


Abstract
Purpose: O6-alkylguanine-DNA alkyltransferase (AGAT) is modulated by methylating agents, which, in turn, abrogates nitrosourea resistance in preclinical studies. The feasibility of administering various sequences of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and temozolomide (TEM) in patients with advanced solid neoplasms was evaluated in this Phase I and pharmacological study to assess this premise in the clinical setting. The study also sought to determine the maximum tolerated dose (MTD) levels of BCNU and TEM as a function of Seq, to characterize the pharmacokinetic (PK) behavior of TEM administered both before and after BCNU, assess AGAT fluctuations in peripheral blood mononuclear cells (PBMCs), and seek preliminary evidence of anticancer activity.
Experimental Design: Sixty-three patients were randomized to receive treatment with oral TEM daily on days 1–5 and BCNU administered i.v., either on day 1 before TEM [Sequence (Seq) B→T] or day 5 after TEM (Seq T→B). Treatment was repeated every 6 weeks. Blood sampling for PK studies was performed on both days 1 and 5 of course one. PBMCs were sampled to evaluate major sequence-dependent effects on AGAT levels.
Results: Neutropenia and thrombocytopenia were the principal dose-limiting toxicities of the BCNU/TEM regimen. These effects were more prominent in patients receiving Seq T→B, resulting in a much lower MTD of 80/100 mg/m2/day compared with 150/110 mg/m2/day for Seq B→T. Notable antitumor activity was observed in patients with glioblastoma multiforme, sarcoma, and ovarian carcinoma. No sequence-dependent PK effects were noted to account for sequence-dependent toxicological effects. At the MTD level, AGAT activity in PBMCs decreased 3-fold, on average, and AGAT fluctuations did not appear to be sequence-dependent.
Conclusions: The principal toxicities of the BCNU/TEM regimen were neutropenia and thrombocytopenia, which were consistent and predictable, albeit sequence-dependent. Seq T→B was substantially more myelosuppressive, resulting in disparate MTDs and dose levels recommended for subsequent disease-directed evaluations (150/110 and 80/100 mg/m2/day for Seq B→T and T→B, respectively). Sequence-dependent differences in TEM PK do not account for this clinically relevant magnitude of sequence-dependent toxicity. The characteristics of the myelosuppressive effects of BCNU/TEM, the paucity of severe nonhematological toxicities, and antitumor activity at tolerable doses warrant disease-directed evaluations on this schedule.
INTRODUCTION
1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU), a bifunctional alkylating agent with a unique mechanism of action and antitumor spectrum, including utility in the therapy of brain neoplasms, lymphomas, and melanoma, is metabolized to chloroethyl moieties that form lethal DNA cross-links unless repaired and removed (1, 2, 3). Whereas DNA—DNA-interstrand cross-link formation is relatively slow, DNA-protein cross-links form rapidly (1), resulting in prolonged inhibition of DNA synthesis (2). However, O6-alkylguanine-DNA alkyltransferase (AGAT) repairs nitrosourea-induced DNA damage before potentially lethal cross-links are formed by removing alkyl adducts at the O6-position of guanine from its substrate O6-chloroethylguanine (3, 4, 5, 6). This process, commonly referred to as “suicide repair,” irreversibly inactivates AGAT, and its activity must be regenerated by de novo synthesis (3, 4).
The degree of in vitro and in vivo tumor resistance to chloroethylnitrosoureas is directly related to intracellular AGAT expression (3, 5, 6, 7, 8). Methyl excision repair negative (Mer−) tumor cells that lack AGAT are relatively incapable of repairing alkylation damage, rendering them sensitive to chloroethylnitrosoureas, whereas Mer-positive (Mer+) cells that express AGAT are relatively resistant to alkylating agents because of their ability to remove monoadducts from DNA and to repair alkylation damage (9). The strong relationship between AGAT expression and BCNU resistance suggests that it may be reasonable to pursue therapeutic strategies that modulate nitrosourea resistance by depleting cellular AGAT. One such strategy involves concurrent administration of other chemotherapeutics that deplete AGAT. For example, the methylating agents streptozotocin (STZ) and dacarbazine (DTIC) form O6-methylguanine (MeG), which, if unrepaired, results in aberrant mismatch repair (MMR) followed by apoptosis via the intrinsic pathway (10, 11, 12). In fact, experimental data indicate that the efficiency of STZ and DTIC methylating properties is directly related to AGAT activity that counters the apoptosis-initiating signal (11, 12, 13). Repair of the MeG adduct requires transfer of the methyl group to AGAT, which ultimately restores DNA to its intact state and depletes AGAT (14, 15, 16). It has been proposed that depletion of tumor AGAT activity by methylating agents may enhance tumor susceptibility to BCNU-induced cytotoxicity (17).
The administration of single-agent STZ and DTIC depletes AGAT in peripheral blood mononuclear cells (PBMCs) and other normal tissues in vivo (18, 19). After treatment with DTIC, AGAT activity progressively decreases; this effect plateaus with increasing doses, most likely because of the saturation of metabolic activation of DTIC (19). In studies evaluating combinations of methylating agents and nitrosourea (20, 21, 22), treatment with STZ before BCNU results in more substantial decrements in AGAT activity in both PBMCs and tumors than does single agent treatment (22). However, the use of DTIC in strategies aimed at modulating BCNU resistance and enhancing combined drug activity through AGAT depletion has been precluded by the high interpatient variability in the metabolic activation of DTIC (23) and its limited spectrum of activity (24, 25, 26).
The oral imidazotetrazine derivative TEM has many characteristics that render it more advantageous as a single agent and in combination with other therapeutic modalities. These features include rapid and broad tissue distribution, stability under acidic conditions that confers complete oral absorption, and the ability to penetrate into the central nervous system (27). Similar to DTIC, TEM is a prodrug of 5-(3-methyltriazen-1-yl)imidazole-4-carboximide (28, 29), the active species that methylates DNA (30). However, metabolic activation of DTIC is exclusively by hepatic p450 metabolism, which is highly unpredictable and variable, whereas the metabolic activation of TEM occurs spontaneously and completely at physiological pH (29, 30). Furthermore, TEM has a unique range of clinical activity including recurrent high-grade astrocytoma, melanoma, and mycosis fungoides (29, 31, 32, 33, 34). TEM also has prominent activity in Mer+ human brain tumor xenografts resistant to BCNU (35).
Early preclinical evaluations of BCNU and TEM (BCNU/TEM) combination regimens sought to establish the optimal sequence of drug administration, but the results have been conflicting (36, 37). In nude mice bearing human SF-295 glioblastoma xenografts, tumor growth inhibition was significantly greater when BCNU dosing preceded TEM (Seq B→T) compared with the alternate sequence (Seq T→B). Interestingly, the superior sequence, Seq B→T, also resulted in less toxicity; both BCNU/TEM regimens demonstrated greater antitumor activity compared with each single agent (36). However, the results of experiments involving HT-29 colon carcinoma indicated that the administration of TEM or DTIC before BCNU greatly enhanced cytotoxicity compared with the alternate sequence, but the degree of enhancement was greater in vitro than in an HT-29 xenograft model, presumably because of enhanced toxicity (37).
The aforementioned studies served as a rationale for clinical evaluation of the feasibility of administering sequence iterations of BCNU/TEM to patients with advanced solid malignancies. The principal objectives of this study were to: characterize the principal toxicities and determine the maximum tolerated dose (MTD) levels of BCNU administered as a brief (i.v.) infusion either before treatment with oral TEM daily for 5 days (Seq B→T) or after 5 days of TEM treatment (Seq T→B) in patients with advanced solid malignancies; to determine the modulatory effects, if any, of BCNU on TEM exposure and clearance; to detect and characterize the sequence-dependent effects of BCNU/TEM on AGAT activity in PBMCs; and seek preliminary evidence of antineoplastic activity.
PATIENTS AND METHODS
Eligibility.
Patients with histologically confirmed advanced solid malignancies refractory to conventional therapy or for whom no effective therapy existed were candidates for this study. Eligibility criteria also included: age ≥18 years; a Southwestern Oncology Group (SWOG) performance status ≤2; life-expectancy ≥3 months; no surgery or chemotherapy within 4 weeks of treatment; radiation therapy ≤20% of the bone marrow; no prior nitrosourea or mitomycin C; adequate hematopoietic [absolute neutrophil count (ANC) ≥1500/μL, platelet count ≥100,000/μL, hemoglobin ≥9.0 g/dl], hepatic (total bilirubin within normal limits, aspartate amino transaminase ≤2 times institutional normal upper limits), and renal (serum creatinine within normal limits or calculated creatinine clearance ≥70 ml/min according to the method of Cockcroft and Gault; Ref. 38) functions; no prior resection of the stomach or small intestines; and no medical condition that could potentially interfere with oral drug administration or limit full compliance with the study. Patients gave written informed consent according to federal and institutional guidelines before treatment.
Dosage and Drug Administration.
