


Abstract
Purpose: Endometrial cancer is a common gynecologic cancer whose incidence is increasing annually worldwide. Current methods to detect endometrial cancer are unreliable and biomarkers are unsatisfactory for screening. Cervical scrapings were reported as a potential source of material for molecular testing. DNA methylation is a promising cancer biomarker, but limited use for detecting endometrial cancer.
Experimental Design: We analyzed two methylomics databases of endometrioid-type endometrial cancer. Using nonnegative matrix factorization algorithm clustered the methylation pattern and reduced the candidate genes. We verified in pools DNA from endometrial cancer tissues and cervical scrapings, and validated in 146 cervical scrapings from patients with endometrioid-type endometrial cancer (n = 50), uterine myoma (n = 40), and healthy controls (n = 56) using quantitative methylation–specific PCR (QMSP). The logistic regression was used to evaluate the performance of methylation signal and gene combination.
Results: We filtered out 180 methylated genes, which constituted four consensus clusters. Serial testing of tissues and cervical scrapings detected 14 genes that are hypermethylated in endometrial cancer. Three genes, BHLHE22, CDO1, and CELF4, had the best performance. Individual genes were sensitivity of 83.7%–96.0% and specificity of 78.7%–96.0%. A panel comprising any two of the three hypermethylated genes reached a sensitivity of 91.8%, specificity of 95.5%, and odds ratio of 236.3 (95% confidence interval, 56.4–989.6). These markers were also applied to cervical scrapings of type II endometrial cancer patients, and detected in 13 of 14 patients.
Conclusions: This study demonstrates the potential use of methylated BHLHE22/CDO1/CELF4 panel for endometrial cancer screening of cervical scrapings. Clin Cancer Res; 23(1); 263–72. ©2016 AACR.
Endometrial cancer is increasing worldwide and mortality rate has not declined over the past several decades. There are currently no satisfactory methods for endometrial cancer screening. Although the application of DNA methylation as a biomarker for cancer detection is promising, research on methylomics of endometrial cancer, especially for screening purposes, is limited. The present study investigated two methylomics databases of endometrioid-type endometrial cancer, identified a panel of methylation biomarkers, and verified the feasibility of using the methylation profile of cervical scrapings to detect endometrial cancer. BHLHE22, CDO1, and CELF4 methylation of cervical scrapings had a sensitivity of 91.8%, specificity of 95.5%, and odds ratio of 236.3 (95% confidence interval, 56.4–989.6) for endometrial cancer detection despite the interference of benign uterine tumors. These sensitive molecular biomarkers expand the scope of the Pap smear. These results reveal a bright future of molecular endometrial cancer screening.
Introduction
Endometrial cancer is the most frequently diagnosed cancer of the female genital tract and ranks as the sixth-most diagnosed female cancer worldwide (1). In 2012, the incidence and mortality were about 310,000 and 76,000, respectively. More than 380,000 new cases and 93,000 deaths from endometrial cancer are expected in 2020 (http://globocan.iarc.fr/). Histopathologic assessment is used to classify endometrial cancer broadly into two types. Type I endometrial cancers comprise well to moderately differentiated (grades 1 to 2) endometrioid adenocarcinomas. Type II endometrial cancers comprise serous type, clear cell, and poorly differentiated (grade 3) endometrioid-type forms. Most type I endometrial cancers are diagnosed at an early stage (I or II) and have a favorable outcome, whereas type II endometrial cancers are typically diagnosed at an advanced stage and have a poor prognosis (2). Although most patients with an endometrioid-type endometrial cancer are diagnosed at the early stage, 15% of patients with a late-stage endometrioid endometrial cancer have a poor 5-year survival rate of less than 50% (40%–50% for stage III, 15%–20% for stage IV; ref. 3). Early detection remains the best way to improve outcome. However, there are currently no satisfactory methods for endometrial cancer screening.
Abnormal uterine bleeding is the most frequent symptom of endometrial cancer, but many other disorders give rise to the same symptom. Even when bleeding occurs in postmenopausal women, only 10% of cases are caused by an endometrial cancer. The choice of the ideal detection strategy depends upon the sensitivity, specificity, probability of accuracy, and cost. Transvaginal ultrasound (TVU) is used to exclude endometrial cancer (4). The cut-off value for TVU in symptomatic premenopausal women and those taking hormone replacement therapy is lower because of variations in endometrial thickness under the influence of circulating female steroid hormones (5). Endometrial samples obtained by suction curettage in an outpatient setting may have a higher sensitivity and specificity compared with TVU. However, the failure rate of this invasive procedure can be up to 54% (6). The clinical guidelines recommend fractional dilatation and curettage (D & C) under anesthesia if the endometrial sampling is inadequate or inclusive for diagnosis (7). Thus, many patients undergo repeated invasive evaluations by endometrial sampling or D & C, which is inconvenient, stressful, and costly. The diagnostic accuracy of hysteroscopy can achieve an overall sensitivity of 86.4% and specificity of 99.2% in both pre- and postmenopausal women (8). However, there is debate over the best cut-off value for endometrial thickness diagnosed with TVU that should warrant endometrial sampling or hysteroscopy. Noninvasive methods to detect the presence or absence of endometrial cancer are measurement of the serum and cervicovaginal concentrations of cancer antigen 125 (CA-125; ref. 9). The accuracy of using CA-125 as an indicator is easily confounded by benign conditions such as adenomyosis or uterine myomas and endometriosis (10, 11). Therefore, CA-125 is not recommended for cancer screening. Cytology from cervical scrapings have also been used for detecting endometrial cancers, but the rate of abnormal results range from 32.3% to 86.0% for type I endometrial cancer and 57.1% to 100% for type II endometrial cancer (12). There is a need for a better method for endometrial cancer screening.
