lines were plated in growth medium at a density of 2 × 105 cells/5 mL in 60 mm dishes or 1 × 105 cells/2 mL DMEM/F-12 on glass coverslips in 6-well plates. At ∼50% confluence, cells were transfected with 200 nmol/L siRNA in Lipofectamine 2000 reagent according to the manufacturer's instructions. Briefly, 20 μL Lipofectamine 2000 was diluted in 500 μL Opti-MEM and incubated for 5 min at room temperature. In a separate tube, 10 μL of 50 μmol/L siRNA was diluted in 450 μL Opti-MEM. Diluted Oligofectamine (250 μL) was added to the diluted siRNA and the complex was incubated for 20 min at room temperature. Cells were washed in Opti-MEM and 1.5 mL Opti-MEM was added to each 60 mm dish and 500 μL Opti-MEM was added to each well of the 6-well plates. siRNA + Opti-MEM complexes (500 μL) were added to each dish/well. The final concentration of the siRNA was 200 nmol/L. After 6 h, 2 mL DMEM/F-12 with 10% fetal bovine serum was added without removing the transfection medium. Transfections were allowed to continue for 48 h. Transfection controls included medium controls (cells in Opti-MEM), vehicle controls (20 μL Lipofectamine 2000 in 500 μL Opti-MEM), and negative control siRNA (Stealth RNAi-negative control scrambled sequence with medium GC content) as described. Following RNAi, knockdown was confirmed with immunofluorescence and Western blot analysis using an anti-CD36 antibody (Pharmingen). Control and CD36-knockdown ID8, SKOV3, OVCAR3, and CAOV3 cells were treated with ABT-510 (1-50 nmol/L) and apoptosis experiments were done as described above. Western blot analysis (described above) was also done with ID8 cell lysates for FasL using an anti-FasL antibody (Cell Signaling Technologies).
Statistical Analysis
Three replicates of all data collected were done and used to determine statistical significance. For immunoblot analysis, VEGF, VEGFR-2, and PCNA densitometric values compared ABT-510-treated versus PBS-treated ovarian primary lesions using one-way ANOVA followed by Bonferroni's test for differences between means. For immunohistochemical and immunofluorescence experiments, a minimum of 5 views at ×250 magnification were used for cell counts. P values are listed in the figure legends.
Results
ABT-510 Reduces Ovarian Tumor Growth in Wild-type and TSP-1-Null Mice
To determine the effect of ABT-510 on ovarian tumor progression, 1 × 106 ID8 cells were injected under the ovarian bursa of both ovaries as described above. Mice received daily i.p. injections of 100 mg/kg ABT-510 in 200 μL PBS or 200 μL PBS alone and were allowed to progress for 30, 60, or 90 days post-tumor induction at which time they were euthanized via CO2 asphyxiation and the ovarian tumors were removed and weighed. By 60 days post-tumor induction, there was a significant (P < 0.05) reduction in ovarian tumor weight in mice receiving ABT-510. By 90 days post-tumor induction, there was a more dramatic reduction (P < 0.01) in ovarian tumor weight in ABT-510-treated animals compared with PBS-treated controls (Fig. 1A).
