
Abstract
Plasma electrolysis, where a solid electrode in an electrolytic cell is replaced by a plasma (or gas discharge), differs from conventional electrolysis by not being dictated by the surface characteristics of an electrode, but by the chemical species injected into the solution from the plasma. Reduction in a plasma cathode configuration occurs mostly by plasma-injected solvated electrons (e−aq), which may engage in side reactions, such as the second order recombination of e−aq, that ultimately reduce the faradaic efficiency for the production of a desired product. In this work, we show that the depletion of reactants at the plasma-liquid interface due to insufficient transport can reduce the predicted faradaic efficiency for a plasma cathode at low concentrations. Measurements of the faradaic efficiency using the dissociative electron attachment to chloroacetate and the ferri/ferrocyanide redox couple confirm this behavior. The effect of other mechanisms on the faradaic efficiency, such as competing oxidation reactions with the hydroxyl radical, are also evaluated and found to be far less significant. Unlike conventional electrolysis, stirring the solution does not increase the faradaic efficiency, but increasing the species concentration does.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Plasma electrolysis is the use of a plasma (or gas discharge) to drive electrons or ions into a liquid solution to initiate reduction and oxidation (redox) reactions. Generally, the system operates under direct current (DC) bias where the plasma replaces one of the metal electrodes from an electrolytic cell and, at the same time, the liquid solution works as one of the electrodes necessary to sustain the plasma.1 Typically, an electrode is suspended above the solution surface and biased negatively (called a plasma cathode) or positively (plasma anode) relative to the solution, causing the interstitial gas to breakdown and form a plasma. Whereas in conventional electrolysis, the reduction and oxidation reactions occur at solid (often metal) electrodes, in plasma electrolysis the redox chemistry is driven not by the surface properties of a solid electrode, but by the highly reactive species introduced into the solution by the plasma.
Perhaps the first plasma electrolysis experiment was conducted by Gubkin in 1887, where he used a plasma cathode to reduce several metal ions in salt solutions.2 Subsequently, several works in the early twentieth century studied these systems for chemical synthesis and evaluated the charge-transfer-to-product yield.3 The field was greatly advanced by Hickling and collaborators, who extensively studied the synthesis of hydrogen peroxide in aqueous solutions4 and hydrazine in ammonia,5 identifying the hydroxyl (•OH) radical as one of the most important reaction initiators. Using a chloroacetate (ClCH2CO2−) solution, they concluded that solvated electrons must be one of the accompanying radicals responsible for the reduction chemistry, making the first analogy between plasma electrolysis and radiation chemistry.6
More recently, it was argued that in the plasma cathode configuration, electrons from the plasma must be injected into the liquid in order for current to be maintained,7 and this was ultimately confirmed by optically detecting plasma-injected solvated electrons via absorption spectroscopy.8,9 Notably, it was found that plasma-injected solvated electrons have essentially the same properties as those produced by radiolysis, such as the rate constants for various reactions reported in the radiation chemistry literature.10,11