The development of molecular biomarkers is encouraging. Studies have identified many specific alterations in endometrial cancer, including gene mutations, microsatellite instability (MSI), copy-number alterations, mRNA and miRNA expression patterns, and DNA methylation (13). The Cancer Genome Atlas (TCGA) classified endometrial cancer according to genetic events such as POLE ultramutation, MSI, and copy-number alterations; this classification supports the idea of diverse molecular phenotypes in types I and II endometrial cancers (13). Type I endometrial cancer frequently has POLE, PTEN, CTNNB1, PIK3CA, ARID1A, KRAS, and ARID5B mutations, whereas type II endometrial cancer has more TP53 mutations. Building on this genetic knowledge, Kinde and colleagues (14) reported that the genetic mutations of endometrial cancer might be detected in cancer tissues and Papanicolaou (Pap) smear specimens; thus, this raises the possibility of molecular screening for endometrial cancers using cervical scrapings.
DNA methylation may occur early in carcinogenesis and is sufficiently stable for analysis (15). The application of DNA methylation as a biomarker for cancer detection or patient stratification has been increasing (16). However, research on endometrial cancer epigenomics, especially for screening purposes, is relatively limited (17–19). A comprehensive methylomics approach to the development of DNA methylation biomarkers for endometrial cancer is lacking. The aims of the present study were to investigate the methylomics of endometrial cancer and to identify a panel of methylation biomarkers present in cervical scrapings that could be used for endometrial cancer detection.
Materials and Methods
Analysis of DNA methylation arrays and MethylCap-sequencing data of endometrial tissues
The first methylomics analysis of 370 endometrial tissues was performed using a Methylation 450K BeadChip array (Illumina, Inc.). We used 270 endometrioid-type endometrial cancers and 28 adjacent normal tissues to identify the differential methylation of endometrial cancer. The demographics are listed in Supplementary Table S1. We downloaded the level 3 data from the TCGA data portal. The methylation score for each CpG was represented as a β-value and normalized, and the details are described in the Supplementary Methods S7 of a TCGA research study (13). We removed the sex chromosome probes, and the remaining 379,054 CpG sites were analyzed in this study. The cancer carcinoma samples exhibited ≥40% neoplastic cellularity. The significantly different methylated CpG sites were analyzed using the Mann–Whitney U test and P ≤ 0.001. We selected the top 5% of genes with differences in hypermethylation from the median β-values of endometrial cancer minus the normal values. We focused on CpG sites located closest (≤1,000 bp) to the TSS of the coding genes. Finally, we selected hypermethylated candidates with ≥5 remaining CpG sites in the coding gene.
The second methylomics analysis of endometrial tissues was performed using MethylCap-seq provided by Dr. Tim Hui-Ming Huang and visualized at The Cancer Methylome System website, http://cbbiweb.uthscsa.edu/KMethylomes/ (20, 21). We selected the MethylCap-seq data with uniquely mapped reads ≥40% of the total mapped reads in the remaining data from 78 endometrioid-type endometrial cancer and 11 adjacent normal tissues (Supplementary Table S1). We analyzed the methylation level for a 2,000 bp region spanning 1,000 bp upstream and downstream of the TSS of the coding genes (reference genome of UCSC version hg18). We excluded the TSS regions in the sex chromosome, and the remaining 23,215 TSS regions were analyzed in this study. Significantly different methylation patterns were analyzed using the Mann–Whitney U test using P ≤ 0.001. We selected the top 5% hypermethylated differences from the median reads of endometrial cancer values minus the normal value.
Clinical samples
Our case–control study set comprised tissues from 175 women: 29 endometrial cancer tissues, one endometrial cancer tissue and paired cervical scrapings, unpaired cervical scrapings from 49 patients with endometrioid-type endometrial cancer, 40 with myoma, and 56 cervical scrapings from normal healthy women. The demographics and clinical data are listed in Supplementary Table S2. These specimens were placed immediately in RNAlater Stabilization Solution (Ambion, Thermo Fisher Scientific) and stored at −80°C until analysis. All specimens were collected between October 2014 and December 2015 at Taipei Medical University-Shuang Ho Hospital, New Taipei, Taiwan. The criteria for inclusion stipulated that the study participants needed to be women ages 30 to 80 years. Age, histologic type of tumor, the International Federation of Gynecology and Obstetrics stage and histological grade were reviewed from the hospital records for each participant. Studies were conducted with approval from the Institutional Review Board of the Taipei Medical University-Shuang Ho Hospital in accordance with the Declaration of Helsinki 2000. Informed consent was obtained from all participants in this study.
To define the methylation status and reduce the candidate gene list, we used three steps in our specimens. For step 1 verification, we used DNA pools from 30 endometrial cancer tissues and cervical scraping of 15, 15, and 20 endometrial cancer, myoma, and normal endometrium, respectively. Those specimens were selected in the first 5 months of collection. Each pooled DNA sample comprised a mixture of an equal amount of DNA from five specimens. For step 2 verification, we used individual DNA samples from 16, 20, and 28 cervical scrapings from endometrial cancer, myoma, and normal endometrium, respectively. The candidate genes achieving the area under a receiver-operating characteristic curve (AUC) ≥0.9 were used in the next validation (Supplementary Table S3). For step 3 validation, we examined individual DNA samples from total cervical scrapings. We aimed to include 50 cervical scrapings of endometrioid-type endometrial cancer at a 1:1.75 ratio with noncarcinomatous controls. We assumed a 10% failure rate for the control specimens. Thus, we collected 146 cervical scrapings from 50 participants diagnosed with endometrioid endometrial cancer and 96 controls. Patients with the nonendometrioid type of endometrial cancer included five with the serous type, four with the mucinous type, and one with the clear cell type.
Consensus clustering of DNA methylation profile analysis
We performed consensus clustering of DNA methylation profiles using the NMF function of MeV4 version 4.9 (22, 23). We expected to identify 2 to 5 hypermethylated gene clusters with a high complementarity because we had calculated the values of cophenetic correlation as 2 to 5 clusters. To estimate the best consensus clustering groups, we performed cophenetic correlation at the maximum iteration of 1,000 to 5,000 using a rank range of 2 to 5 groups (Supplementary Table S4). We selected the optimum consensus clusters according to the highest cophenetic correlation value for 4 groups. We selected candidates from 12% of the genes in each cluster, and followed established criteria including top differences in methylation levels in endometrial cancer and normal endometrium for each gene, changes in methylation levels of ≥1.5-fold, and ≥200 bp regions.