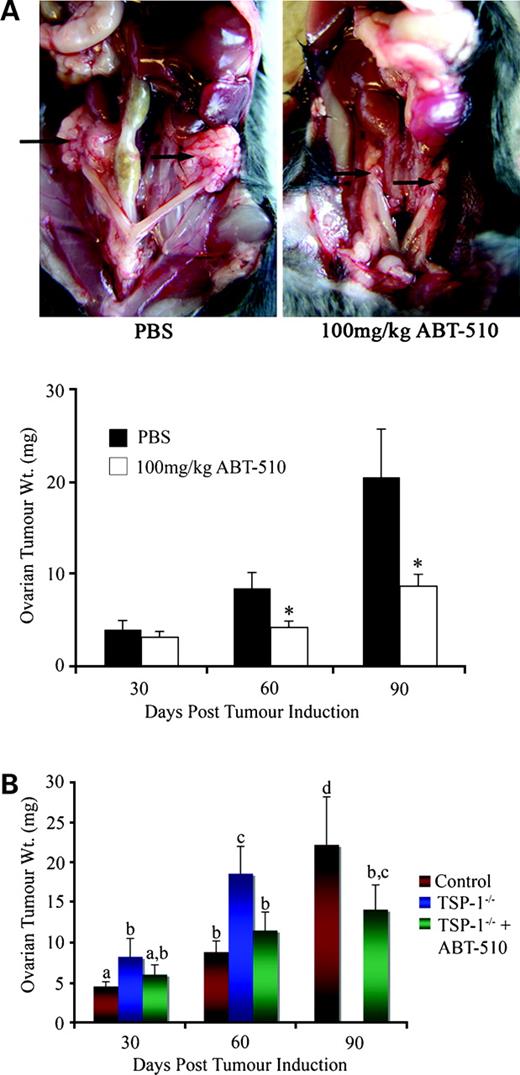
A, ABT-510 inhibits ovarian tumor formation. Ovarian tumor formation was induced by injecting 1 × 106 spontaneously transformed murine ovarian surface epithelial cells under the ovarian bursa of both ovaries of syngeneic C57BL/6 mice. In PBS-treated animals, large bilateral primary tumors were evident at 90 d post-tumor induction, whereas 100 mg/kg ABT-510 significantly decreased the size of ovarian masses. Ovarian tissue was collected at 30, 60, and 90 d post-tumor induction and ABT-510 significantly reduced ovarian tumor weight at 60 and 90 d compared with PBS-treated controls. (n = 5 mice per group). *, P < 0.05, statistical difference between PBS-treated and ABT-510-treated groups. B, ovarian tumors grow larger in mice lacking TSP-1. There was a significant increase in ovarian tumor weight in TSP-1−/− mice at 30 and 60 d post-tumor induction (blue columns) compared with wild-type controls (red columns). Before 90 d post-tumor induction, TSP-1−/− animals had to be euthanized due to morbidity. The addition of ABT-510 (green columns) significantly reduced ovarian tumor size in TSP-1−/− mice, increasing their survival time to beyond 90 d post-tumor induction (n = 5 animals per group). Columns with different letters are statistically different (P < 0.05).
A, ABT-510 inhibits ovarian tumor formation. Ovarian tumor formation was induced by injecting 1 × 106 spontaneously transformed murine ovarian surface epithelial cells under the ovarian bursa of both ovaries of syngeneic C57BL/6 mice. In PBS-treated animals, large bilateral primary tumors were evident at 90 d post-tumor induction, whereas 100 mg/kg ABT-510 significantly decreased the size of ovarian masses. Ovarian tissue was collected at 30, 60, and 90 d post-tumor induction and ABT-510 significantly reduced ovarian tumor weight at 60 and 90 d compared with PBS-treated controls. (n = 5 mice per group). *, P < 0.05, statistical difference between PBS-treated and ABT-510-treated groups. B, ovarian tumors grow larger in mice lacking TSP-1. There was a significant increase in ovarian tumor weight in TSP-1−/− mice at 30 and 60 d post-tumor induction (blue columns) compared with wild-type controls (red columns). Before 90 d post-tumor induction, TSP-1−/− animals had to be euthanized due to morbidity. The addition of ABT-510 (green columns) significantly reduced ovarian tumor size in TSP-1−/− mice, increasing their survival time to beyond 90 d post-tumor induction (n = 5 animals per group). Columns with different letters are statistically different (P < 0.05).