Solvated electrons (e−aq), commonly called hydrated electrons when in aqueous solutions, are one of the most powerful reductants with a reduction potential of −2.88 V vs. NHE,12 and can be used to reduce a wide variety of species. For this reason, there has been significant growth in the study of plasma electrolysis, leading to emerging new opportunities such as the synthesis of colloidal nanoparticles13 with narrow size distributions and without the use of surfactants or other stabilizers14 and the catalyst-free processing of dissolved carbon dioxide (CO2) into formate and oxalate15 or nitrogen (N2) into ammonia (NH3).16 Furthermore, the engineering of scalable, high-throughput systems shows the potential translational promise of these approaches.17,18
The main challenge in a plasma cathode configuration is to selectively and efficiently reduce a chemical species. Injected e−aq can either reduce the target reactant, which acts as an electron reactant (R),
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0001.gif)
or self-recombine via the diffusion-limited second-order recombination process,
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0002.gif)
which is responsible for hydrogen evolution. That is, the second-order recombination in Reaction 2 competes with Reaction 1 to reduce the equivalent faradaic efficiency of the intended reaction.19 Part of the problem comes from the extraordinarily fast rate of second-order recombination, with a reaction rate constant of 2k2 = 1.1 × 1010 M−1s−1,20 which inherently sets a limit on the maximum concentration of e−aq. Thus, when the reaction rates of Reactions 1 and 2 are comparable, some of the generated e−aq will electrolyze water rather than reduce R. For example, reduction of silver ions into silver with a plasma cathode yielded low faradaic efficiencies at bulk AgNO3 concentrations of 5 mM and 15 mM, but almost 100% faradaic efficiency at 150 mM.19
Another potential limitation in plasma cathode systems comes from competing oxidizing reactions carried out by other radical species produced by the plasma-liquid interaction. When a noble gas plasma, such as argon (Ar), is placed in contact with an aqueous solution, highly energetic plasma species like free electrons, ions, or electronically-excited atoms, may dissociate either surface or evaporated water molecules and generate hydroxyl (•OH) radicals.21–23 These dissolved •OH radicals may then compete with other species (Reaction 1) to react with e−aq or even re-oxidize the reduced chemical species back to the starting compound or to a different product.
In this work, we explore the nature of reduction by a plasma cathode and assess how various mechanisms inherently limit the faradaic efficiency. The dissociative electron attachment of e−aq to ClCH2CO2− was used as a model reaction system, and for comparison, the ferri/ferrocyanide redox couple was used to understand the case of reversible reactions. Experiments are compared to theoretical models, and the results show that the depletion of the reactant at the plasma-liquid interface severely limits the faradaic efficiency at low reactant concentrations. The effect of competing reactions by •OH and the effect of reactant depletion at the interface are analyzed and discussed.
Experimental
Chemical reaction system
Chloroacetate (sodium chloroacetate, 98%, Sigma Aldrich) and ferricyanide (potassium ferricyanide (III), powder or chunks, <10 μm, 99%, Sigma Aldrich) solutions were reduced in a glass H-cell electrochemical reactor (Adams & Chittenden) separated by a glass frit of P4 (ISO 4793) porosity, shown in Figure 1. In the ferricyanide (Fe(CN)63−) experiments, the catholyte consisted of 0.1−1 mM Fe(CN)63− solutions dissolved in deionized (DI) water, using 82 mM of sodium perchlorate (NaClO4 for molecular biology; DNase, RNase, and protease, none detected, 98%, Sigma Aldrich) as a background, non-reactive electrolyte. For ClCH2CO2− experiments, solutions of 0.1−1 mM ClCH2CO2− were prepared in 41 mM sodium tetraborate (Na2Ba4O7), which provided the same conductivity (10.3 mS) as 82 mM NaClO4, while maintaining the pH of the solution below 9; the pH was kept below 9 to prevent the interference of OH−, generated by Reaction 2, with Cl− measurements (see Chemical analysis). The anolyte for both experiments was 82 mM NaClO4 dissolved in DI water.