DNA extraction, bisulfite conversion, and quantitative methylation–specific PCR
Genomic DNA was extracted from cervical scraping cells and tissues using the QIAmp DNA Mini Kit (QIAGEN). We used 1 μg genomic DNA to do bisulfite modified using the EZ DNA Methylation Kit (Zymo Research Corp.), according to the manufacturer's recommendations, and dissolved in 70 μL of nuclease-free water. PCR products were amplified with the LightCycler 480 SYBR Green I Master (Roche) and performed using LightCycler 480. A 20 μL reaction contained 2 μL bisulfite-converted DNA, 250 nmol/L each primer, and 10 μL Master Mix. The reactions were conducted on LightCycler 480 under the following thermal profiles: 95°C for 5 minutes, 50 cycles consisted of 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, and final extension at 72°C for 5 minutes. All specimens conducted duplicate testing in each gene. To normalize the input DNA, we designed primers to measure amount of a non-CpG region of COL2A1 in each methylation-independent assay. The DNA methylation level was estimated the methylation index (M-index) using the formula: 10,000 × 2[(Cp of COL2A) − (Cp of Gene)] (24). Test results with Cp-values of COL2A >36 were defined as detection failure. The primers were designed by Oligo 7.0 Primer Analysis software (Molecular Biology Insights, Inc.).
Statistical analysis
The Mann–Whitney nonparametric U test was used to identify differences in methylation level between two categories, and the Kruskal–Wallis test was used to identify differences in methylation level between more than two categories. Correlations between categorical clinical variables and methylation level were identified by the Fisher exact test for 2-by-2 categories and the Freeman–Halton extension of Fisher exact probability test for 3-by-2 categories. Logistic regression was used to estimate OR and 95% confidence interval (CI) to describe the endometrial cancer risk associated with the methylation status of each candidate gene and gene combination. All significant differences were assessed using a two-tailed P ≤ 0.05. The receiver-operating characteristic curve was used to select the optimal threshold for distinguishing the absence and presence of disease. The optimum threshold was calculated as the maximal sum of sensitivity and specificity (Youden's method) and 200 bootstrapping iterations in the pROC package in R. Above analyses and plots were performed using the statistical package in R (version 3.1.2) and MedCalc (version 14.12.0). We used pairwise Fisher exact post hoc tests and an assumed alpha error proportion of 0.01 to evaluate statistic power by G*Power version 3.1.9.2.
Results
Differential methylomics analysis between endometrial cancer and the normal endometrium
We analyzed two public sets of methylomics data from endometrial tissues to try to identify candidate genes that are hypermethylated in endometrial cancer. The logistics of the present study are illustrated in Fig. 1A. Set A was derived from the methylomics of endometrial cancer generated using a Methylation 450K BeadChip of samples from the TCGA database, which included 270 samples of endometrioid endometrial cancer and 28 adjacent normal tissues (13). Set B included the methylomics of endometrial cancer generated in our previous study using methyl–DNA-binding domain capture coupled with next-generation sequencing (MethylCap-seq), which included 78 samples of endometrial cancer and 11 adjacent normal tissues (20). The demographics of Set A and Set B were listed in Supplementary Table S1. In set A, we identified 2,869 genes with significant differences in the DNA methylation level [≥5 significant probes spanning ± 1 kb of the transcription start site (TSS; Fig. 1B)]. In Set B, we identified 1,527 genes with significant differences in the DNA methylation level at the promoter region (spanning ± 1 kb of the TSS, Fig. 1C). To reduce the gene lists, we merged the top 5% of genes of the two datasets, which resulted in 180 candidate genes.
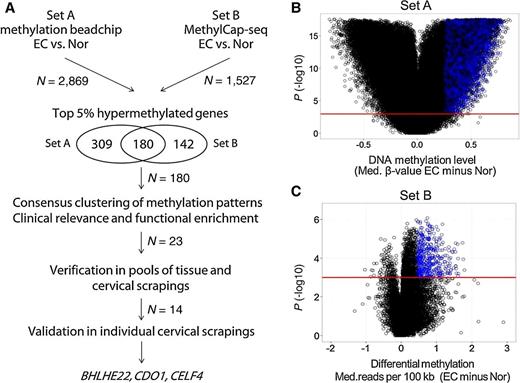
Discovery using methylomics in endometrial cancer tissues. A, Logistics of the discovery of new genes that are hypermethylated in endometrial cancer shown in a flow chart. B and C, Volcano plot showing the distribution of differential methylation between endometrial cancer tissues and adjacent normal (Nor) tissues in Set A (performed using Methylation 450K BeadChip) and Set B (performed using methylation capture sequencing, MethylCap-seq). In Set A, one dot represents the differential methylation status of a CpG site. In Set B, one dot represents the differential methylation status of regions spanning ±1 kb of the TSS. We selected significantly different methylation levels at P ≤ 0.001. The blue dots represent the top 5% difference in hypermethylation in regions spanning ±1 kb of the TSS. N, gene numbers. Med, median.
Discovery using methylomics in endometrial cancer tissues. A, Logistics of the discovery of new genes that are hypermethylated in endometrial cancer shown in a flow chart. B and C, Volcano plot showing the distribution of differential methylation between endometrial cancer tissues and adjacent normal (Nor) tissues in Set A (performed using Methylation 450K BeadChip) and Set B (performed using methylation capture sequencing, MethylCap-seq). In Set A, one dot represents the differential methylation status of a CpG site. In Set B, one dot represents the differential methylation status of regions spanning ±1 kb of the TSS. We selected significantly different methylation levels at P ≤ 0.001. The blue dots represent the top 5% difference in hypermethylation in regions spanning ±1 kb of the TSS. N, gene numbers. Med, median.
Identification of a subgroup of endometrial cancer patients defined using consensus methylation clustering
We generated a methylation heat map of these 180 genes (Fig. 2A and B). The heat map revealed heterogeneity of the methylation patterns such as the long-debated concept of the CpG island methylator phenotype in colon cancer (25). We hypothesized that patients with the same histologic type may be clustered further according to different methylation patterns, which may confer a different prognosis or provide complementary biomarkers for screening. We analyzed the correlations between methylation patterns using a nonnegative matrix factorization (NMF) algorithm for Set B data, which resulted in four consensus methylation clusters (Fig. 2C and Supplementary Table S4). There were 58, 56, 25, and 48 genes grouped into clusters a, b, c, and d, respectively (listed in Supplementary Table S5). The methylation patterns of metagenes are displayed in Fig. 2D, and the methylation patterns around the transcriptional start site of representative genes from each cluster are shown in Fig. 2E.