Epithelial ovarian tumor formation was also induced in TSP-1-null mice. TSP-1−/− mice were split into PBS-treated and ABT-510-treated groups as described above. In TSP-1−/− mice treated with PBS, ovarian weight was significantly (P < 0.05) increased compared with PBS-treated wild-type control animals (Fig. 1B). By 60 days post-tumor induction, ovarian tissue weight was dramatically increased over controls (P < 0.05). We were unable to collect tissue at 90 days post-tumor induction, as the animals developed excessively large tumors and ascites accumulation and had to be sacrificed before this time point. Treatment of tumor-bearing TSP-1−/− mice with ABT-510 significantly decreased ovarian tumor weight at 30 and 60 days post-tumor induction (P < 0.05) and morbidity associated with ovarian tumor development was decreased sufficiently such that we were able to obtain data at 90 days post-tumor induction (Fig. 1B). At 90 days post-tumor induction, TSP-1−/− mice treated with ABT-510 had a significant reduction in ovarian tumor weight (P < 0.05) compared with PBS-treated controls with tumor similar to that observed at 60 days post-tumor induction (Fig. 1B). Mice treated with ABT-510 were all able to reach day 90 without premature sacrifice.
ABT-510 Inhibits I.p. Lesion Formation and Ascites Accumulation
At 90 days post-tumor induction, in wild-type mice, ascites fluid was aspirated with a syringe and 25-gauge needle and the volume was measured. PBS-treated mice exhibited large distended abdomens containing an average of 6 ± 2.7 mL ascites fluid (Fig. 2). ABT-510 treatment significantly (P < 0.01) reduced the volume of ascites with an average fluid volume of 0.8 ± 1.3 mL (Fig. 2). PBS-treated and ABT-510-treated mice were also evaluated for the extent of secondary lesion infiltration into the peritoneal cavity. ABT-510 treatment completely abolished the formation of peritoneal lesions and no masses were observed in any of the peritoneal cavities of ABT-510-treated mice (Fig. 2). This was a significant (P < 0.01) reduction compared with PBS-treated mice, which had an average secondary lesion score of 9.4 ± 4.4 (Fig. 2).

ABT-510 reduces ascites production and the formation of secondary lesions in the peritoneal cavity. At 90 d post-tumor induction, ascites fluid was collected and measured and the extent of secondary lesion formation was scored. There was significant reduction in the formation of ascites (black columns) and a complete absence of secondary lesions (gray columns) in mice treated with ABT-510 (n = 5 mice per group). *, P < 0.01, statistical difference between PBS-treated and ABT-510-treated groups.
ABT-510 reduces ascites production and the formation of secondary lesions in the peritoneal cavity. At 90 d post-tumor induction, ascites fluid was collected and measured and the extent of secondary lesion formation was scored. There was significant reduction in the formation of ascites (black columns) and a complete absence of secondary lesions (gray columns) in mice treated with ABT-510 (n = 5 mice per group). *, P < 0.01, statistical difference between PBS-treated and ABT-510-treated groups.
ABT-510 Reverses Ovarian Tumor Hypervascularization and Increases the Proportion of Mature Blood Vessels
We have shown previously that epithelial ovarian tumors in our mouse model of EOC are significantly hypervascularized (27). Mice receiving daily i.p. injections of PBS showed large, irregularly shaped blood vessels by 90 days post-tumor induction (Fig. 3A). ABT-510 treatment significantly (P < 0.01) reduced ovarian blood vessel area and perimeter (Fig. 3A), returning the tissue to a more normal appearance of vascularity. Mature blood vessels are characterized by the presence of luminal epithelial cells with a pericyte covering. When tissues were stained for smooth muscle actin, which localizes vessel pericytes, ABT-510 had a 6-fold increase (P < 0.01) in the percentage of mature blood vessels within the ovarian tumor tissue (Fig. 3B). Tissue sections from ABT-510-treated and control ovaries were analyzed for areas of hypoxia using hypoxyprobe-1. At low oxygen tension (<10 mm Hg), hypoxyprobe-1 forms chemical adducts with macromolecules within the cell (28). These chemical adducts were localized to ovarian carcinoma tissue sections by using anti-hypoxyprobe-1MaAb1 antibody. Comparison of ABT-510-treated and control ovaries illustrated a difference in tissue perfusion between the two groups (Fig. 3C). Within the PBS-treated group, many sections contained structures resembling large diameter vessels surrounded by areas of hypoxia (Fig. 3C). We infer from this relationship that the large, tortuous vessels are largely nonfunctional, which results in large areas of hypoxia. In contrast, ABT-510-treated ovarian sections were characterized by numerous, small diameter vessels that exhibited little positive immunostaining, which we hypothesize resulted from increased tissue perfusion. Sections from PBS-treated mice had a widespread, generalized increase in staining when compared with ABT-510-treated mice.