The starting doses of BCNU and TEM, 55 mg/m2 and 35 mg/m2/day for 5 days, respectively, were selected because they produce minimal toxicity as single agents (36, 39, 40). A modified Fibonacci scheme, in which both BCNU and TEM were escalated successively in each cohort of new patients from an initial dose level of BCNU/TEM (mg/m2) 55/35 to 75/55, 100/80, 150/110, and 200/150, was used to guide dose escalation. If required, further dose escalation of both agents would proceed at 20% increments. TEM was administered as a single oral (p.o.) daily dose for 5 days; fasting was required 4 h (h) before and 2 h after treatment. BCNU was administered i.v. on day 1, 2 h before, and on day 5, 2 h after TEM on Seq B→T and T→B, respectively. Treatment was repeated every 6 weeks. Cumulative BCNU doses up to 1400 mg/m2 were permitted. Dose reduction to the previous dose level was permitted for patients experiencing dose-limiting toxicities (DLTs). If a patient experienced DLT at the first dose level, then retreatment criteria permitted a 25% dose reduction of both agents. At least three new patients were treated at each escalated dose level. Intrapatient dose escalation was permitted if a patient had at least stable disease and if two patients had completed treatment at the next higher dose without DLT. If one of three new patients at any dose level experienced DLT, then a maximum of six new patients were treated at that dose level. The MTD or recommended Phase II dose for each sequence was defined as the highest dose level that induced DLT in less than two of six new patients treated with each sequence. DLT was defined during the first course as grade ≥3 nonhematological toxicity (except nausea and vomiting associated with suboptimal pharmacological prophylaxis and/or management); grade 3 transaminitis >2 weeks; ANC <500/μl lasting more than 5 days and/or associated with fever; hemoglobin <6.5 g/dl; and platelet count <25,000/μl. Toxicity was graded according to the National Cancer Institute Common Toxicity Criteria (Version 1.0; Ref. 41).
TEM was supplied as 20 and 100 mg capsules by the National Cancer Institute (Bethesda, MD). The calculated dose of TEM was rounded to the nearest 20 mg to accommodate capsule strength. BCNU (Carmustine; Bristol-Myers Squibb, Princeton, NJ) was supplied in 5-ml vials containing 100 mg BCNU (lyophilized) and 3 ml dehydrated alcohol, USP. After reconstitution, the solution was diluted in 500 ml of 5% dextrose solution and was infused over 1 h. Patients routinely received dexamethasone 10 mg i.v. or p.o. and a 5-hydroxytryptamine-3 antagonist i.v. or p.o. 30-min before treatment on days 1–5. The use of hematopoietic growth factor support was not permitted.
Randomization Process.
The first patient in each dose level was randomized to treatment with BCNU/TEM on either Seq B→T or Seq T→B; the second patient was treated with the reverse sequence. After treatment of the first two patients, additional treatment assignments were determined by randomization. If one Seq cohort was open to accrual, then the next patient was treated with that Seq.
Pretreatment and Follow-Up Studies.
Histories, physical examinations, performance status assessments, and routine laboratory studies were performed pretreatment and weekly. A chest X-ray and electrocardiogram were obtained pretreatment and before each course. Pulmonary function tests with diffusion capacity were performed pretreatment, at study exit, and when cumulative BCNU dose exceeded 800 mg/m2. A bone marrow biopsy was required in any patient experiencing >10 days of grade 4 neutropenia or thrombocytopenia. Tumor measurements were performed pretreatment and after every other course. Patients were able to continue treatment if they did not develop progressive disease. A complete response was scored if there was disappearance of all active disease on two measurements separated by a minimum period of 4 weeks. A partial response required at least a 50% reduction in the sum of the product of the bidimensional measurements of all documented lesions separated by at least 4 weeks. Progressive disease was defined as at least a 25% increase in the sum of the products of the bidimensional measurements of all measurable disease.
Plasma Sampling and Assay.
Five-ml blood samples were collected in prechilled heparinized (nonseparator) tubes immediately before TEM administration and at 10, 20, 30, 60, and 90 min and 2, 4, 6, 8, 10, and 24 h posttreatment on days 1 and 5 of the first course. The blood was centrifuged for 10 min at 4°C. A 1-ml aliquot of plasma was transferred to polyethylene tubes containing 0.1 ml of 1.0 n HCl and was stored at −20°C or colder until analysis by high-performance liquid chromatography.
A modified high-performance liquid chromatography procedure previously reported by Newlands et al. (42) was used to determine TEM plasma concentrations. Analytical standards of TEM and the internal standard, ethazalastone, were obtained from the National Cancer Institute. Briefly, a sample of 500 μl of acidified patient plasma or quality control samples containing 20 μl (400 ng) of internal standard was double extracted with ethylacetate and evaporated to dryness under nitrogen. The percentage recovery for both TEM and internal standard was greater than 70%. The residue was redissolved with 300 μl of mobile phase (acetonitrile/0.1% acetic acid), was vortexed, and was filtered; and 75 μl was injected onto an high-performance liquid chromatography system using an autosampler. The system consisted of a Waters Sperisorb S5 ODS2 column (4.6 × 250 mm; Milford, MA) preceded by a Novapak C18 precolumn (Waters) with the mobile phase pumped at a flow rate of 1 ml/min and UV detection set at 316 nm. Chromatograms and peak areas were stored and analyzed on a Waters Maxima data acquisition software system. The assay was linear over the range of 0.01–50 μg/ml using a weighting factor of 1/concentration. The coefficients of determination were greater than 0.999 for all standard curves.
PK Analysis.
The pharmacokinetic (PK) parameters for TEM were calculated using model-independent methods (43). Peak plasma concentrations (Cpmax) and the time at which they occurred (Tmax) were determined by inspection of the individual patient’s concentration-time data. Elimination rate constants were estimated by linear regression of the last two to three data points on the terminal log linear portion of the concentration-time curves. Terminal half-lives (t1/2) were calculated by dividing 0.693 by the terminal elimination rate constant. The area under the concentration-time curve (AUC) was determined by the trapezoidal method up to the last datum point and extrapolated to infinity. Clearance (CL/F) was estimated by dividing TEM dose (mg/m2) by AUC. Differences in PK parameters between the two dosing sequences were assessed using a paired t test with a P ≤ 0.05 as the a priori level of significance.
Pharmacodynamic Analysis.
The relationships between TEM systemic exposure and toxicity were explored. Total TEM exposure, calculated as the mean AUC values on days 1 and 5 multiplied by 5, was related to grades of neutropenia, thrombocytopenia, and transaminitis. The percentage decrement in blood cell counts was calculated as follows: 100% × [(pretreatment counts − nadir counts)/pretreatment counts]. The magnitude of the percentage decrement in blood cells was measured by using the sigmoid Emax model of drug effect (PK Analyst, Version 1.0, Model 25; MicroMath, Salt Lake City, UT).
Statistical Analysis.
A two-way ANOVA was used to analyze the effects of BCNU/TEM sequence and dose on ANC and platelet count nadirs. The main effects for dose (values formatted as BCNU/TEM dose levels: 50/35, 75/55, 100/80, 150/110, 200/150, and 240/180) and sequence (B→T and T→B), as well as dose-sequence interactions were included. Tukey’s Studentized Range (HSD) test was used to determine which pairs of dose levels differed significantly. Linear regression was used to assess the relationship between TEM dose and peak concentrations and AUC values. Differences in PK parameters between the dosing sequences were assessed using a paired t test with a P ≤ 0.05 as the a priori level of significance. Spearman’s rank correlation was used to determine the relationship between TEM exposure and toxicity.
AGAT Analysis.
At the MTD, 60-ml blood was sampled pretreatment and at 24, 48, 72, 96, and 120 h after TEM administration in three patients from each sequence. PBMCs were isolated by density gradient separation and incubated with [3H]methyl DNA in HEPES buffer in a total volume of 300 μl for 60 min at 37°C, as described previously (44). The reaction mixture contained excess substrate DNA to determine total AGAT activity. The reaction was stopped with 7.5% trichloroacetic acid at 4°C for 30 min. The precipitate was collected by centrifugation at 13,000 rpm for 2 min and was washed with 300 μl of 80% ethanol. Methylated purines were liberated from precipitated DNA during hydrolysis with 150 μl of 0.1N HCl at 80°C for 1-h. O6-[3H]methylguanine and N7-[3H]methylguanine, which were constant during incubation and served as the internal standard, were separated in supernatant by reverse-phase high-performance liquid chromatography and were quantitated by liquid scintillation counting. Radioactivity (dpm) in each peak (N7-methylguanine and N6-methylguanine), derived from the substrate, ranged from 350 to 800 dpm for the MeG peak (21–48 fmol) and were 12-fold higher for N7-methylguanine. Peak values were corrected for background radioactivity based on positive and negative controls. AGAT activity was expressed as fmol of MeG removed/μg DNA. Single, duplicate, and triplicate assays were performed on 7, 23, and 5 samples, respectively, as protein quantity permitted. The limit of detection, which was based on the linear portion of the assay, was defined as AGAT activity (fmol/μg DNA) that corresponded to 12% of the MeG present in the substrate [3H]methyl DNA used in each assay. The limit of detection value was 0.05 fmol/μg of sample DNA.