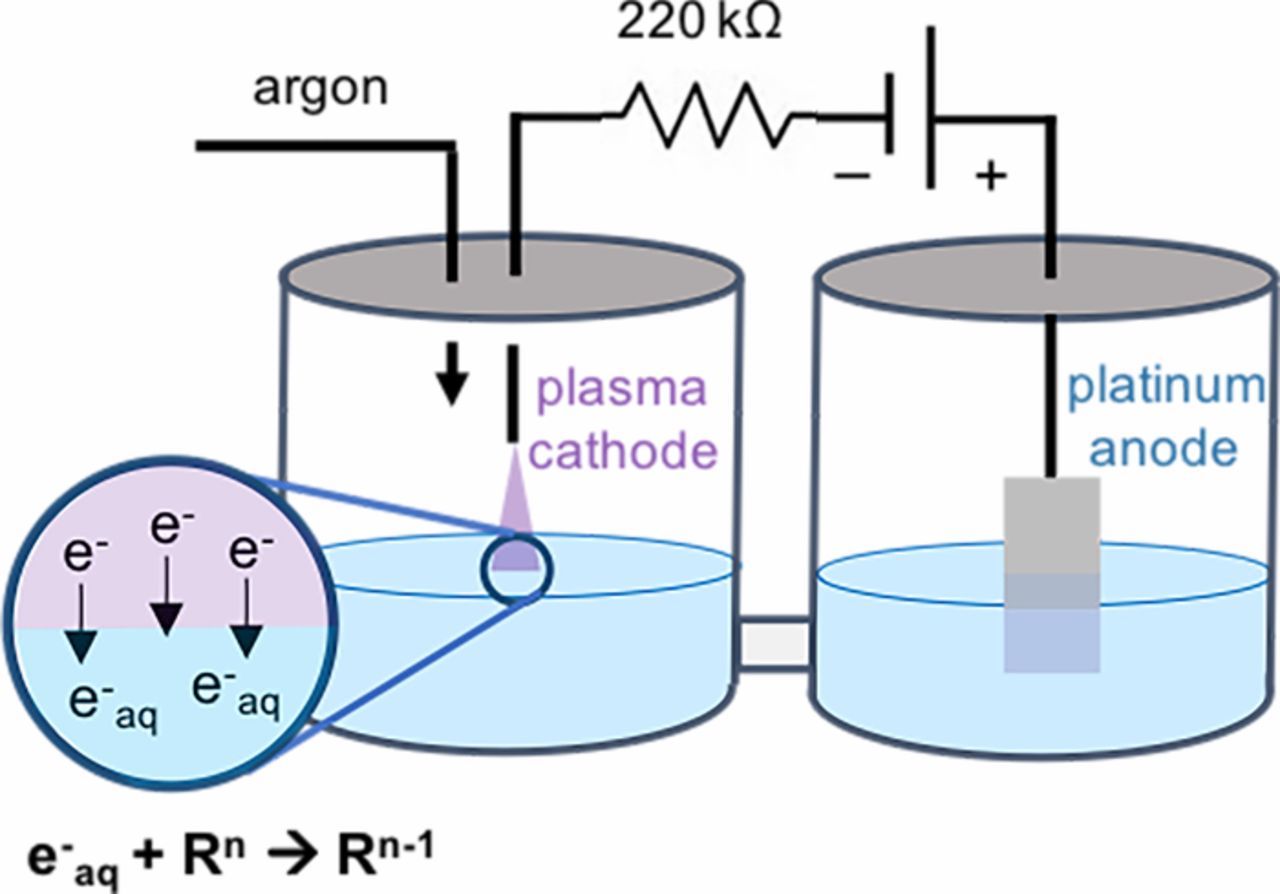
Figure 1. Schematic of the plasma electrolytic H-cell with a plasma cathode used in this work. The inset shows how electrons in the plasma dissolve to form solvated electrons and subsequently reduce reactants in the solution.
In this configuration, an argon plasma functioned as a cathode, and a platinum electrode submerged in the solution functioned as the electrochemical anode, similar to our prior work.15 Air was purged out of the cathodic half-cell using a hose inserted through the lid of the cell, and a constant 250 cm3/min flow of Ar (UHP, Airgas) was used to pressurize the head space. An argon plasma was ignited between a stainless-steel capillary (Restek, 1.58 mm outer diameter, 0.017 mm inner diameter) and the catholyte by applying a negative bias of ∼3 kV using a high-voltage power supply (Glassman EH Series). A 220 kΩ ballast resistor was used to limit the current and prevent the transition of the plasma to an arc. Because the cathode and anode are in separate compartments, the oxidation products at the anode, mainly oxygen, did not interfere with the plasma chemistry. Similarly, neither ClCH2CO2− nor Fe(CN)63− were oxidized at the anode. Experiments were run for 10 minutes at a constant current of 10 mA, with a distance between the capillary and the liquid surface of ∼1 mm that lead to a current density that varied less than 5% from experiment to experiment. Unless otherwise specified, the solutions were not stirred during experiments. To determine the effect of forced convection on the faradaic efficiency, some experiments were run with a stirred solution using a magnetic stirrer and 1 cm stir bar revolving at a rate of 300 rpm.