ABT-510 reverses the hypervascularization observed in epithelial ovarian tumor, increases the proportion of mature blood vessels, and reduces tissue hypoxia. A, at 90 d post-tumor induction, ovarian tissue was immunostained for isolectin (green) and counterstained with 4′,6-diamidino-2-phenylindole (blue). ABT-510 treatment significantly reduced blood vessel area and perimeter and returned the blood vessels to a more normal vascular phenotype. B, ovarian tumors collected at 90 d post-tumor induction were stained for CD31 (red), which stains endothelial cells, and smooth muscle actin (SMA; green), which stains pericytes. Colocalization (yellow) shows the presence of endothelial cells with pericyte coverage indicative of mature blood vessels. ABT-510 treatment resulted in a significant increase in the proportion of tumor cells with pericyte coverage. C, ABT-510 reduced tissue hypoxia at 90 d post-tumor induction. Hypoxyprobe labeling (brown) indicates area of low oxygen tension (<10 mm Hg), which is prevalent in PBS control ovaries and reduced in ABT-510-treated mice (n = 5 animals per group). *, P < 0.01, statistical difference between PBS-treated and ABT-510-treated groups.
ABT-510 reverses the hypervascularization observed in epithelial ovarian tumor, increases the proportion of mature blood vessels, and reduces tissue hypoxia. A, at 90 d post-tumor induction, ovarian tissue was immunostained for isolectin (green) and counterstained with 4′,6-diamidino-2-phenylindole (blue). ABT-510 treatment significantly reduced blood vessel area and perimeter and returned the blood vessels to a more normal vascular phenotype. B, ovarian tumors collected at 90 d post-tumor induction were stained for CD31 (red), which stains endothelial cells, and smooth muscle actin (SMA; green), which stains pericytes. Colocalization (yellow) shows the presence of endothelial cells with pericyte coverage indicative of mature blood vessels. ABT-510 treatment resulted in a significant increase in the proportion of tumor cells with pericyte coverage. C, ABT-510 reduced tissue hypoxia at 90 d post-tumor induction. Hypoxyprobe labeling (brown) indicates area of low oxygen tension (<10 mm Hg), which is prevalent in PBS control ovaries and reduced in ABT-510-treated mice (n = 5 animals per group). *, P < 0.01, statistical difference between PBS-treated and ABT-510-treated groups.
ABT-510 Induces Tumor Cell Apoptosis and Inhibits Survival Pathways
The mechanism of action of TSP-1 as an antiangiogenic agent involves the binding of CD36. We have shown previously that CD36 is expressed by ovarian cells (19) and its expression in a human ovarian carcinoma cell line A2780 was reported (29). We confirmed CD36 expression in the tumorigenic ID8 line before injection and post-tumor induction. Ninety days post-ID8-injected ovarian tumors exhibit widespread immunostaining in both stromal and endothelial cell compartments (data not shown). ABT-510 treatment resulted in a significant reduction in primary tumor size and the number of secondary lesions in our EOC model. As TSP-1 is known to induce cell death in tumor endothelial cells, thus depriving tumor cells of endothelial cell survival factors (30), we analyzed apoptosis in ABT-510-treated and PBS-treated primary ovarian tumors in wild-type and TSP-1−/− mice. TSP-1−/− mice had a significantly lower (P < 0.05) basal level of tumor cell apoptosis compared with wild-type controls (Fig. 4A). In both groups, ABT-510-treated ovaries had a significantly (P < 0.05) higher number of TUNEL-positive cells compared with ovaries from PBS-treated animals (Fig. 4A). Immunoblot analysis of ABT-510 tissue lysates from wild-type animals was done to determine expression of phosphorylated forms of Akt, phosphatidylinositol 3-kinase, and p38 MAPK, which are all members of important cell survival pathways (31). A decrease in relative phospho-phosphatidylinositol 3-kinase and its downstream mediator Akt reveal a loss of cytoprotective signal, which predisposes cells to apoptosis (Fig. 4B). The stress-activated p38 MAPK was also deactivated in response to ABT-510 treatment (Fig. 4B). ABT-510 treatment resulted in an increase in relative expression of the proapoptotic FAS (Fig. 4B).