RESULTS
General.
Sixty-three patients, whose pertinent characteristics are displayed in Table 1, received 148 courses of BCNU/TEM sequences through seven dose levels (Table 2). The first courses of three patients were not evaluable because of rapidly progressive malignant disease. The numbers of new and total patients and total courses at each dose level, as well as the cumulative incidence of DLT, are shown in Table 3. Thirty-five and 28 patients received 90 courses of Seq B→T (median, 2; range, 1–16) and 63 courses of Seq T→B (median, 2; range, 1–5, respectively).
Patient accrual to both sequence cohorts at the first and second dose levels was expanded because of dose-limiting transaminitis and two patient deaths due to progressive disease and myelosuppression. Further dose escalation on Seq B→T proceeded without DLT to 100/80, 150/110, and 200/150 mg/m2. Although the first patient treated at the 240/180 mg/m2 dose level did not experience DLT, subsequent patients were treated at the next lower dose level, 200/150 mg/m2, based on concern that repetitive treatment at the higher dose would not be feasible. Indeed, the incidence of DLT was demonstrated to be unacceptably high in patients treated with Seq B→T at the 200/150 dose level because three of nine new patients developed DLT in the first course. In contrast, the next lower dose level, 150/110 mg/m2, was determined to be the MTD for Seq B→T because no DLT was experienced by seven new patients.
After grades 3 and 4 thrombocytopenia were noted in courses 1 and 2, respectively, in the first patient treated on Seq T→B 80/100 mg/m2, the cohort size was increased, because of concerns about the tolerability of cumulative dosing. However, DLT was not experienced by any of the next five patients, permitting further dose escalation. In the course of dose escalation, one patient was inadvertently treated on Seq T→B 150/200 mg/m2 and experienced severe dose-limiting neutropenia and thrombocytopenia. Furthermore, the incidence of DLT at the previous dose level, Seq T→B 110/150, was determined to be unacceptably high because DLT occurred in two of five new patients. Therefore, further patient accrual proceeded on Seq T→B at 80/100 mg/m2, which was determined to be the MTD, with only one DLT occurring in the first courses of 10 new patients.
Hematological Toxicity.
The rates of relevant hematological toxicities as functions of the total numbers of patients and courses at each BCNU/TEM sequence cohort are displayed in Tables 2 and 3, respectively. The cumulative rate of dose-limiting hematological toxicities, which principally were comprised by grade 4 thrombocytopenia and neutropenia, were unacceptably high for patients treated with Seq B→T and T→B at 200/150 and 110/150 mg/m2, respectively. Overall, the incidences of severe hematological sequelae were felt to be acceptable, with severe neutropenia complicated by fever occurring in one (2%) patient, grade 4 neutropenia >5 days experienced by five (8%) patients, grade 4 thrombocytopenia occurring in eight (13%) patients, and platelet transfusions required in 13 (9%) of 145 courses. One course of thrombocytopenia was complicated by a self-limited gastrointestinal hemorrhage. Treatment delay due to unresolved neutropenia or thrombocytopenia lasting from 1 to 4 weeks was required in 10 (7%) courses.
A two-way ANOVA showed no statistically significant effect on ANC nadir for either the dose-sequence interaction or sequence (P = 0.2701 and 0.9501, respectively). The main effect for dose was overall significant at the 5% level (P < 0.0001). Similarly, there was no statistically significant effect on platelet count nadir for either the dose-sequence interaction or sequence (P = 0.1157 and 0.5356, respectively). The main effect for dose was overall significant at the 5% level (P < 0.0001).
Severe effects of BCNU/TEM on RBCs were rarely observed. Grades 3 and 4 anemia were noted in six (4%) and three (2%) courses, respectively. Only one dose-limiting event occurred during a first course. RBC transfusions were performed in 15 (10%) courses. Although there was a small number of serious events, effects on RBCs did not appear to be related to drug administration sequence.
Nonhematological Toxicity.
The nonhematological toxicities associated with administration of BCNU/TEM sequences were generally mild to moderate in severity (Table 4). No relationships between the incidence and severity of any of the nonhematological effects and the sequence of BCNU/TEM administration were apparent. Nausea, vomiting and fatigue were the most common nonhematological toxicities. Nausea and vomiting generally occurred in the peritreatment period and were rarely grade 3–4 in severity, possibly because of routine premedication with corticosteroids and 5-hydroxytryptamine-3 antagonists, and never precluded TEM administration. On rare occasions, nausea and vomiting required use of prochloroperazine, 5-hydroxytryptamine-3 antagonists, and/or corticosteroids.
Fifteen (25%) of the patients experienced a treatment-related transaminitis typically on day 8 that usually resolved within 2 weeks. Grades 3 and 4 transaminitis were observed in two and one patient, respectively, none of whom had hepatic metastases. The first patient experienced grade 3 transaminitis in each of 14 courses (Seq B→T 75/55 mg/m2). Transaminase values peaked on approximately day 8 and resolved to pretreatment levels before the next scheduled treatment. The second patient treated with Seq T→B 35/50 mg/m2 experienced transient grade 4 transaminitis that did not recur after dose reduction to 26/37 mg/m2. Transaminitis in the third patient peaked to grade 3 on day 30 after treatment with Seq B→T 50/35 mg/m2 but resolved 14 days later; the development of rapidly progressive malignant disease precluded retreatment.
Two patients experienced progressive dyspnea associated with decrements in pulmonary function tests in the setting of progressive pulmonary metastases. Review of pulmonary function test results for the entire population revealed no consistent changes as a function of dose level and cumulative therapy. Other mild to moderate (grade 1–2) nonhematological events included constipation, anorexia, diarrhea, malaise, mucositis, rash, headache, and alkaline phosphatase elevations. However, these effects were noted across the entire dosing range, and definite temporal relationships could not be discerned for any of these potential toxicities, indicating that the underlying malignant process may have been contributory.
Antitumor Activity.
Eleven patients experienced objective antitumor activity or had no evidence of progression for at least four courses. No particular treatment sequence predominated in these individuals; the small numbers of patients, wide range of malignancies, and prior treatment prohibited a rigorous analysis of the effects of drug sequence on antitumor activity. Two patients had major objective responses. One patient, a 29-year-old male with multifocal glioblastoma multiforme previously treated with radiation/paclitaxel, experienced complete resolution of diplopia and ataxia and an 83% reduction in measurable central nervous system disease (partial response) after two courses of Seq B→T 75/55 mg/m2. This response persisted for 19 months (13 total courses), at which time he expired because of complications from central venous access placement. A postmortem examination revealed malignant disease limited to the rim of a residual cerebellar lesion. Additionally, a 54-year-old female with advanced ovarian cancer refractory to paclitaxel, carboplatin, and topotecan had complete resolution of all evaluable disease, as well as normalization of CA-125 from a pretreatment value of 382 units/ml (complete response), after three courses of Seq B→T 100/80 mg/m2. The patient elected to discontinue investigational treatment for personal reasons and, instead, received three courses of DTIC before developing progressive disease.
Three patients with soft tissue sarcoma or malignancies with sarcomatous components also experienced objective, albeit minor, disease regression. A 51-year-old female with peritoneal liposarcoma that had progressed during treatment with doxorubicin experienced a 25% reduction in measurable disease with improvement in abdominal symptoms lasting 20 months after 16 total courses of Seq B→T 50/35 and 75/55 mg/m2. A second subject, a 52-year-old male with metastatic osteosarcoma that progressed after four doxorubicin- and/or ifosfamide-based regimens, one of which contained DTIC, had a 29% reduction in the size of lung lesions that lasted 6 months after treatment with Seq T→B 55/75 mg/m2. Finally, a 49-year-old female with advanced uterine carcinosarcoma, previously treated with ifosfamide/mesna, experienced a 34% reduction in measurable disease lasting 8 months after treatment with Seq B→T 100/80 mg/m2.
Six other patients, including one each with glioblastoma multiforme, astrocytoma, mesothelioma and colorectal, renal, and adenoid cystic carcinomas, had stable disease after at least four courses of BCNU/TEM.
PK/Pharmacodynamic Studies.
Forty-nine patients had complete plasma blood sampling for PK. The pertinent TEM PK parameters for each of the two BCNU/TEM sequences are summarized in Table 5. TEM was rapidly absorbed, with peak concentrations achieved at 1.2 ± 0.67 h and 1.3 ± 0.90 h after treatment on days 1 and 5, respectively. Respective harmonic mean t1/2 on days 1 and 5 were 1.83 ± 0.42 h and 1.74 ± 0.39 h and systemic clearance averaged 6.54 ± 1.67 and 6.69 ± 1.57 liter/h/m2. PK parameters reflecting systemic exposure increased proportionately with increasing doses of TEM; relationships between dose (mg/m2) and both peak concentrations and AUC values were moderately strong (r2 = 0.55 and 0.84, respectively). None of the values for Tmax, t1/2, or CL/F on days 1 and 5 were affected by the sequence of drug administration (P > 0.45 for all values).