Clustering and survival analysis of the top hypermethylated genes. A and B, Heat maps show the hypermethylation signature of 180 genes in endometrial cancers of Sets A and B. The methylation level of tumor samples showed heterogeneous methylation patterns. C, The correlation matrix shows four consensus clustering of methylation patterns in endometrial cancer samples for Set B, because the methylation status was calculated from the effects of total CpG sites. Strong and weak correlations are shown in red and blue, respectively. The selected candidates are labeled on the right. D, Metagene patterns are plotted in four clusters. The dark blue line shows the median methylation status of each metagene. E, This gene shows the median methylation of endometrial cancer and normal endometrial tissue in Set B in the region spanning ±1 kb of the TSS. The blue box shows coding regions (CDS). The triangle shows the transcriptional orientation.
Clustering and survival analysis of the top hypermethylated genes. A and B, Heat maps show the hypermethylation signature of 180 genes in endometrial cancers of Sets A and B. The methylation level of tumor samples showed heterogeneous methylation patterns. C, The correlation matrix shows four consensus clustering of methylation patterns in endometrial cancer samples for Set B, because the methylation status was calculated from the effects of total CpG sites. Strong and weak correlations are shown in red and blue, respectively. The selected candidates are labeled on the right. D, Metagene patterns are plotted in four clusters. The dark blue line shows the median methylation status of each metagene. E, This gene shows the median methylation of endometrial cancer and normal endometrial tissue in Set B in the region spanning ±1 kb of the TSS. The blue box shows coding regions (CDS). The triangle shows the transcriptional orientation.
To explore further the clinical relevance of these clusters, we tested the correlations between these clusters and clinicopathologic parameters (Table 1). Clusters a and b were significantly associated with tumor grade (P < 0.05 and P < 0.01, respectively). Low methylation of cluster a was associated with a lower rate of genetic events: MLH1 mutations (P = 0.01) and MSI (P < 0.01). This is consistent with previously published results (13, 26).
Relationships between DNA methylation status and clinical variables in four gene clusters
| Cluster a | Cluster b | Cluster c | Cluster d | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Number of cases | H (%) | L (%) | P | H (%) | L (%) | P | H (%) | L (%) | P | H (%) | L (%) | P |
| Age, y | |||||||||||||
| <55 | 8 | 6 (16.2) | 2 (8.3) | 0.46 | 8 (16.7) | 0 (0.0) | 0.18 | 3 (20.0) | 5 (10.9) | 0.39 | 4 (20.0) | 4 (9.8) | 0.42 |
| ≥≧55 | 53 | 31 (83.8) | 22 (91.7) | 40 (83.3) | 13 (100.0) | 12 (80.0) | 41 (89.1) | 16 (80.0) | 37 (90.0) | ||||
| Stage | |||||||||||||
| I–II | 57 | 35 (94.6) | 22 (91.7) | 0.64 | 46 (95.8) | 11 (84.6) | 0.20 | 15 (100.0) | 42 (73.7) | 0.56 | 17 (85.0) | 40 (97.6) | 0.10 |
| III | 4 | 2 (5.4%) | 2 (8.3) | 2 (4.2) | 2 (15.4) | 0 (0.0) | 4 (8.7) | 3 (15.0) | 1 (2.4) | ||||
| Gradea | |||||||||||||
| G1 | 28 | 18 (48.6) | 10 (41.7) | <0.05 | 26 (54.2) | 2 (15.4) | <0.01 | 7 (46.7) | 21 (45.7) | 1.00 | 10 (50.0) | 18 (43.9) | 0.52 |
| G2 | 29 | 19 (52.4) | 10 (41.7) | 21 (43.7) | 8 (61.5) | 7 (46.7) | 22 (47.8) | 10 (50.0) | 19 (46.3) | ||||
| G3 | 4 | 0 (0.0) | 4 (16.7) | 1 (2.1) | 3 (23.1) | 1 (6.7) | 3 (6.5) | 0 (0.0) | 4 (9.8) | ||||
| MLH1 mutation | |||||||||||||
| No | 39 | 19 (51.4) | 20 (83.3) | 0.01 | 28 (58.3) | 11 (84.6) | 0.11 | 8 (53.3) | 31 (67.4) | 0.36 | 13 (65.0) | 26 (63.4) | 1.00 |
| Yes | 22 | 18 (48.6) | 4 (16.7) | 20 (41.7) | 2 (15.4) | 7 (46.7) | 15 (32.6) | 7 (35.0) | 15 (36.6) | ||||
| MSI | |||||||||||||
| No | 38 | 18 (48.6) | 20 (83.3) | <0.01 | 27 (56.2) | 11 (84.6) | 0.10 | 8 (53.3) | 30 (65.2) | 0.54 | 27 (56.2) | 11 (84.6) | 0.10 |
| Yes | 23 | 19 (51.4) | 4 (16.7) | 21 (43.7) | 2 (15.4) | 7 (46.7) | 16 (34.8) | 21 (43.7) | 2 (15.4) | ||||
| Cluster a | Cluster b | Cluster c | Cluster d | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Number of cases | H (%) | L (%) | P | H (%) | L (%) | P | H (%) | L (%) | P | H (%) | L (%) | P |
| Age, y | |||||||||||||
| <55 | 8 | 6 (16.2) | 2 (8.3) | 0.46 | 8 (16.7) | 0 (0.0) | 0.18 | 3 (20.0) | 5 (10.9) | 0.39 | 4 (20.0) | 4 (9.8) | 0.42 |
| ≥≧55 | 53 | 31 (83.8) | 22 (91.7) | 40 (83.3) | 13 (100.0) | 12 (80.0) | 41 (89.1) | 16 (80.0) | 37 (90.0) | ||||
| Stage | |||||||||||||
| I–II | 57 | 35 (94.6) | 22 (91.7) | 0.64 | 46 (95.8) | 11 (84.6) | 0.20 | 15 (100.0) | 42 (73.7) | 0.56 | 17 (85.0) | 40 (97.6) | 0.10 |
| III | 4 | 2 (5.4%) | 2 (8.3) | 2 (4.2) | 2 (15.4) | 0 (0.0) | 4 (8.7) | 3 (15.0) | 1 (2.4) | ||||
| Gradea | |||||||||||||
| G1 | 28 | 18 (48.6) | 10 (41.7) | <0.05 | 26 (54.2) | 2 (15.4) | <0.01 | 7 (46.7) | 21 (45.7) | 1.00 | 10 (50.0) | 18 (43.9) | 0.52 |