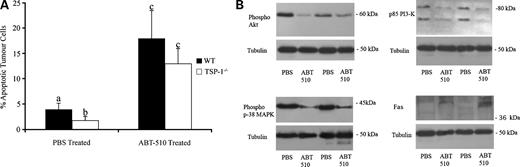
ABT-510 induces apoptosis in ovarian tumors and alters phosphorylation status and expression of various proapoptotic and antiapoptotic factors. A, in comparable sized tumors, PBS-treated TSP-1−/− mice had fewer (P < 0.05) apoptotic tumor cells than wild-type mice. ABT-510 treatment caused an increase (P < 0.05) in tumor cell apoptosis in both groups. B, proteins isolated from 90 d tumors from wild-type animals showed phosphorylation status of Akt, phosphatidylinositol 3-kinase, and p38 MAPK are decreased, indicating a proapoptotic role for ABT-510. ABT-510 also up-regulates expression of the proapoptotic Fas (n = 5 animals per group). Columns with different letters are statistically different (P < 0.05).
ABT-510 induces apoptosis in ovarian tumors and alters phosphorylation status and expression of various proapoptotic and antiapoptotic factors. A, in comparable sized tumors, PBS-treated TSP-1−/− mice had fewer (P < 0.05) apoptotic tumor cells than wild-type mice. ABT-510 treatment caused an increase (P < 0.05) in tumor cell apoptosis in both groups. B, proteins isolated from 90 d tumors from wild-type animals showed phosphorylation status of Akt, phosphatidylinositol 3-kinase, and p38 MAPK are decreased, indicating a proapoptotic role for ABT-510. ABT-510 also up-regulates expression of the proapoptotic Fas (n = 5 animals per group). Columns with different letters are statistically different (P < 0.05).
ABT-510 Induces Tumor Cell Apoptosis via CD36 and FasL
ID8 cells were immunopositive for the TSP-1 receptor CD36 in vitro (Fig. 5A). Using RNAi, we were able to significantly reduce the expression of CD36 protein in cultured ID8 cells (Fig. 5A). Western blot analysis showed that the RNAi knockdown of CD36 was ∼80% compared with controls (data not shown). ABT-510 was able to significantly increase tumor cell apoptosis (Fig. 5B) and this apoptotic effect was inhibited in ID8 cells that had reduced CD36 levels due to RNAi (Fig. 5B). In the presence of 20 nmol/L ABT-510, ID8 cells had an increase in expression of proapoptotic FasL (Fig. 5C). In CD36 RNAi ID8 cells, however, the expression of FasL was reduced (Fig. 5C). In addition to murine ovarian cancer cells, ABT-510 induced a significant (P < 0.05) increase in apoptosis in the human EOC cell lines SKOV3, OVCAR3, and CAOV3 (Fig. 5D). Knockdown of CD36 in the human ovarian cancer cell lines significantly (P < 0.05) reduced the ABT-510-induced apoptosis, but the rate of apoptosis was still higher than untreated controls (Fig. 5D).

ABT-510 induces apoptosis in murine and human ovarian epithelial cancer cells. A, ID8 cells expressed CD36 and RNAi reduced CD36 values by ∼80%. B, ABT-510 induced apoptosis in ID8 cells and this effect was inhibited in ID8 CD36 RNAi cells. a, P < 0.05, statistically different from control (1 nmol/L ABT-510 treatment); b, P < 0.05, statistically different between ID8 and ID8 + CD36 RNAi treatment groups. C, ABT-510 (20 nmol/L) caused an increase in expression of FasL, whereas cotreatment with CD36 RNAi significantly inhibited this increase in FasL expression. D, ABT-510 increased the incidence of apoptosis in the human epithelial cancer cell lines SKOV3, OVCAR3, and CAOV3. CD36 RNAi reduced ABT-510-induced apoptosis. Columns with different symbols are statistically different (P < 0.05).