A weak relationship between the percentage decrements in ANC and the cumulative duration of TEM exposure was evident (r2 = 0.39); whereas the relationship between the percentage decrements in platelets and cumulative TEM exposure was somewhat stronger (r2 = 0.59; Fig. 1). The AUC associated with a 50% reduction in platelets was 54.76 μg·h/ml. No relationship between the grade of hepatic transaminitis and TEM AUC was apparent.
AGAT Activity in PBMCs.
Serial measurements of AGAT activity (fmol/μg DNA) in the PBMCs of six patients, including three subjects treated with both BCNU/TEM sequences at their respective MTDs, are displayed in Table 6. The material from a pretreatment sample from one subject treated with Seq B→T was insufficient for analysis. In the five assessable patients, a mean decrement in AGAT by ∼3-fold was noted after treatment with BCNU/TEM. Fluctuations in AGAT in patients treated with Seq B→T (200/150 mg/m2) and Seq T→B (80/100 mg/m2) are detailed in Figs. 2and 3, respectively. AGAT inhibition was incomplete in all six patients. Maximal mean AGAT inhibition (mean, 59.9 fmol/μg DNA; range, 40–76%) occurred at 24 h (one patient), 48 h (three patients), and 96 h (two patients). No major sequence-dependent effects were apparent.
DISCUSSION
The rationale for evaluating the combination of BCNU/TEM is based in part on preclinical studies indicating that TEM and other methylating agents enhance BCNU cytotoxicity, presumably by augmenting AGAT depletion (20, 21, 22). The development of TEM, an oral methylating agent that is spontaneously and completely metabolically activated to 5-(3-methyltriazen-1-yl)imidazole-4-carboximide, in contrast to DTIC, which undergoes variable and unpredictable hepatic p450 activation, offered a more straightforward evaluation of this strategy (29, 30). However, preclinical studies have yielded conflicting results regarding the cytotoxic and sequence-dependent effects of the combination of BCNU/TEM on tumor and normal tissues (36, 37). Therefore, the present study was designed to evaluate the toxicological, pharmacological, and biological effects of alternate sequences of BCNU/TEM.
The principal toxicities of the BCNU/TEM regimen were neutropenia and thrombocytopenia. Seq T→B was substantially more myelosuppressive than Seq B→T, resulting in disparate MTD levels. On the basis of these data, the dose levels recommended for subsequent disease-directed evaluations are 150/110 and 80/100 mg/m2 for Seq B→T and T→B, respectively. Antitumor responses were also noted in several patients, the two most impressive of which occurred in individuals treated on Seq B→T at doses below the MTD. The results of the present study also confirm those of a previous Phase I trial that evaluated an alternate dosing schedule in which sequences of BCNU/TEM were administered in a single day (45). The MTD levels were 150/550 and 400/100 mg/m2 for Seq B→T and Seq T→B (45), respectively, and are identical to the total doses for each drug and sequence in the present study. In fact, the same BCNU/TEM sequence (Seq B→T) and total dose (150/550 mg/m2) as in the present study were recommended for Phase II evaluations.
The relatively high incidence of drug-related hepatic transaminitis, observed in 25% of patients, was not unexpected due to high levels of AGAT activity in hepatic tissues (46), and is comparable with the 26% incidence of transaminitis reported with single-agent BCNU (40). Although veno-occlusive disease leading to death was reported in one patient treated with BCNU/TEM administered on a single day (45), hepatic toxicity was asymptomatic, isolated, and inconsequential in the present study that used a more protracted administration schedule. However, clinical experience with BCNU/TEM at the recommended Phase II dose is quite limited, and it would be prudent to monitor patients closely for hepatotoxicity in subsequent clinical evaluations.
The modulation of TEM clearance by BCNU does not seem to be responsible for the vivid sequence-dependent toxicological effects observed in the present study. Relevant PK parameters indicative of TEM exposure and clearance were similar on both sequences. The absence of major PK interactions between TEM and BCNU is further supported by the similarity of TEM PK parameters obtained on treatment days 1 and 5. In fact, the TEM PK parameter estimates in the present study, in which plasma was sampled on both days 1 and 5, are in close agreement with those derived from studies of single-agent TEM (36, 39), as well as from studies of TEM administered in combination with BCNU on a sequential single-day administration schedule (45). Schold et al. (45) reported similar non-sequence-dependent PK parameters in patients treated with BCNU/TEM sequences on an alternate day-1 dose-sequencing schedule. In contrast to the results of Schold et al., in which strong pharmacodynamic relationships between TEM exposure and the grade of neutropenia (r2 = 0.62; P < 0.002) and thrombocytopenia (r2 = 0.74; P < 0.002) were apparent, these pharmacodynamic relationships were much weaker in the present study in which BCNU/TEM were administered on a more prolonged days-1-to-5 schedule.
The lack of major sequence-dependent PK interactions suggests that the sequence-dependent toxicological effects are attributable to inherent biological and/or biochemical phenomena. However, this deduction must be tempered by the fact that this study did not explore BCNU PK modulation by TEM. The increased toxicity observed in Seq T→B may be explained by TEM-induced AGAT inhibition, which may potentially sensitize hematopoietic cells to the myelosuppressive effects of BCNU (35, 47). Indeed, Panella et al. (21) reported that the administration of an alternate methylating agent, STZ, on day 1–4 in combination with BCNU on day 3 led to AGAT inhibition in PBMCs of a magnitude similar to that of in vitro studies in which STZ enhanced the cytotoxicity of BCNU. Interestingly, the reverse sequence, BCNU before STZ on a day-1-to-5 schedule, induced AGAT depletion without enhancing systemic toxicity (48).
Sequences of BCNU/TEM led to a mean 3-fold decrement in AGAT activity in PBMCs at the MTD level. In the present study, a predefined limit on the total volume of blood sampling precluded PK assessment in those patients undergoing PBMC AGAT studies, prohibiting assessment of potential pharmacodynamic relationships between PBMC AGAT levels and TEM PK parameters. In any case, the validity of AGAT activity in PBMCs as a surrogate marker of AGAT activity in tumors has not been clearly established. In contrast to malignant cells, PBMCs are not proliferating and do not rapidly regenerate AGAT (4). Furthermore, after treatment with single-agent BCNU or O6-benzylguanine, the resynthesis of AGAT in PBMCs is much slower than in tumor cells, suggesting that AGAT activity in PBMCs may not be a useful surrogate in this setting (4, 18, 45, 49, 50, 51). In addition, wide interindividual variability in AGAT PBMC depletion, in response to DTIC and TEM/fotemustine sequences has been observed (52). Although these results suggest that AGAT activity in PBMCs may not adequately reflect AGAT fluctuations in tumors, such measurements may be useful in establishing proof of principle from a mechanistic standpoint. In fact, the magnitude of AGAT decrements in PBMCs is similar to that achieved in in vitro studies in which BCNU cytotoxicity is enhanced by methylating alkylators and may, in fact, be sufficient to sensitize tumor cells to BCNU (17). A number of studies suggest that once a threshold level of AGAT activity is reached, further modulation does not improve cytotoxicity, indicating that complete inactivation of AGAT may not be necessary (53, 54). However, other studies relating AGAT activity to antitumor activity suggest that the situation is much more complex (55). In patients with brain tumors treated with BCNU, for example, progression-free and overall survival (P = 0.008 and P < 0.0002, respectively) were significantly longer in low-intermediate (6 and 29 months) compared with high (3 and 8 months) AGAT expressing neoplasms (55).
Other strategies that maximize AGAT depletion by preventing its regeneration entail administration of TEM on either a more protracted dosing schedule or a more frequent dosing interval. To date, protracted dosing schedules demonstrate a fairly consistent 2.1-fold increase in TEM exposure over 4 weeks (56, 57), whereas twice and thrice-daily TEM dosing schedules have resulted in greater than a 90% reduction in AGAT activity (58). In fact, an every-4-h TEM dosing interval is currently under evaluation to further maximize AGAT depletion (59). Further modulation of AGAT may be accomplished by O6-benzylguanine, which enhances the responsiveness to alkyl nitrosoureas in vitro and in vivo (60, 61).
The challenge remains to further define the patient population in which AGAT modulation is the optimal therapeutic strategy. Approximately 5% of tumors (62) and 30% of gliomas (63, 64) lack AGAT and may be highly sensitive to methylating agents. However, if low-AGAT-expressing tumors are equisensitive to BCNU and BCNU/TEM, then BCNU/TEM combinations may be optimally used in treating resistant tumors. Methylation of cytidine phosphate guanosine (CpG) islands in the promoter region of AGAT is associated with loss of AGAT expression, responsiveness to BCNU, and an increase in overall survival and time to tumor progression in patients with gliomas (65). Further confirmation of the relationship between AGAT promoter methylation and responsiveness to BCNU may potentially define other tumor populations responsive to BCNU/TEM dosing strategies.