| G2 | 29 | 19 (52.4) | 10 (41.7) | 21 (43.7) | 8 (61.5) | 7 (46.7) | 22 (47.8) | 10 (50.0) | 19 (46.3) | ||||
| G3 | 4 | 0 (0.0) | 4 (16.7) | 1 (2.1) | 3 (23.1) | 1 (6.7) | 3 (6.5) | 0 (0.0) | 4 (9.8) | ||||
| MLH1 mutation | |||||||||||||
| No | 39 | 19 (51.4) | 20 (83.3) | 0.01 | 28 (58.3) | 11 (84.6) | 0.11 | 8 (53.3) | 31 (67.4) | 0.36 | 13 (65.0) | 26 (63.4) | 1.00 |
| Yes | 22 | 18 (48.6) | 4 (16.7) | 20 (41.7) | 2 (15.4) | 7 (46.7) | 15 (32.6) | 7 (35.0) | 15 (36.6) | ||||
| MSI | |||||||||||||
| No | 38 | 18 (48.6) | 20 (83.3) | <0.01 | 27 (56.2) | 11 (84.6) | 0.10 | 8 (53.3) | 30 (65.2) | 0.54 | 27 (56.2) | 11 (84.6) | 0.10 |
| Yes | 23 | 19 (51.4) | 4 (16.7) | 21 (43.7) | 2 (15.4) | 7 (46.7) | 16 (34.8) | 21 (43.7) | 2 (15.4) | ||||
Abbreviations: H, high methylation; L, Low methylation; MLH1, mutL homolog 1 gene; MSI, microsatellite instability. The high and low methylation dichotomized by the Youden's method of pROC for distinguishing recurrence.
aUsing Freeman–Halton extension of the Fisher exact probability test. Other P values calculated by the Fisher exact test.
Verification of DNA methylation in pools of tissues and cervical scrapings
Using the results of consensus clustering and methylation pattern analysis, we selected 23 candidates for verification using DNA pooled from cancer tissues and cervical scrapings (Supplementary Fig. S1). These 23 genes exhibited significantly greater methylation in all cancer tissues. We found that the presence of uterine myoma, a common benign uterine tumor, confounded the detection of methylation in cervical scrapings. We included a pool of cervical scrapings from symptomatic myomas to select specific candidate genes. Fourteen genes demonstrated specific DNA methylation in endometrial cancer samples and were subjected to further validation.
Validation of DNA methylation in individual cervical scrapings
We tested DNA methylation in a pilot set of cervical scrapings and calculated AUC for cancer detection (Supplementary Table S3 and Supplementary Fig. S2). BHLHE22, CELF4, CDO1, and ZNF662 were selected for further testing of 146 cervical scrapings, including samples of endometrial cancer (n = 50), myoma (n = 40), and normal tissue (n = 56; Fig. 3A and Supplementary Table S6). The AUCs of BHLHE22, CELF4, CDO1, and ZNF662 ranged from 0.89 to 0.95 (Fig. 3B). The clinical performance is shown in Table 2 and Fig. 3C. CELF4 had the best sensitivity (96.0%) with specificity of 78.7% and OR of 88.4 (95% CI, 19.7–397.3). CDO1 had the best specificity (93.8%) with sensitivity of 82.0% and OR of 68.3 (95% CI, 22.8–204.7). We selected the best AUC values of three genes for gene combination analysis. Combined testing of BHLHE22, CELF4, and CDO1 had 91.8% sensitivity, 95.5% specificity, and a 236.3 times elevated risk (95% CI, 56.4–989.6) for any two positive tests. These results were similar for the detection of endometrial cancer at earlier stages (sensitivity, 89.5%; specificity, 95.5%; OR, 178.5, 95% CI, 42.2–754.9). To test the performance of these genes in detecting type II endometrial cancer, we detected the methylation of BHLHE22, CELF4, and CDO1 in scrapings from 14 type II endometrial cancer patients, including 5 patients with the serous type, 4 with a high-grade endometrioid type, 4 with the mucinous type, and 1 with the clear cell type (Table 3). The samples from all patients except for one with the serous type (13/14; 92.9%) exhibited hypermethylation in this three-gene panel.
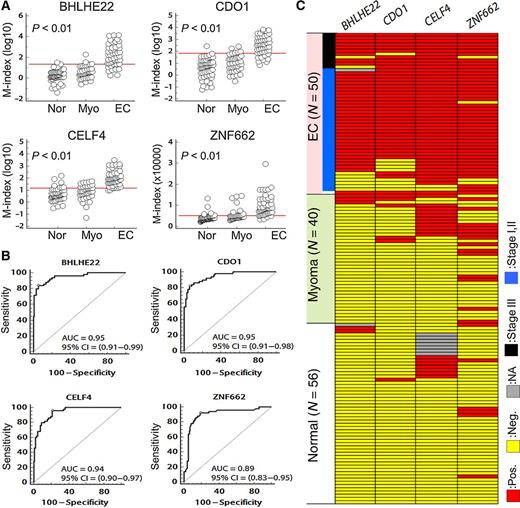
Validation of methylation levels in cervical scrapings. A, The dot plots show the distribution of methylation status for BHLHE22, CDO1, CELF4, and ZNF662. Using individual cervical scrapings, we validated that the DNA methylation status was higher in endometrial cancer samples than in samples of normal endometrium and myoma. The red lines show the cut-off values listed in Table 2. P values calculated by the Kruskal–Wallis test. B, Area under the receiver-operating characteristic curve for DNA methylation status of four candidate genes in total cervical scrapings. P values for all the analyses were <0.001 for comparison of the area equal 0.5. The optimal cut-off values were calculated by the maximal sensitivity and specificity, and listed in Table 2. C, The DNA-methylated phenotypes of BHLHE22, CDO1, CELF4, and ZNF662. Each row represents one specimen. According to the methylation status of BHLHE22, CDO1, and CELF4, of the 146 cervical scraping samples, almost all endometrial cancer samples and a few myoma samples were positive for methylation of at least two genes. In contrast, none of the normal endometrial tissue samples were positive for methylation of two genes. Positive methylation is indicated in red, negative methylation in yellow, and none available in gray. EC, endometrial cancer; N, case number; Pos., positive; Neg., negative; NA, none available.