Glycerol (≥99.5%, Sigma Aldrich) and methanol (for HPLC, ≥99.9%, Sigma Aldrich), were used as •OH scavengers in some experiments to analyze the effect of •OH radicals on the liquid-phase chemical reduction. Glycerol was chosen as an •OH scavenger because it reacts with •OH with a reaction rate constant of k = 1.9 × 109 M−1s−1,20 but it does not react with e−aq.24 In a similar manner, methanol readily reacts with •OH (k = 9.7 × 108 M−1s−1),20 but not e−aq and was used to confirm the glycerol results. Both were mixed in the catholyte only, glycerol at a concentration of 500 mM and methanol at 200 mM. Due to higher reduction efficiency in the presence of the alcohols, •OH scavenger experiments with Fe(CN)63− were run for 3 minutes instead of 10 minutes to prevent bulk concentration depletion.
Chemical analysis
A chloride-selective electrode (double junction, Cole-Parmer) was used to measure the chloride concentration produced by the reaction of e−aq with ClCH2CO2− via
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0003.gif)
A calibration curve was built prior to each measurement because the voltage response of the electrode tended to slowly drift from day to day. Solutions of sodium chloride (NaCl), adjusted for ionic strength with NaClO4, were used to relate known concentrations of chloride (Cl−) to voltage measurements obtained by the chloride-selective electrode connected to a multimeter (Agilent 34401A 6 ½ Digit Multimeter). More details of the calibration can be found in Section S.1 of the Supplementary Material. Additionally, given that recent publications have made a case for the importance of the hydrogen radical (H•) as a reducing species both in RF-driven plasmas,25 where H• may be introduced into the liquid by convection, as well as in plasma cathodes,26 where H• can be formed in the liquid phase, the use of ClCH2CO2− as a model system made it simple to distinguish the reduction by e−aq as opposed to H•, which reacts with ClCH2CO2− via
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0004.gif)
That is, by measuring only the Cl− yield, we were able to distinguish only the dissociative attachment of e−aq, Reaction 3, for accurate calculation of the faradaic efficiency, as discussed below.
For the ferri/ferrocyanide experiments, an UV-Vis-ES spectrometer (Ocean Optics USB2000+) was used to measure the absorbance of Fe(CN)63− at ∼ 420 nm in a quartz cuvette (3.5 mL, 10 mm, Vernier). Fe(CN)63− reacts with e−aq via
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0005.gif)
producing ferrocyanide (Fe(CN)64−). Because Fe(CN)64− does not absorb light at 420 nm,27 the absorbance measurements were then used to obtain the amount of Fe(CN)63− reacted after each plasma treatment. A calibration curve was built and used to measure the Fe(CN)63− concentration after plasma experiments (see Section S.2 in the Supplementary Material).
Additionally, because reactions between ions are inevitably affected by electrostatic interactions with the solvent and thus depend on the ionic strength of the solution, the reaction rate constants between e−aq and ClCH2CO2−, and e−aq and Fe(CN)63−, were obtained as a function of ionic strength. A Titan-Beta 8 MeV pulsed electron Linear Accelerator (LINAC) in the Radiation Laboratory at the University of Notre Dame was used to obtain the reaction rate constants via transient absorption spectroscopy of e−aq with ClCH2CO2− and Fe(CN)63− dissolved in DI water, as well as in 82 and 163 mM NaClO4 solutions. The reaction rate constant between e−aq and ClCH2CO2− at an ionic strength of ∼82 mM is 1.48 × 109 M−1s−1, and the reaction rate constant between e−aq and Fe(CN)63− at an ionic strength of ∼82 mM is 1.16 × 1010 M−1s−1. The details of these experiments are explained in Section S.3 of the Supplementary Material.