Although AGAT appears to play a principal role in resistance to TEM, alternate mechanisms of resistance must be considered. Intact MMR function is critical for the cytotoxicity of methylating drugs. Recent evaluation of pediatric tumor xenografts found sensitivity to TEM correlated with both AGAT deficiency and MMR proficiency (66, 67). Treatment of human tumor cells with TEM induces an increase in the activity of poly(ADP-ribose) polymerase, which is involved in nucleotide excision repair (68, 69); inhibition of poly(ADP-ribose) polymerase enhances methylating agent cytotoxicity (70, 71). Friedman et al. (35) prospectively correlated response to TEM with AGAT and MMR status in patients with malignant glioma. Optimal sensitivity and specificity for detection of nonresponders occurred when the percent cellular reactivity to MMR and AGAT was >60 and >20%, respectively. The difference in response in high- and low-AGAT-expressing gliomas, 9.1 and 60%, respectively, was statistically significant (P = 0.009; Ref. 35).
Further definition of tumor populations that lack DNA MMR capacity may spare the use of methylating agents in this scenario, unless a method to restore MMR is developed. Identification of tumors with elevated AGAT activity and normal MMR capacity would permit intervention with an agent such as O6-benzylguanine, which depletes AGAT and restores sensitivity to methylating agents and nitrosoureas in preclinical studies (72, 73). Additionally, in view of the principal role of Bcl-2 in MeG-induced apoptosis, an agent that inhibits Bcl-2 expression in tumor cells may be of value. A future combination regimen with agents that inhibit DNA repair and amplification of apoptotic signaling in tumors may be warranted.
Further pursuit of nitrosourea and methylating agent combinations have not garnished much enthusiasm to date, because of residual AGAT activity after equivocal results of studies with agents such as STZ and the toxic nature of these combinations (74). However, the debilitating neurological manifestations of primary brain tumors, the single-agent activity of TEM and BCNU in this tumor type, the potential activity of the combination of these agents, the more predictable metabolic conversion of TEM compared with earlier methylating agents, and the lack of alternative active agents in this tumor type make BCNU/TEM attractive for further development. In fact, the lack of neurotoxicity, the ability to effectively prevent nausea and vomiting, and the lack of modulation by corticosteroids and antieleptics make the combination of BCNU/TEM feasible for clinical use, pending additional Phase II/III studies recommending a new, less myelosuppressive, and more active regimen in this patient population. Administration of Seq B→T facilitates treatment, requiring patient visits only on day 1 as opposed to day 1 and 5 and allows administration of higher doses of both agents, possibly resulting in a greater therapeutic index. The lack of sequence-dependent activity, PK profiles, and AGAT levels, in combination with sequence-dependent toxicity, favor Seq B→T at 150/110 mg/m2 as the recommended sequence and dose for further Phase II/III evaluation.
Grant support: Supported in part by CA69853 and CA54174, and the Frederic C. Bartter Clinical Research Unit of the Audie Murphy Veterans Administration Hospital through NIH Grant MO1 RR01346.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Requests for reprints: Eric K. Rowinsky, Institute for Drug Development, Cancer Therapy and Research Center, 7979 Wurzbach Road, Zellars Building, San Antonio, TX 78229. Phone: (210) 616-5945; Fax: (210) 692-7502; E-mail: Erowinsk@idd.org

Scatterplots of total temozolomide area under the concentration-time curve (AUC) days 1–5 (μg × h/ml) versus (A) absolute neutrophil count (% change) and (B) platelet count (% change).
Scatterplots of total temozolomide area under the concentration-time curve (AUC) days 1–5 (μg × h/ml) versus (A) absolute neutrophil count (% change) and (B) platelet count (% change).
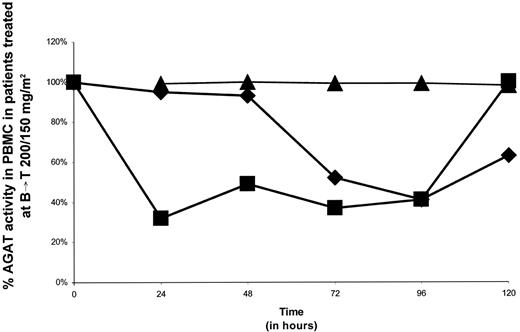
Scatterplots of O6-alkylguanine-DNA alkyltransferase (AGAT) activity in successive samples of peripheral blood mononuclear cells (PBMC) of patients treated with the following tumor types at one dose level above the maximum tolerated dose for temozolomide (TEM) administered p.o. daily on days 1–5 and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) administered i.v. on day 1 before TEM (Seq B→T) 200/150 mg/m2 as a percentage of baseline prechemotherapy levels: ♦, soft tissue (synovial cell) sarcoma; ▪, liver carcinoma; ▴, soft tissue sarcoma (leiomyosarcoma).
Scatterplots of O6-alkylguanine-DNA alkyltransferase (AGAT) activity in successive samples of peripheral blood mononuclear cells (PBMC) of patients treated with the following tumor types at one dose level above the maximum tolerated dose for temozolomide (TEM) administered p.o. daily on days 1–5 and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) administered i.v. on day 1 before TEM (Seq B→T) 200/150 mg/m2 as a percentage of baseline prechemotherapy levels: ♦, soft tissue (synovial cell) sarcoma; ▪, liver carcinoma; ▴, soft tissue sarcoma (leiomyosarcoma).

Scatterplots of O6-alkylguanine-DNA alkyltransferase (AGAT) activity in successive samples of peripheral blood mononuclear cells (PBMC) of patients treated with the following tumor types at the maximum tolerated dose for p.o. TEM daily on days 1–5 and BCNU administered i.v. on day 5 after TEM (Seq T→B) 80/100 mg/m2 as a percentage of baseline prechemotherapy levels: ♦, osteogenic sarcoma; •, glioblastoma multiforme; ▴, soft tissue (synovial cell) sarcoma.
Scatterplots of O6-alkylguanine-DNA alkyltransferase (AGAT) activity in successive samples of peripheral blood mononuclear cells (PBMC) of patients treated with the following tumor types at the maximum tolerated dose for p.o. TEM daily on days 1–5 and BCNU administered i.v. on day 5 after TEM (Seq T→B) 80/100 mg/m2 as a percentage of baseline prechemotherapy levels: ♦, osteogenic sarcoma; •, glioblastoma multiforme; ▴, soft tissue (synovial cell) sarcoma.
Patient characteristics
| Characteristic | No. of patients |
|---|---|
| Number of patients (evaluable)a | 63 (60) |
| Median number of courses/patient (range) | 2 (1–16) |
| Gender (Male/Female) | 40/23 |
| Median age (range) | 49 (19–80) |
| Median performance status (SWOG)b | 1 |
| 0 | 26 |
| 1 | 35 |
| 2 | 2 |
| Prior therapy: | |
| Chemotherapy only | 38 |
| Chemotherapy and radiation | 18 |
| Radiation only | 5 |
| None | 2 |
| Primary tumor types: | |
| Sarcoma | 20 |
| Melanoma | 11 |
| Colorectal | 12 |
| Brain (GBM) | 6 |
| Endometrium | 2 |
| Mesothelioma | 2 |
| Ovary | 2 |
| Other [pancreas, bladder, breast, brain (astrocytoma), stomach, head and neck, liver, kidney] | 1 each |
| Characteristic | No. of patients |
|---|---|
| Number of patients (evaluable)a | 63 (60) |
| Median number of courses/patient (range) | 2 (1–16) |
| Gender (Male/Female) | 40/23 |
| Median age (range) | 49 (19–80) |
| Median performance status (SWOG)b | 1 |
| 0 | 26 |
| 1 | 35 |
| 2 | 2 |
| Prior therapy: | |
| Chemotherapy only | 38 |
| Chemotherapy and radiation | 18 |
| Radiation only | 5 |
| None | 2 |
| Primary tumor types: | |
| Sarcoma | 20 |
| Melanoma | 11 |
| Colorectal | 12 |
| Brain (GBM) | 6 |
| Endometrium | 2 |
| Mesothelioma | 2 |
| Ovary | 2 |
| Other [pancreas, bladder, breast, brain (astrocytoma), stomach, head and neck, liver, kidney] | 1 each |
Three patients were not fully evaluable because of progression of malignant disease and death in course 1.
SWOG, Southwestern Oncology Group; GBM, glioblastoma multiforme.