Validation of methylation levels in cervical scrapings. A, The dot plots show the distribution of methylation status for BHLHE22, CDO1, CELF4, and ZNF662. Using individual cervical scrapings, we validated that the DNA methylation status was higher in endometrial cancer samples than in samples of normal endometrium and myoma. The red lines show the cut-off values listed in Table 2. P values calculated by the Kruskal–Wallis test. B, Area under the receiver-operating characteristic curve for DNA methylation status of four candidate genes in total cervical scrapings. P values for all the analyses were <0.001 for comparison of the area equal 0.5. The optimal cut-off values were calculated by the maximal sensitivity and specificity, and listed in Table 2. C, The DNA-methylated phenotypes of BHLHE22, CDO1, CELF4, and ZNF662. Each row represents one specimen. According to the methylation status of BHLHE22, CDO1, and CELF4, of the 146 cervical scraping samples, almost all endometrial cancer samples and a few myoma samples were positive for methylation of at least two genes. In contrast, none of the normal endometrial tissue samples were positive for methylation of two genes. Positive methylation is indicated in red, negative methylation in yellow, and none available in gray. EC, endometrial cancer; N, case number; Pos., positive; Neg., negative; NA, none available.
Performance of candidate genes and gene combinations in cervical scrapings
| Positive number | ||||||
|---|---|---|---|---|---|---|
| Gene name | Cut-off valuea | Case | Control | Se (%) | Sp (%) | ORb (95% CI) |
| Overall stage of EC | ||||||
| BHLHE22 | 21.9 | 41/49 | 6/95 | 83.7 | 93.7 | 76.0 (24.8–233.3) |
| CDO1 | 67.5 | 41/50 | 6/96 | 82.0 | 93.8 | 68.3 (22.8–204.7) |
| CELF4 | 14.0 | 48/50 | 19/89 | 96.0 | 78.7 | 88.4 (19.7–397.3) |
| ZNF662 | 5034.8 | 46/50 | 19/96 | 92.0 | 80 | 46.6 (14.9–145.5) |
| BHLHE22 + CDO1 + CELF4 | Any two Pos. | 45/49 | 4/88 | 91.8 | 95.5 | 236.3 (56.4–989.6) |
| Early stage of EC | ||||||
| BHLHE22 + CDO1 + CELF4 | Any two Pos. | 34/38 | 4/88 | 89.5 | 95.5 | 178.5 (42.2–754.9) |
| Positive number | ||||||
|---|---|---|---|---|---|---|
| Gene name | Cut-off valuea | Case | Control | Se (%) | Sp (%) | ORb (95% CI) |
| Overall stage of EC | ||||||
| BHLHE22 | 21.9 | 41/49 | 6/95 | 83.7 | 93.7 | 76.0 (24.8–233.3) |
| CDO1 | 67.5 | 41/50 | 6/96 | 82.0 | 93.8 | 68.3 (22.8–204.7) |
| CELF4 | 14.0 | 48/50 | 19/89 | 96.0 | 78.7 | 88.4 (19.7–397.3) |
| ZNF662 | 5034.8 | 46/50 | 19/96 | 92.0 | 80 | 46.6 (14.9–145.5) |
| BHLHE22 + CDO1 + CELF4 | Any two Pos. | 45/49 | 4/88 | 91.8 | 95.5 | 236.3 (56.4–989.6) |
| Early stage of EC | ||||||
| BHLHE22 + CDO1 + CELF4 | Any two Pos. | 34/38 | 4/88 | 89.5 | 95.5 | 178.5 (42.2–754.9) |
Abbreviations: CI, confidence interval; EC, endometrial cancer; OR, odds ratio; Pos., positive; Se, sensitivity; Sp, specificity; Early stage includes stage I and stage II.
aDichotomization of methylation levels is based on the distribution in endometrial cancer versus noncarcinomatous participants using the Youden's method.
bOR is calculated using univariate logistic regression. We computed the achieved powers (1-β) >0.99 using post hoc pairwise Fisher exact tests. In combination with gene analysis, the missing data of one gene were excluded.