ABT-510 induces apoptosis in murine and human ovarian epithelial cancer cells. A, ID8 cells expressed CD36 and RNAi reduced CD36 values by ∼80%. B, ABT-510 induced apoptosis in ID8 cells and this effect was inhibited in ID8 CD36 RNAi cells. a, P < 0.05, statistically different from control (1 nmol/L ABT-510 treatment); b, P < 0.05, statistically different between ID8 and ID8 + CD36 RNAi treatment groups. C, ABT-510 (20 nmol/L) caused an increase in expression of FasL, whereas cotreatment with CD36 RNAi significantly inhibited this increase in FasL expression. D, ABT-510 increased the incidence of apoptosis in the human epithelial cancer cell lines SKOV3, OVCAR3, and CAOV3. CD36 RNAi reduced ABT-510-induced apoptosis. Columns with different symbols are statistically different (P < 0.05).
Expression of Vascular and Proliferative Markers Is Decreased following Treatment with ABT-510
Tissue homogenates were collected from ovarian tissue 90 days post-tumor induction from PBS and ABT-510 treated mice and subjected to Western blot analysis for the proliferation marker PCNA and the angiogenic markers VEGF and its receptor VEGFR-2. PCNA protein levels were significantly (P < 0.05) decreased in mice treated with ABT-510. Similarly VEGF and VEGFR-2 protein levels were significantly (P < 0.05) decreased following ABT-510 treatment (Fig. 6).

ABT-510 treatment reduces the expression of angiogenic and proliferative factors. A, tumor tissue homogenates collected at 90 d post-tumor induction showed reduced protein levels for VEGF and its receptor and the proliferation marker PCNA. B, densitometry was done on a minimum of three different immunoblot experiments. Results are expressed relative to α-tubulin. Mean ± SE expression of the different factors for each treatment group. *, P < 0.05, statistical difference between PBS-treated and ABT-510-treated groups.
ABT-510 treatment reduces the expression of angiogenic and proliferative factors. A, tumor tissue homogenates collected at 90 d post-tumor induction showed reduced protein levels for VEGF and its receptor and the proliferation marker PCNA. B, densitometry was done on a minimum of three different immunoblot experiments. Results are expressed relative to α-tubulin. Mean ± SE expression of the different factors for each treatment group. *, P < 0.05, statistical difference between PBS-treated and ABT-510-treated groups.
Discussion
In order for any solid tumor to grow beyond 1-2mm, it must recruit a new blood supply, which typically occurs through angiogenic processes (32). In this study, we provide evidence that ABT-510 may be effective in treating EOC through both antiangiogenic and antitumor effects. Daily i.p. administration of ABT-510 inhibited tumor growth, reduced ascites formation and decreased dissemination of secondary tumors in an orthotopic injected syngeneic mouse model of ovarian cancer.