Rates of dose-limiting toxicities (DLTs) as functions of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)/temozolomide (TEM) treatment sequence and dose level
| Dose level | Treatment | No. of pts with DLTs in the first course | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCNU (mg/m2 on day 1 or 5) | TEM (mg/m2/day × 5)a | Sequence | No. of pts (new) | No. of evaluable coursesb | ANC<500/μl >5 days | ANC<500/μl + fever | Platelets <25,000/μl | Grade 3–4 nonheme toxicity | New pts with any DLT/no. of new pts | |||||||||
| −1 | 37 | 26 | T→B | 2 (0) | 2 | 0 | 0 | 0 | 0 | 0/0 | ||||||||
| 1 | 50 | 35 | B→T | 6 (6) | 17 | 0 | 0 | 0 | 0 | 1/6 | ||||||||
| 1 | 50 | 35 | T→B | 7 (6) | 12 | 0 | 0 | 1 | 1 | 1/6 | ||||||||
| 2 | 75 | 55 | B→T | 7 (6) | 28 | 0 | 0 | 0 | 0 | 0/6 | ||||||||
| 2 | 75 | 55 | T→B | 6 (6) | 10 | 1 | 0 | 1 | 0 | 1/6 | ||||||||
| 3 | 100 | 80 | B→T | 5 (3) | 13 | 0 | 0 | 0 | 0 | 0/3 | ||||||||
| 3 | 100 | 80 | T→B | 10 (10) | 20 | 0 | 0 | 1 | 0 | 1/10 | ||||||||
| 4 | 150 | 110 | B→T | 9 (7) | 16 | 0 | 0 | 0 | 0 | 0/7 | ||||||||
| 4 | 150 | 110 | T→B | 5 (5) | 10 | 2 | 0 | 3 | 0 | 2/5 | ||||||||
| 5 | 200 | 150 | B→T | 9 (9) | 15 | 1 | 0 | 3 | 0 | 3/9 | ||||||||
| 5 | 200 | 150 | T→B | 1 (1) | 1 | 1 | 1 | 1 | 1c | 1/1 | ||||||||
| 6 | 240 | 180 | B→T | 1 (1) | 1 | 0 | 0 | 0 | 0 | 0/1 | ||||||||
| Dose level | Treatment | No. of pts with DLTs in the first course | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCNU (mg/m2 on day 1 or 5) | TEM (mg/m2/day × 5)a | Sequence | No. of pts (new) | No. of evaluable coursesb | ANC<500/μl >5 days | ANC<500/μl + fever | Platelets <25,000/μl | Grade 3–4 nonheme toxicity | New pts with any DLT/no. of new pts | |||||||||
| −1 | 37 | 26 | T→B | 2 (0) | 2 | 0 | 0 | 0 | 0 | 0/0 | ||||||||
| 1 | 50 | 35 | B→T | 6 (6) | 17 | 0 | 0 | 0 | 0 | 1/6 | ||||||||
| 1 | 50 | 35 | T→B | 7 (6) | 12 | 0 | 0 | 1 | 1 | 1/6 | ||||||||
| 2 | 75 | 55 | B→T | 7 (6) | 28 | 0 | 0 | 0 | 0 | 0/6 | ||||||||
| 2 | 75 | 55 | T→B | 6 (6) | 10 | 1 | 0 | 1 | 0 | 1/6 | ||||||||
| 3 | 100 | 80 | B→T | 5 (3) | 13 | 0 | 0 | 0 | 0 | 0/3 | ||||||||
| 3 | 100 | 80 | T→B | 10 (10) | 20 | 0 | 0 | 1 | 0 | 1/10 | ||||||||
| 4 | 150 | 110 | B→T | 9 (7) | 16 | 0 | 0 | 0 | 0 | 0/7 | ||||||||
| 4 | 150 | 110 | T→B | 5 (5) | 10 | 2 | 0 | 3 | 0 | 2/5 | ||||||||
| 5 | 200 | 150 | B→T | 9 (9) | 15 | 1 | 0 | 3 | 0 | 3/9 | ||||||||
| 5 | 200 | 150 | T→B | 1 (1) | 1 | 1 | 1 | 1 | 1c | 1/1 | ||||||||
| 6 | 240 | 180 | B→T | 1 (1) | 1 | 0 | 0 | 0 | 0 | 0/1 | ||||||||
× 5, for 5 days; pts, patients; ANC, absolute neutrophil count; B→T, BCNU on day 1 followed by TEM days 1–5; T→B, TEM on days 1–5 followed by BCNU on day 5.
Three courses not fully evaluable for toxicity because of patient deaths from progressive malignancy during first courses.
Patient inadvertently treated at this dose level and expired because of neutropenic sepsis.
Hematological toxicity as functions of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) temozolomide (TEM) treatment sequence and dose level
| Dose level | Dose (mg/m2/day) | Seqa | No. of courses | Incidence of hematological effects in all courses (course 1) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neutrophils | Platelets | Hgb | ||||||||||||||||||||||||
| Nadir | Toxicity grade | Nadir | Toxicity grade | Grade | ||||||||||||||||||||||
| BCNU | TEM | Median (range)(/μl) | Mean (/μl) | 3 | 4 | 4>5 days | 4 + Fever | Median (range) (×103/μl) | Mean (×103/μl) | 3 | 4 | 4 | ||||||||||||||
| −1 | 37 | 26 | T→B | 2 | 3,409 (2,948–3,869) | 3409 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 144 (37–251) | 144 | 1 (0) | 0 (0) | 0 (0) | |||||||||||
| 1 | 50 | 35 | B→T | 17 | 4,070 (1,361–5,466) | 3,909 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 340 (248–456) | 331 | 0 (0) | 0 (0) | 0 (0) | |||||||||||
| 1 | 50 | 35 | T→B | 12 | 5,300 (1,400–16,856) | 4,709 | 0 (0) | 0 (0) | 0 (0) | (0) | 247 (15–428) | 248 | 0 (0) | 1 (0) | 0 (0) | |||||||||||
| 2 | 75 | 55 | B→T | 28 | 2,306 (931–8,056) | 2,710 | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 88 (38–371) | 171 | 2 (0) | 0 (0) | 0 (0) | |||||||||||
| 2 | 75 | 55 | T→B | 10 | 3,087 (171–5,908) | 3,998 | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 207 (13–244) | 185 | 0 (0) | 1 (1) | 0 (0) | |||||||||||
| 3 | 100 | 80 | B→T | 13 | 2,135 (790–3,800) | 4,087 | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 124 (33–192) | 129 | 1 (0) | 0 (0) | 0 (0) | |||||||||||
| 3 | 100 | 80 | T→B | 20 | 2,700 (966–4,930) | 2,578 | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 121 (19–220) | 119 | 1 (1) | 2 (0) | 1 (0) | |||||||||||
| 4 | 150 | 110 | B→T | 16 | 3,387 (306–4,600) | 4,087 | 0 (0) | 1 (0) | 0 (0) | 0 (0) | 128 (21–225) | 107 | 1 (0) | 1 (0) | 0 (0) | |||||||||||
| 4 | 150 | 110 | T→B | 10 | 1,501 (19–3,900) | 1,909 | 1 (1) | 0 (0) | 2 (2) | 0 (0) | 80 (6–520) | 177 | 0 (0) | 4 (3) | 0 (0) | |||||||||||
| 5 | 200 | 150 | B→T | 15 | 1,558 (77–2,700) | 1,377 | 2 (0) | 1 (1) | 3 (3) | 0 (0) | 42 (7–122) | 51 | 5 (2) | 5 (3) | 1 (0) | |||||||||||
| 5 | 200 | 150 | T→B | 1 | 0 (0) | 0 | 0 (0) | 0 (0) | 1 (1) | 1 (1) | 3 (3) | 3 | 0 (0) | 1 (1) | 1 (1) | |||||||||||
| 6 | 240 | 180 | B→T | 1 | 1,470 (1,470) | 1,470 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 46 (46) | 46 | 1 (1) | 0 (0) | 0 (0) | |||||||||||
| Dose level | Dose (mg/m2/day) | Seqa | No. of courses | Incidence of hematological effects in all courses (course 1) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neutrophils | Platelets | Hgb | ||||||||||||||||||||||||
| Nadir | Toxicity grade | Nadir | Toxicity grade | Grade | ||||||||||||||||||||||
| BCNU | TEM | Median (range)(/μl) | Mean (/μl) | 3 | 4 | 4>5 days | 4 + Fever | Median (range) (×103/μl) | Mean (×103/μl) | 3 | 4 | 4 | ||||||||||||||
| −1 | 37 | 26 | T→B | 2 | 3,409 (2,948–3,869) | 3409 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 144 (37–251) | 144 | 1 (0) | 0 (0) | 0 (0) | |||||||||||
| 1 | 50 | 35 | B→T | 17 | 4,070 (1,361–5,466) | 3,909 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 340 (248–456) | 331 | 0 (0) | 0 (0) | 0 (0) | |||||||||||
| 1 | 50 | 35 | T→B | 12 | 5,300 (1,400–16,856) | 4,709 | 0 (0) | 0 (0) | 0 (0) | (0) | 247 (15–428) | 248 | 0 (0) | 1 (0) | 0 (0) | |||||||||||
| 2 | 75 | 55 | B→T | 28 | 2,306 (931–8,056) | 2,710 | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 88 (38–371) | 171 | 2 (0) | 0 (0) | 0 (0) | |||||||||||
| 2 | 75 | 55 | T→B | 10 | 3,087 (171–5,908) | 3,998 | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 207 (13–244) | 185 | 0 (0) | 1 (1) | 0 (0) | |||||||||||
| 3 | 100 | 80 | B→T | 13 | 2,135 (790–3,800) | 4,087 | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 124 (33–192) | 129 | 1 (0) | 0 (0) | 0 (0) | |||||||||||
| 3 | 100 | 80 | T→B | 20 | 2,700 (966–4,930) | 2,578 | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 121 (19–220) | 119 | 1 (1) | 2 (0) | 1 (0) | |||||||||||
| 4 | 150 | 110 | B→T | 16 | 3,387 (306–4,600) | 4,087 | 0 (0) | 1 (0) | 0 (0) | 0 (0) | 128 (21–225) | 107 | 1 (0) | 1 (0) | 0 (0) | |||||||||||
| 4 | 150 | 110 | T→B | 10 | 1,501 (19–3,900) | 1,909 | 1 (1) | 0 (0) | 2 (2) | 0 (0) | 80 (6–520) | 177 | 0 (0) | 4 (3) | 0 (0) | |||||||||||
| 5 | 200 | 150 | B→T | 15 | 1,558 (77–2,700) | 1,377 | 2 (0) | 1 (1) | 3 (3) | 0 (0) | 42 (7–122) | 51 | 5 (2) | 5 (3) | 1 (0) | |||||||||||
| 5 | 200 | 150 | T→B | 1 | 0 (0) | 0 | 0 (0) | 0 (0) | 1 (1) | 1 (1) | 3 (3) | 3 | 0 (0) | 1 (1) | 1 (1) | |||||||||||
| 6 | 240 | 180 | B→T | 1 | 1,470 (1,470) | 1,470 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 46 (46) | 46 | 1 (1) | 0 (0) | 0 (0) | |||||||||||
Seq, sequence; Hgb, hemoglobin; B→T, BCNU on day 1 followed by TEM on days 1–5; T→B, TEM on days 1–5 followed by BCNU on day 5.