Results
Analytical expression for the faradaic efficiency
The faradaic efficiency for the reaction of solvated electrons with the reactant (Reaction 1) is defined as the rate of reduction of the reactant by solvated electrons divided by the creation rate of solvated electrons. For a plasma cathode system, a steady state species rate balance for solvated electrons equates their generation from the current of the plasma and their consumption via Reaction 1 (for any reactant R) and Reaction 2 (second-order recombination). The resulting balance is
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0006.gif)
where j is the current density of the plasma, l is the interfacial penetration depth of the electrons, and F is Faraday's constant. At the extreme of an abundance of reactants, when kr[R] ≫ 2k2[e−aq], there is no competition for solvated electrons and the faradaic efficiency, η, is equivalently 100% (i.e., all of the electron current is consumed through reduction of the reactant). However, in some practical conditions such as the reduction of dissolved gases,15 the reaction rate kr[R][e−aq] is much slower than the second order recombination, and the faradaic efficiency becomes
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0007.gif)
That is, there is a linear relationship between η and [R] when η is small.
Competition with •OH has minimal effect on the faradaic efficiency
Figure 2 shows both measured and theoretical (using Equation 7) faradaic efficiencies as a function of the reactant concentration [R] for both the ClCH2CO2− (Figure 2a) and Fe(CN)63− (Figure 2b) systems. The theoretical estimation is based on a second-order recombination reaction rate constant of 2k2 = 1.1 × 1010 M−1 s−1, a current density of j = 15,600 A m−2, and a penetration depth of l = 24 nm. The current density was estimated by dividing the current by the area of the plasma, as previously reported,28 and the penetration depth was obtained from an analytical approximation,29 which is on the order of previously reported penetration depths.30,31 For both reactant systems, the measured faradaic efficiencies (closed circles) are far lower than what is predicted from the theory in Equation 7 (solid lines). One possible explanation is that competition with •OH, which is not accounted for in the analytical expression, reduces the measured faradaic efficiency.
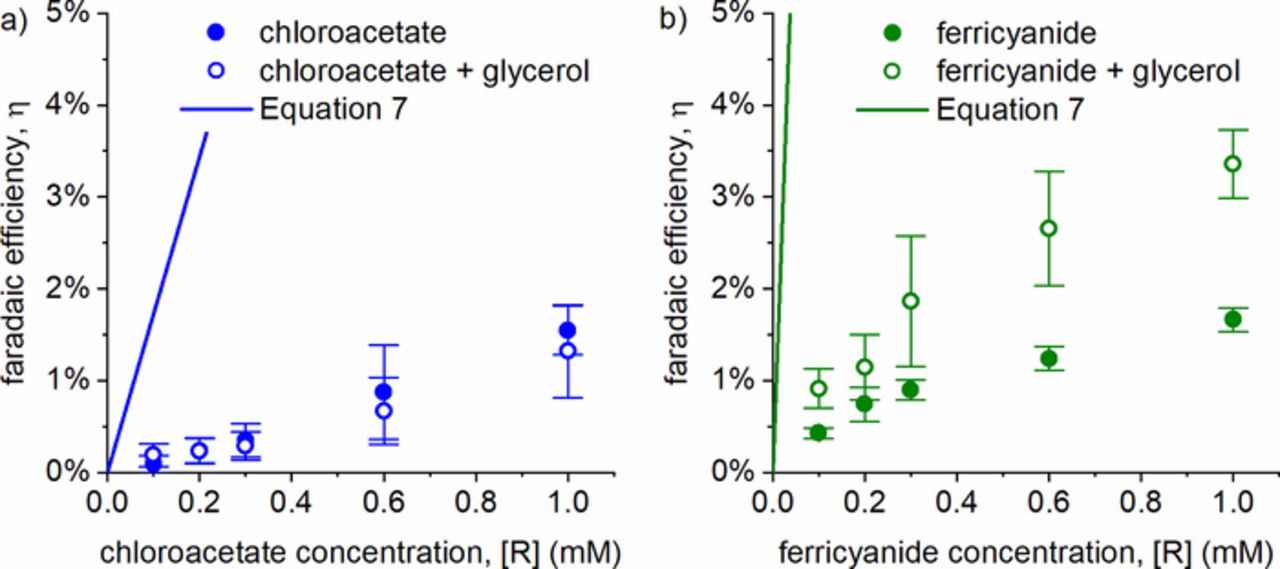
Figure 2. Faradaic efficiency without (●) and with (○) glycerol as an •OH scavenger, and the theoretical efficiency as predicted by Equation 7 for (a) ClCH2CO2−, and (b) Fe(CN)63−. Error bars reflect 95% confidence intervals for N ≥ 3 experiments.