The vasculature of rapidly growing tumors has been shown to be abnormal, with the presence of large, tortuous vessels, lack of anastomosis and numerous blind ends (33). Endothelial cells of tumor vessels often are not stabilized by adjacent pericytes, which provide vessel support and are an indicator of mature blood vessels (34). As a result of this rapidly-growing abnormal vasculature, tumor tissue has high interstitial pressure, reduced perfusion and areas of tissue hypoxia (35). It has been proposed that combinational therapy of antiangiogenic agents combined with cytotoxic drugs can enhance tumor growth inhibition (34). Antiangiogenic compounds appear to prune distorted tumor vessels, normalizing the vasculature, which improves tissue perfusion, allowing for improved delivery of cytotoxic chemotherapy drugs to the tumor tissue (34). ABT-510 is more effective at inhibiting growth of neuroblastoma xenografts in combination with valproic acid than as a single agent (36). Administration of ABT-510 in our model reduced the presence of large tortuous vessels, increased the proportion of mature vessels with pericyte coverage and decrease tissue hypoxia. These morphological changes by ABT-510 included smaller diameter vessels and an overall decrease in tumor vascular area, leading to a somewhat normalized vasculature. Although the tumor tissue of ABT-510 treated animals appeared to be better perfused, the decreased proliferation and increased apoptosis seen in these animals was likely due to the direct pro-apoptotic effect of ABT-510 on the ovarian tumor cells. Thus ABT-510 appears to affect ovarian tumors by stimulating tumor cell apoptosis and by limiting abnormal vessel growth and reducing tissue hypoxia. By reducing tumor hypoxia, ABT-510 also likely contributed to the reduced VEGF levels seen in the mice treated with the peptide. VEGF is potently expressed in response to tissue hypoxia in an attempt to stimulate angiogenesis and provide adequate oxygenation (37). With reduced hypoxia, there would be lower stimulus for VEGF expression and a reduction in the abnormally high VEGF levels seen in untreated mice. Used in combination therapy, ABT-510 may be an effective agent in normalizing blood vessels and increasing tissue perfusion, which would facilitate intratumor delivery of chemotherapeutic drugs. ABT-510 has been shown to inhibit VEGF-induced endothelial cell migration (21) and can stimulate endothelial apoptosis (38). ABT-510 has also been shown to restore low wild type levels of circulating endothelial cells in TSP KO mice, where these are elevated ∼5-fold (39) when injected over 5 days. TSP-1-induced endothelial apoptosis is mediated by Fyn, p38 MAPK and caspases-3 pathways (16) that leads to increased of FasL expression (40). Furthermore, TSP-1 has been shown to block VEGF mediated nitric oxide synthesis, which is essential for angiogenesis (41). The effects of TSP-1 appear ultimately to lead to reduced microvascular density (MVD). We have previously shown that full length TSP-1 can bind and internalize VEGF through the low-density lipoprotein receptor-related protein 1 (17). Other reports have shown that other members of the TSR superfamily can inhibit VEGF induced angiogenesis by direct protein to protein interactions. Heparin affin regulatory peptide (HARP) and connective tissue growth factor (CTGF) both contain the TSR motif and this domain binds the 165 splice variant of VEGFA (42, 43). In human bladder cancer and Lewis lung carcinoma models (36, 38), ABT-510 increased endothelial cell apoptosis and reduced tumor vessel density (38). Our results are consistent with these observations as mice receiving 100 mg/kg ABT-510 exhibited a significant reduction in vasculature area and perimeter.
As EOC progresses into later stages, the primary tumor sheds cells, which disseminate throughout the abdominal cavity to form secondary lesions. Although the mechanism behind dissemination is unclear (44), ABT-510 treatment significantly inhibited the formation of secondary lesions. As the formation of secondary lesions are usually only seen at later stages, ABT-510 may have prevented their formation by delaying progression and development of the primary tumors, not allowing it to reach an advanced stage.
A hallmark in EOC is the accumulation of malignant ascites, which accounts for many of the non-specific symptoms experienced during late stage disease. Generally, increased morbidity is associated with the building of fluid within the abdominal and pelvic cavities. Ascites may arise from decreased clearance from abdominal lymphatics that become blocked with secondary lesions and increased vascular permeability that correlates with neovascularization of primary tumors (45). Tumor production of VEGF is implicated in this increased permeability (46) as VEGF protein levels are dramatically increased in malignant ascites fluid of EOC patients (47). If the primary tumor is responsible for elevated VEGF levels, then reducing tumor volume would potentially lead to lower VEGF expression, a decrease in tumor vasculature and reduced ascites. In addition to decreasing tumor volume, ABT-510 may have reduced ascites formation by normalizing the tumor vasculature. Tumor vessels are known to be leaky, with increased fenestrations and fluid extravasation, which contributes to the accumulation of abdominal ascites in EOC (48, 49). The reduction in proportion of these large, leaky vessels likely contributed to decreased ascites in the ABT-510 treated animals.