Nonhematological toxicity as a function of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)/temozolomide (TEM) treatment sequence (course 1)
| Toxicity | Sequence B→Ta (n = 32) | Sequence T→B (n = 28) | Both sequences (n = 60) (%) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade | Grade | ||||||||||||||||||
| 1 | 2 | 3 | 4 | 1–4 (%) | 1 | 2 | 3 | 4 | 1–4 (%) | All | |||||||||
| Fatigue | 14 | 3 | 0 | 0 | 53 | 4 | 2 | 1 | 0 | 25 | 40 | ||||||||
| Nausea | 10 | 3 | 1 | 0 | 44 | 8 | 2 | 0 | 0 | 36 | 40 | ||||||||
| Vomiting | 6 | 4 | 1 | 0 | 34 | 5 | 1 | 0 | 0 | 21 | 28 | ||||||||
| Transaminitis | 4 | 2 | 3 | 0 | 28 | 4 | 2 | 0 | 1 | 25 | 27 | ||||||||
| Constipation | 6 | 0 | 0 | 0 | 19 | 6 | 0 | 0 | 0 | 21 | 20 | ||||||||
| Anorexia | 3 | 1 | 0 | 0 | 12 | 5 | 0 | 0 | 0 | 18 | 15 | ||||||||
| Diarrhea | 4 | 0 | 0 | 0 | 12 | 2 | 1 | 0 | 0 | 11 | 12 | ||||||||
| Malaise | 3 | 1 | 0 | 0 | 12 | 2 | 0 | 0 | 0 | 7 | 10 | ||||||||
| Alkaline phosphatase elevation | 2 | 0 | 0 | 0 | 6 | 4 | 1 | 0 | 0 | 18 | 10 | ||||||||
| Rash | 1 | 2 | 0 | 0 | 9 | 3 | 0 | 0 | 0 | 11 | 10 | ||||||||
| Infection | 2 | 0 | 0 | 0 | 6 | 3 | 0 | 0 | 1b | 14 | 10 | ||||||||
| Headache | 3 | 0 | 0 | 0 | 9 | 2 | 0 | 0 | 0 | 7 | 8 | ||||||||
| Creatinine elevation | 1 | 0 | 0 | 0 | 3 | 3 | 0 | 0 | 0 | 11 | 7 | ||||||||
| Mucositis | 3 | 1 | 0 | 0 | 12 | 0 | 0 | 0 | 0 | 0 | 7 | ||||||||
| Hyperbilirubinemia | 0 | 0 | 0 | 1c | 3 | 0 | 0 | 0 | 2c | 7 | 5 | ||||||||
| Toxicity | Sequence B→Ta (n = 32) | Sequence T→B (n = 28) | Both sequences (n = 60) (%) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade | Grade | ||||||||||||||||||
| 1 | 2 | 3 | 4 | 1–4 (%) | 1 | 2 | 3 | 4 | 1–4 (%) | All | |||||||||
| Fatigue | 14 | 3 | 0 | 0 | 53 | 4 | 2 | 1 | 0 | 25 | 40 | ||||||||
| Nausea | 10 | 3 | 1 | 0 | 44 | 8 | 2 | 0 | 0 | 36 | 40 | ||||||||
| Vomiting | 6 | 4 | 1 | 0 | 34 | 5 | 1 | 0 | 0 | 21 | 28 | ||||||||
| Transaminitis | 4 | 2 | 3 | 0 | 28 | 4 | 2 | 0 | 1 | 25 | 27 | ||||||||
| Constipation | 6 | 0 | 0 | 0 | 19 | 6 | 0 | 0 | 0 | 21 | 20 | ||||||||
| Anorexia | 3 | 1 | 0 | 0 | 12 | 5 | 0 | 0 | 0 | 18 | 15 | ||||||||
| Diarrhea | 4 | 0 | 0 | 0 | 12 | 2 | 1 | 0 | 0 | 11 | 12 | ||||||||
| Malaise | 3 | 1 | 0 | 0 | 12 | 2 | 0 | 0 | 0 | 7 | 10 | ||||||||
| Alkaline phosphatase elevation | 2 | 0 | 0 | 0 | 6 | 4 | 1 | 0 | 0 | 18 | 10 | ||||||||
| Rash | 1 | 2 | 0 | 0 | 9 | 3 | 0 | 0 | 0 | 11 | 10 | ||||||||
| Infection | 2 | 0 | 0 | 0 | 6 | 3 | 0 | 0 | 1b | 14 | 10 | ||||||||
| Headache | 3 | 0 | 0 | 0 | 9 | 2 | 0 | 0 | 0 | 7 | 8 | ||||||||
| Creatinine elevation | 1 | 0 | 0 | 0 | 3 | 3 | 0 | 0 | 0 | 11 | 7 | ||||||||
| Mucositis | 3 | 1 | 0 | 0 | 12 | 0 | 0 | 0 | 0 | 0 | 7 | ||||||||
| Hyperbilirubinemia | 0 | 0 | 0 | 1c | 3 | 0 | 0 | 0 | 2c | 7 | 5 | ||||||||
B→T, BCNU on day 1 followed by TEM on days 1–5; T→B, TEM on days 1–5 followed by BCNU on day 5.
Pseudomonas sepsis.
Two patients with melanoma with rapidly progressive liver metastases documented on imaging studies and one patient with transient (24 h) hyperbilirubinemia after packed red blood cell transfusion; none of the three episodes were considered a dose-limiting toxicity.