It is well-known that in plasma electrolysis, •OH are readily produced by the dissociation of water, and that these rapidly react with solvated electrons,
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0008.gif)
Consequently, a large presence of •OH should reduce the faradaic efficiency by pure competition with Reaction 1. In addition to lowering the e−aq concentration, when the reactant has a reversible oxidation reaction, the presence of •OH has the potential to further decrease the reduction efficiency by oxidizing the product back into the reactant. Such is the case for Fe(CN)64−, which can be reduced back to Fe(CN)63− by
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0009.gif)
The effects of Reaction 8 and Reaction 9 on the reduction efficiency can be quantified using an •OH scavenger, such as glycerol. When glycerol is present at high concentrations, the reaction of •OH with glycerol (k10 = 1.8 × 109 M−1 s−1),32
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0010.gif)
dominates over Reaction 8 and Reaction 9, and minimizes the reduction of •OH by e−aq and the oxidation of Fe(CN)64− by •OH. The product glycerol radical, like any other carbon-centered alcohol radical, does not react with e−aq.11
The reduction efficiency in the presence of 500 mM of glycerol (open circles) is also shown in Figure 2 and can be compared to the measurements without glycerol (closed circles). The competition with •OH for solvated electrons (Reaction 8) seems to have little effect on the faradaic efficiency in the ClCH2CO2− experiments in Figure 2a, where the measurements with and without glycerol overlap. However, in the Fe(CN)63− experiment, the presence of •OH does affect the reduction efficiency significantly, as shown in Figure 2b. This observation can be reconciled with that of Figure 2a by realizing that the product glycerol radical is in fact a reducing species, capable of reducing Fe(CN)63− to Fe(CN)64-. This reduction reaction is known for other alcohol radicals with Fe(CN)63−,33 and transient absorption experiments at the Notre Dame LINAC have confirmed this process in the case of glycerol, with a measured rate constant 2.2 × 109 M−1 s−1 (Section S.4 of the Supplementary Material). As a point of comparison, very similar results to Figure 2b were obtained using methanol as an •OH scavenger (Section S.5 of the Supplementary Material). If we presume that the •OH flux is only a few percent relative to the e−aq, the alcohol might have very little effect on the steady state concentration of e−aq, in agreement with results of Figure 2a. However, the glycerol product radicals, though few in number, will be relatively long-lived species, and will eventually find and react with Fe(CN)63−. The absolute increase of faradaic efficiency shown in Figure 2b is at most 1.5%, even though the relative increase is a factor of two.
It seems safe to conclude that •OH radicals play a very minor role in the chemistry induced by the plasma cathode in this system. Critically, the measured faradaic efficiencies shown in Figure 2, both with and without an •OH scavenger, are much lower than the theoretical kinetics-based prediction of Equation 7. These results indicate that the measured low efficiencies cannot be explained solely by competing chemical reactions.