Characteristics and methylation status of candidate genes in cervical scrapings from type II endometrial cancer
| Participant no. | Age (y) | Histology | Stage | Grade | M-index of BHLHE22 | M-index of CELF4 | M-index of CDO1 | Any two positivea |
|---|---|---|---|---|---|---|---|---|
| GYCA-03-2001 | 57 | Ser | 1A | G3 | 2579.2 | 577.1 | 1533.6 | Yes |
| GYCA-03-2002 | 59 | En | 3C | G3 | 2.0 | 14.8 | 142.3 | Yes |
| GYCA-03-2003 | 64 | Ser | 3C | G2 | 153.0 | 37.7 | 559.4 | Yes |
| GYCA-03-2004 | 54 | En | 1B | G3 | 365.2 | 14.2 | 320.2 | Yes |
| GYCA-03-2005 | 58 | En | 3A | G3 | 810.5 | 106.0 | 796.6 | Yes |
| GYCA-03-2006 | 43 | En | 3A | G3 | 13707.8 | 563.3 | 3110.0 | Yes |
| GYCA-03-2007 | 60 | Mu | 2 | G1 | 9.0 | 340.8 | 1353.7 | Yes |
| GYCA-03-2008 | 73 | CC | 4B | G3 | 311.4 | 82.3 | 1725.4 | Yes |
| GYCA-03-2009 | 74 | Ser | 1A | G3 | 37.9 | 6.4 | 1220.0 | Yes |
| GYCA-03-2010b | 45 | Ser | 1 | 5.9 | 10.4 | 6.4 | No | |
| GYCA-03-2011 | 68 | Ser | 2 | G3 | 2044.8 | 10.8 | 2102.2 | Yes |
| GYCA-03-2012 | 56 | Mu | 1B | G1 | 240.1 | 85.8 | 172.8 | Yes |
| GYCA-03-2013 | 55 | Mu | 1A | G1 | 53.4 | 22.4 | 12.6 | Yes |
| GYCA-03-2014 | 57 | Mu | 1A | G2 | 375.5 | 172.2 | 217.2 | Yes |
| Participant no. | Age (y) | Histology | Stage | Grade | M-index of BHLHE22 | M-index of CELF4 | M-index of CDO1 | Any two positivea |
|---|---|---|---|---|---|---|---|---|
| GYCA-03-2001 | 57 | Ser | 1A | G3 | 2579.2 | 577.1 | 1533.6 | Yes |
| GYCA-03-2002 | 59 | En | 3C | G3 | 2.0 | 14.8 | 142.3 | Yes |
| GYCA-03-2003 | 64 | Ser | 3C | G2 | 153.0 | 37.7 | 559.4 | Yes |
| GYCA-03-2004 | 54 | En | 1B | G3 | 365.2 | 14.2 | 320.2 | Yes |
| GYCA-03-2005 | 58 | En | 3A | G3 | 810.5 | 106.0 | 796.6 | Yes |
| GYCA-03-2006 | 43 | En | 3A | G3 | 13707.8 | 563.3 | 3110.0 | Yes |
| GYCA-03-2007 | 60 | Mu | 2 | G1 | 9.0 | 340.8 | 1353.7 | Yes |
| GYCA-03-2008 | 73 | CC | 4B | G3 | 311.4 | 82.3 | 1725.4 | Yes |
| GYCA-03-2009 | 74 | Ser | 1A | G3 | 37.9 | 6.4 | 1220.0 | Yes |
| GYCA-03-2010b | 45 | Ser | 1 | 5.9 | 10.4 | 6.4 | No | |
| GYCA-03-2011 | 68 | Ser | 2 | G3 | 2044.8 | 10.8 | 2102.2 | Yes |
| GYCA-03-2012 | 56 | Mu | 1B | G1 | 240.1 | 85.8 | 172.8 | Yes |
| GYCA-03-2013 | 55 | Mu | 1A | G1 | 53.4 | 22.4 | 12.6 | Yes |
| GYCA-03-2014 | 57 | Mu | 1A | G2 | 375.5 | 172.2 | 217.2 | Yes |
Abbreviations: CC, clear cell; En, endometrioid; G, grade; M-index, methylation index; Mu, mucinous; Ser, serous. Stage is following the International Federation of Gynecology and Obstetrics principle.
aThe cut-off values based on the results calculated in endometrioid type. Cut-off values of M-index are 21.9, 14.0, and 69.5 for BHLHE22, CELF4, and CDO1, respectively. Bold italics show that the M-index is higher than cut-off values.
bThe patient was diagnosed as serous type of endometrial cancer by dilatation and curettage in an outside clinic, but the hysterectomy specimens disclosed complex atypical hyperplasia only in our hospital.
Discussion
The present study used a methylomics approach to identify methylated genes in endometrial cancer samples and verified the feasibility using the methylation profile of cervical scrapings to detect endometrial cancer. Several observations support the validity of our results. In the list of 180 genes, CDH13, COL14A1, HAAO, HAND2, HS3ST2, HTR1B, NPY, and ZNF177, have been reported in endometrial cancer samples in independent studies (17–19, 21, 27). Because different cancers may have common pathways, the hypermethylated genes identified in our gene list might also be methylated in other cancers, and ADHFE1, CDO1, DPP6, EFS, FEZF2, GHSR, GRIA4, NEFL, PARP15, PAX6, SSTR1, and TRH have been reported (28–40). We have identified for the first time hypermethylation of BHLHE22, CCDC140, CELF4, GALNTL6, ZNF334, and ZNF662 in cancers. The functions of these genes in cancer biology remain unknown. Hypermethylation of BHLHE22, CDO1, and CELF4 in cervical scrapings had the best performance for detecting endometrial cancer. BHLHE22 (also named BHLHB5) encodes a helix–loop–helix transcription factor, which suggests that it is involved in cell differentiation (41, 42). CDO1 encodes a protein responsible for decreasing cytotoxicity caused by overproduction of cysteine and is associated with drug sensitization by increasing ROS (43, 44). CELF4 (also named BRUNOL4) encodes a protein with three domains for binding RNA-recognition motif that may be involved in RNA alternative splicing during specific cell development (45–47). However, the functions of CDO1, CELF4, and BHLHE22 in tumorigenesis are unknown. Further studies are warranted to understand the roles of these genes in endometrial cancer.
Recent progress in the detection of endometrial cancer and ovarian cancer using genetic mutations in cervical scrapings or uterine lavage are encouraging (14, 48, 49). Kinde and colleagues (14) used a sophisticated “PapGene” platform to interrogate 46 regions of 12 genes in 14 liquid-based cytology materials from endometrial cancer patients. Seven of the 12 gene mutations could be detected, and the detection rate was 100% if any mutation was counted (14). Maritschnegg and colleagues (49) use uterine lavage as materials for genetic testing of 16 genes using next-generation sequencing or singleplex digital droplet polymerase chain reaction (PCR). All five endometrial cancers could be detected. Although promising, there are limitations in many aspects of these methods and improvements are needed. Mutation-based screening is thought to be exquisitely specific because normal cells should not contain mutations. In the study by Kinde and colleagues, there were only 14 healthy controls, and the specificity is a major concern for clinical applications. The study of uterine lavage samples by Maritschnegg and colleagues (49) showed that 29.6% of patients with a benign gynecological condition, including uterine myomas, ovarian cysts, or ovarian teratomas, harbored a genetic mutation. Further clarification of these mutations as markers for screening of various benign conditions is warranted. For screening of endometrial cancer, uterine lavage is not an easy or standard procedure in the outpatient clinic setting, especially for postmenopausal women. Suction curettage is a more direct method for obtaining tissue samples from patients when there is a high suspicion of endometrial cancer. This invasive and inconvenient method may be reserved for specific situations such as ovarian cancer screening in high-risk populations.