The ability of ABT-510 to decrease tumor size, ascites formation and the presence of secondary lesions demonstrates its effectiveness as a single-agent therapy. Metronomic administration of chemotherapeutics along with antiangiogenic compounds has shown that cytotoxic drugs and antiangiogenic agents can have a synergistic effect (22). Metronomic chemotherapy involves the administration of low-dose chemotherapeutics at close intervals and has been shown to inhibit tumor angiogenesis (50). One mechanism of metronomic chemotherapy is to increase expression of pro-apoptotic Fas in endothelial cells (51) and we showed that ABT-510 treatment increases FasL expression in ovarian tumor cells in vitro, and others have demonstrated an increase in FasL in response to ABT-510 (40). Quesada et al., illustrated that frequent low dose of doxorubicin sensitizes endothelial cells to FasL and ABT-510 was able to synergistically inhibit tumor growth in bladder and prostate xenograft models (22). Activation of CD36 by TSP-1 phosphorylates p38 and JNK, which results in an increased expression of FasL and apoptosis (52), which we saw in our tumor cells in response to ABT-510.
Tumors from ABT-510-treated mice had a significant increase in apoptosis in epithelial and endothelial tissues. Decreased phosphorylation of both phosphatidylinositol 3-kinase and Akt would lead to loss of antiapoptotic signals, which sensitizes cells to apoptosis (53). This coupled with increased expression of FasL would create a proapoptotic environment. The stress-activated p38 MAPK was dephosphorylated in response to ABT-510 treatment when compared with PBS-treated mice. Although TSP-1 has been shown to lead to increased in p38 MAPK, which leads to apoptosis (16), other cells types have shown that p38 MAPK can have a cytoprotective effects (54); thus, p38 MAPK may act in cell-specific contexts. We showed in vitro that ABT-510 treatment results in an increased expression of proapoptotic FasL and that expression of CD36 is involved in this pathway. The exact mechanism of tumor cell apoptosis in our model of EOC remains to be elucidated.
Protein collected from ABT-510-treated tumors show a decrease in expression of VEGF, VEGFR-2, and PCNA compared with PBS-treated controls. As VEGF and VEGFR-2 are expressed on both endothelial and tumor cells in our model, reduced expression would be expected to affect both cellular compartments. Reduced VEGF expression would have multiple effects in an ovarian tumor such as decreased angiogenesis, reduced vascular permeability, and increased susceptibility to apoptosis of endothelial and tumor cells. Decreased vascularization and ascites formation were evident in our models as was an increase in epithelial and endothelial apoptosis. Currently, the mechanisms behind the decreased VEGF expression are unknown but likely involve the decrease observed in primary tumor volume, reducing the secretion of VEGF (and likely other growth factors) from the ovarian tumor tissue. In addition to the apoptotic effect of ABT-510, the decrease in VEGF and VEGFR-2 may also have contributed to tumor due to their cytoprotective function in endothelial cells (55). We have shown a strong extravascular cytoprotective effect for VEGF and VEGFR-2 in ovarian cells (56) and have identified a survival role for these molecules in ovarian epithelial tumor cells (data not shown). In the ovarian tumors in this study, ABT-510 significantly increased apoptosis of both endothelial and tumor cells, which would explain the significantly decreased volume in our ABT-510-treated ovaries. In mice lacking TSP-1, the tumor growth rate was accelerated, and there were fewer apoptotic tumor cells. In contrast, when ABT-510 was added, there was a significant increase in tumor cell death, reduced tumor growth rate, and lower tumor burden. These results suggest that TSP-1 is an important component of the tumor microenvironment and acts to combat tumor cell proliferation.
In conclusion, ABT-510 appears to be effective as single-agent therapy in an orthotopic, syngeneic model of EOC. Our model provides evidence that ABT-510 can delay tumor progression, normalize tumor vasculature, and increase survival. Further studies administering ABT-510 in combination with commonly used chemotherapeutic drugs such as valproic acid, paclitaxel, and cisplatin are warranted.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant support: Ontario Institute for Cancer Research grant 047142 and Canadian Institutes for Health Research.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Acknowledgments
We thank Michelle Ross for technical assistance.