Temozolomide (TEM) pharmacokinetic parameters
| Sequence | No. of patients | TEM dose (mg/m2/day) | Cpmax (μg/ml)a,b | Tmax (h)b | T1/2 (h)c | AUC (μg·h/ml)b | CL/F (liter/h/m2)b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | ||||||||
| B→T | 5 | 35 | 2.39 (0.8) | 2.48 (1.1) | 0.56 (0.3) | 0.88 (0.8) | 1.64 (0.4) | 1.71 (0.1) | 5.95 (1.9) | 5.75 (1.6) | 6.93 (3.2) | 6.76 (2.0) | |||||
| T→B | 6 | 35 | 2.27 (0.7) | 2.16 (1.4) | 1.07 (0.6) | 1.49 (1.3) | 1.81 (0.4) | 1.76 (0.2) | 7.09 (2.7) | 6.07 (1.4) | 5.47 (1.6) | 6.02 (1.2) | |||||
| B→T | 8 | 55 | 2.63 (1.2) | 2.20 (1.0) | 1.13 (0.6) | 1.69 (1.1) | 1.18 (0.3) | 1.95 (0.5) | 8.14 (1.1) | 7.91 (1.1) | 6.82 (0.8) | 7.03 (1.0) | |||||
| T→B | 6 | 55 | 3.25 (1.0) | 2.87 (0.8) | 1.17 (0.3) | 1.29 (0.6) | 1.62 (0.1) | 1.48 (0.2) | 9.25 (1.3) | 8.84 (2.0) | 6.14 (0.9) | 6.58 (1.3) | |||||
| B→T | 4 | 80 | 3.13 (0.6) | 5.20 (2.4) | 1.75 (0.3) | 1.10 (0.6) | 1.86 (0.2) | 1.57 (0.7) | 13.15 (1.9) | 13.85 (2.1) | 6.18 (0.9) | 5.87 (1.3) | |||||
| T→B | 6 | 80 | 3.79 (1.3) | 3.30 (1.3) | 1.06 (0.8) | 1.59 (1.2) | 2.22 (0.6) | 1.82 (0.3) | 12.33 (2.8) | 11.80 (3.0) | 6.85 (2.0) | 7.29 (2.6) | |||||
| B→T | 3 | 110 | 5.12 (4.2) | 6.28 (3.8) | 2.50 (2.1) | 0.92 (0.8) | 2.43 (0.4) | 1.46 (0.1) | 18.08 (6.2) | 18.40 (6.6) | 6.46 (2.2) | 6.39 (2.3) | |||||
| T→B | 5 | 110 | 6.21 (2.8) | 6.04 (1.1) | 1.03 (0.6) | 0.73 (0.5) | 1.93 (0.7) | 1.67 (0.2) | 15.99 (4.6) | 14.98 (4.7) | 7.32 (2.3) | 7.78 (2.1) | |||||
| B→T | 4 | 150 | 6.51 (1.1) | 7.81 (2.1) | 1.38 (0.5) | 1.13 (0.6) | 1.85 (0.5) | 1.76 (0.4) | 23.81 (2.6) | 23.34 (1.9) | 6.32 (0.7) | 6.42 (0.6) | |||||
| T→B | 1 | 150 | 6.59 | 6.02 | 1.50 | 2.00 | 1.27 | 6.86 | 25.78 | 28.12 | 5.82 | 5.33 | |||||
| B→T | 1 | 180 | 8.09 | 7.46 | 1.50 | 1.00 | 2.53 | 1.56 | 31.05 | 33.72 | 5.80 | 5.34 | |||||
| Sequence | No. of patients | TEM dose (mg/m2/day) | Cpmax (μg/ml)a,b | Tmax (h)b | T1/2 (h)c | AUC (μg·h/ml)b | CL/F (liter/h/m2)b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | ||||||||
| B→T | 5 | 35 | 2.39 (0.8) | 2.48 (1.1) | 0.56 (0.3) | 0.88 (0.8) | 1.64 (0.4) | 1.71 (0.1) | 5.95 (1.9) | 5.75 (1.6) | 6.93 (3.2) | 6.76 (2.0) | |||||
| T→B | 6 | 35 | 2.27 (0.7) | 2.16 (1.4) | 1.07 (0.6) | 1.49 (1.3) | 1.81 (0.4) | 1.76 (0.2) | 7.09 (2.7) | 6.07 (1.4) | 5.47 (1.6) | 6.02 (1.2) | |||||
| B→T | 8 | 55 | 2.63 (1.2) | 2.20 (1.0) | 1.13 (0.6) | 1.69 (1.1) | 1.18 (0.3) | 1.95 (0.5) | 8.14 (1.1) | 7.91 (1.1) | 6.82 (0.8) | 7.03 (1.0) | |||||
| T→B | 6 | 55 | 3.25 (1.0) | 2.87 (0.8) | 1.17 (0.3) | 1.29 (0.6) | 1.62 (0.1) | 1.48 (0.2) | 9.25 (1.3) | 8.84 (2.0) | 6.14 (0.9) | 6.58 (1.3) | |||||
| B→T | 4 | 80 | 3.13 (0.6) | 5.20 (2.4) | 1.75 (0.3) | 1.10 (0.6) | 1.86 (0.2) | 1.57 (0.7) | 13.15 (1.9) | 13.85 (2.1) | 6.18 (0.9) | 5.87 (1.3) | |||||
| T→B | 6 | 80 | 3.79 (1.3) | 3.30 (1.3) | 1.06 (0.8) | 1.59 (1.2) | 2.22 (0.6) | 1.82 (0.3) | 12.33 (2.8) | 11.80 (3.0) | 6.85 (2.0) | 7.29 (2.6) | |||||
| B→T | 3 | 110 | 5.12 (4.2) | 6.28 (3.8) | 2.50 (2.1) | 0.92 (0.8) | 2.43 (0.4) | 1.46 (0.1) | 18.08 (6.2) | 18.40 (6.6) | 6.46 (2.2) | 6.39 (2.3) | |||||
| T→B | 5 | 110 | 6.21 (2.8) | 6.04 (1.1) | 1.03 (0.6) | 0.73 (0.5) | 1.93 (0.7) | 1.67 (0.2) | 15.99 (4.6) | 14.98 (4.7) | 7.32 (2.3) | 7.78 (2.1) | |||||
| B→T | 4 | 150 | 6.51 (1.1) | 7.81 (2.1) | 1.38 (0.5) | 1.13 (0.6) | 1.85 (0.5) | 1.76 (0.4) | 23.81 (2.6) | 23.34 (1.9) | 6.32 (0.7) | 6.42 (0.6) | |||||
| T→B | 1 | 150 | 6.59 | 6.02 | 1.50 | 2.00 | 1.27 | 6.86 | 25.78 | 28.12 | 5.82 | 5.33 | |||||
| B→T | 1 | 180 | 8.09 | 7.46 | 1.50 | 1.00 | 2.53 | 1.56 | 31.05 | 33.72 | 5.80 | 5.34 | |||||
Cpmax, maximum plasma concentration; Tmax, time to maximum plasma concentration; T1/2, elimination half-life; AUC, area under the plasma concentration time curve; CL/F, clearance; B→T, BCNU on day 1 followed by TEM on days 1–5; T→B, TEM on days 1–5 followed by BCNU on day 5.
Values represent harmonic means (±SD).
Values represent means and (±SD).
O6-alkylguanine-DNA alkyltransferase (AGAT) activity in peripheral blood mononuclear cells (PBMCs) after treatment with sequences of 1,3-bis(2-chloroethyl)-1-nitrosourea/temozolomide (BCNU/TEM)
| BCNU/TEM Dose Level (mg/m2) | Sequence | Age/Gender | Tumor type | Mean AGAT levela fmol/μg DNA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pretreatment | 24 h | 48 h | 72 h | 96 h | 120 h | |||||||||
| 200/150 | B→Tb | 28/M | Soft tissue sarcoma (synovial cell) | 9.20 | 8.70 (−5.4) | 8.60 (−6.5) | 5.10 (−44.7) | 4.00 (−56.5) | 6.20 (−32.6) | |||||
| 200/150 | B→T | 71/M | Liver carcinoma | 12.00 | 3.80 (−68.3) | 5.90 (−50.8) | 4.40 (−63.3) | 4.90 (−59.2) | 12.40 (+3.3) | |||||
| 200/150 | B→T | 49/F | Soft tissue sarcoma (leiomyosarcoma) | —c | 1.10 | 0.10 | 0.60 | 0.60 | 2.00 | |||||
| 80/100 | T→B | 70/M | Glioblastoma multiforme | 10.20 | 4.60 (−46.7) | 2.70 (−73.5) | 2.90 (−71.6) | 2.40 (−76.5) | 3.50 (−65.7) | |||||
| 80/100 | T→B | 32/M | Osteogenic sarcoma | 3.90 | 2.00 (−48.7) | 1.60 (−58.9) | 2.10 (−46.1) | 1.80 (−53.8) | 2.80 (28.2) | |||||
| 80/100 | T→B | 30/F | Soft tissue sarcoma (synovial cell) | 4.80 | 3.30 (−31.2) | 2.90 (−39.6) | 3.70 (−22.9) | 3.60 (−25.0) | 3.30 (−32.1) | |||||
| BCNU/TEM Dose Level (mg/m2) | Sequence | Age/Gender | Tumor type | Mean AGAT levela fmol/μg DNA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pretreatment | 24 h | 48 h | 72 h | 96 h | 120 h | |||||||||
| 200/150 | B→Tb | 28/M | Soft tissue sarcoma (synovial cell) | 9.20 | 8.70 (−5.4) | 8.60 (−6.5) | 5.10 (−44.7) | 4.00 (−56.5) | 6.20 (−32.6) | |||||
| 200/150 | B→T | 71/M | Liver carcinoma | 12.00 | 3.80 (−68.3) | 5.90 (−50.8) | 4.40 (−63.3) | 4.90 (−59.2) | 12.40 (+3.3) | |||||
| 200/150 | B→T | 49/F | Soft tissue sarcoma (leiomyosarcoma) | —c | 1.10 | 0.10 | 0.60 | 0.60 | 2.00 | |||||
| 80/100 | T→B | 70/M | Glioblastoma multiforme | 10.20 | 4.60 (−46.7) | 2.70 (−73.5) | 2.90 (−71.6) | 2.40 (−76.5) | 3.50 (−65.7) | |||||
| 80/100 | T→B | 32/M | Osteogenic sarcoma | 3.90 | 2.00 (−48.7) | 1.60 (−58.9) | 2.10 (−46.1) | 1.80 (−53.8) | 2.80 (28.2) | |||||
| 80/100 | T→B | 30/F | Soft tissue sarcoma (synovial cell) | 4.80 | 3.30 (−31.2) | 2.90 (−39.6) | 3.70 (−22.9) | 3.60 (−25.0) | 3.30 (−32.1) | |||||
Values in parentheses represent percent change from pretreatment value.
B→T, BCNU on day 1 followed by TEM on days 1–5; T→B, TEM on days 1–5 followed by BCNU on day 5.
Insufficient sample.