DNA methylation testing of cervical scrapings or self-collected vaginal lavage could be a better method if the markers are sufficiently sensitive and specific. Candidate DNA methylation testing of CADM1/MAL/miR124-2 in cervical scrapings was reported to have 76.2% sensitivity for endometrial cancer if any positive result of the three genes was counted. However, the panel was developed for detection of cervical lesions. The broader scope of its application for cervical cancer to endometrial cancer does not have satisfactory sensitivity or specificity. Genome-wide attempts to discover new DNA methylation markers have used a limited array containing 807 genes known to be relevant to cancer. Wentzensen and colleagues (18) found eight genes that were significantly hypermethylated in endometrial cancer tissues and tested their ability to discriminate endometrial cancer in endometrial brush materials. The ORs ranged from 3.44 (95% CI, 1.33–8.91) for ASCL2 to 18.61 (95% CI, 5.50–62.97) for HTR1B in tissue samples and from 3.57 (95% CI, 1.35–9.47) for HTR1B to 10.92 (95% CI, 3.23–36.91) for ADCYAP1 in endometrial brush materials. The estimated AUC was 0.85 with a sensitivity >90% and specificity >50% in endometrial brush materials. They did not test these genes in cervical scrapings or assess the effects of common benign tumors such as uterine myoma.
Recently, Bakkum-Gamez and colleagues (17) reported the feasibility of using a more minimally invasive biospecimen sampling technique such as a vaginal tampon coupled with testing of methylation markers for detection of endometrial cancer. They tested 12 genes, including the eight genes identified by Wentzensen and colleagues described above, by bisulfite pyrosequencing to evaluate DNA methylation in vagina tampons for 38 endometrial cancers and 28 benign endometrium samples. HTR1B had the best AUC of 0.82 for distinguishing benign from malignant endometrial tumors (17). Our study used a more comprehensive methylomics approach for detecting endometrioid endometrial cancer and identified new genes in both tissues and cervical scrapings. In this application-orientated approach, BHLHE22, CDO1, and CELF4 methylation of cervical scrapings had a sensitivity of 91.8%, specificity of 95.5%, and OR of 236.3 (95% CI, 56.4–989.6) for endometrial cancer detection despite interference from benign uterine tumors. This is the best performance of molecular biomarkers for endometrial cancer detection reported in the literature to date. The combination of these new biomarkers and refined collection method may be useful in the widespread deployment of endometrial cancer screening, especially for the high-risk populations such as women with postmenopausal bleeding, high BMI (≥30 kg/m2), family history of hereditary nonpolyposis colorectal cancer (HNPCC), and breast cancer patients treated with estrogen therapy (50). The extent to which the changes in methylation of type I endometrial cancer can be applied to type II endometrial cancer remains a concern. In the TCGA report, uterine serous tumors and 25% of high-grade endometrioid tumors had few DNA methylation changes despite extensive copy number alterations and frequent TP53 mutations. However, the present study revealed that BHLHE22, CDO1, and CELF4 derived from type I endometrial cancer tissues were also frequently hypermethylated in cervical scrapings of type II endometrial cancer, including serous, high-grade endometrioid, mucinous, and clear cell types, although the case numbers were limited. The performance of BHLHE22, CDO1, and CELF4 methylation in detecting endometrial cancer should be validated in studies with larger sample sizes and including hyperplasia. Further exploration of the methylomics of type II endometrial cancer is warranted. In addition, we found methylated changes in cervical scrapings from patients with benign uterine myoma. Because uterine myoma is a common benign tumor in women, markers for screening purposes should avoid interference caused by this benign tumor. The mechanism responsible for the similar methylation changes in endometrial cancer and uterine myoma remains to be elucidated.
In summary, using an epigenomics approach, we identified promising DNA methylation biomarkers for endometrial cancer screening. These sensitive molecular biomarkers expand the scope of the Pap smear, which has been used for more than half a century and is used widely in developed countries with a high prevalence of uterine cancer. Future clinical trials of BHLHE22, CDO1, and CELF4 methylation in women with postmenopausal bleeding and in high-risk populations are planned.
Disclosure of Potential Conflicts of Interest
H. Lai is a co-inventor of three patents on methods for cancer diagnosis and prognosis, a method for Tumor detection, and a method for tumor detection II, which are owned by Taipei Medical University.
Authors' Contributions
Conception and design: H.-C. Lai
Development of methodology: R.-L. Huang, T.H.-M. Huang, H.-C. Lai
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): R.-L. Huang, P.-H. Su, T.-I. Wu, Y.-T. Hsu, H.-C. Wang, H.-C. Lai
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): R.-L. Huang, Y.-P. Liao, W.-Y. Lin, H.-C. Lai
Writing, review, and/or revision of the manuscript: Y.-P. Liao, T.-I. Wu, Y.-T. Hsu, H.-C. Lai
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Y.-C. Weng, Y.-C. Ou, T.H.-M. Huang, H.-C. Lai
Study supervision: T.-I. Wu, H.-C. Wang, H.-C. Lai
Acknowledgments
We thank Rohit R. Jadhav of Tim H.M. Huang's Laboratory for collecting the MethylCap-seq data and providing computation services. Additionally, we also thank the Research Information Section, Office of Information Technology, Taipei Medical University for providing computation services.
Grant Support
This work was supported by the Ministry of Health and Welfare grant MOHW105-TDU-PB-212-000007, the Shuang Ho Hospital-Taipei Medical University grant 103TMU-SHH-11, the Taipei Medical University grant TMUTOP103005‐1, the National Health Research Institutes grant NHRI-EX105-10406BI, and the Ministry of Science and Technology grant NSC102-2628-B-038-010-MY3.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.