Reactant transport limit significantly limits faradaic effi-ciency
An alternative explanation for the measured low faradaic efficiencies is the depletion of the target reactant R at the plasma-liquid interface. Recent analytical models show that the penetration of both e−aq and •OH will only be on the order of 10–100 nm due to recombination.29 If the electron reactant is depleted within this small distance near the interface, then the faradaic efficiency will be inherently limited. Figure 3a shows this concept schematically, where the reaction and subsequent diffusion of reactant from the bulk cause a depletion profile similar to that described by Nernst.34
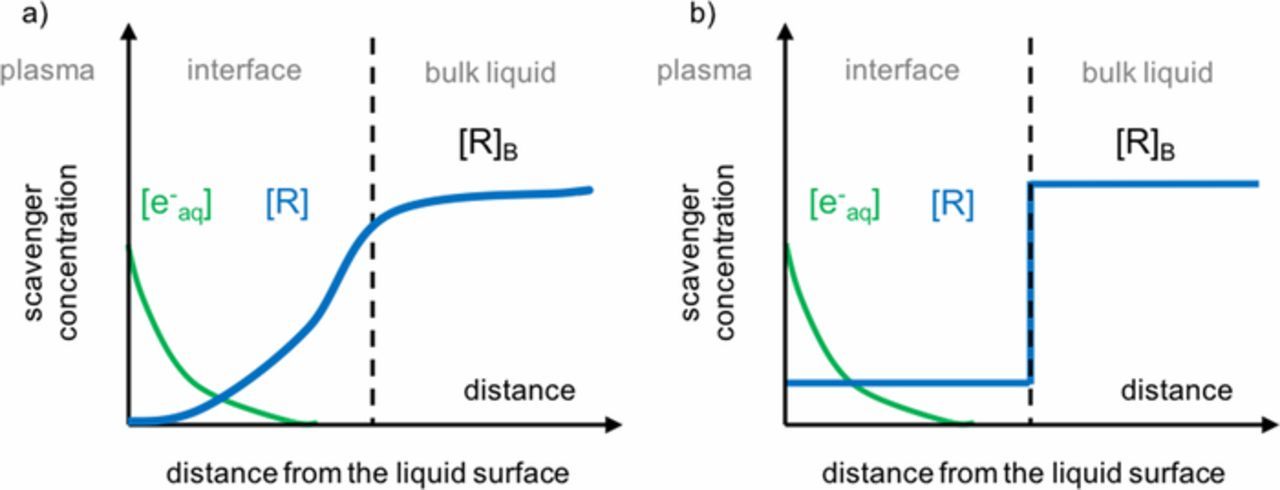
Figure 3. Concentration of a reactant from the plasma-liquid interface to the liquid shown as (a) the expected concentration profile and (b) a simplified model of the concentration differences between the bulk [R]B and the interface [R].
To approximate this, a one-dimensional, steady state reaction-diffusion equation can be derived for the electron reactant
![Equation ([11])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0011.gif)
where  is the diffusion coefficient of R in the catholyte. While Equation 11 describes the diffusive transport of reactants from the bulk to the interface, it is also important to note that the e−aq concentration also decreases with depth from the liquid surface as shown in Figure 3a.29 This will limit the rate of reaction in Equation 11. We account for this reduction by defining an effective concentration [e−aq]eff as some fraction of the e−aq concentration at the catholyte surface [e−aq], that is, [e−aq]eff = β[e−aq], where β takes some value less than 1. To simplify this further, the concentration gradient for R can be approximated as two different constant concentrations, one at the bulk and one at the interface as shown in Figure 3b. Equation 11 can hence be approximated by the difference between the reactant concentration in the bulk [R]B and at the interface [R],
is the diffusion coefficient of R in the catholyte. While Equation 11 describes the diffusive transport of reactants from the bulk to the interface, it is also important to note that the e−aq concentration also decreases with depth from the liquid surface as shown in Figure 3a.29 This will limit the rate of reaction in Equation 11. We account for this reduction by defining an effective concentration [e−aq]eff as some fraction of the e−aq concentration at the catholyte surface [e−aq], that is, [e−aq]eff = β[e−aq], where β takes some value less than 1. To simplify this further, the concentration gradient for R can be approximated as two different constant concentrations, one at the bulk and one at the interface as shown in Figure 3b. Equation 11 can hence be approximated by the difference between the reactant concentration in the bulk [R]B and at the interface [R],
![Equation ([12])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0012.gif)
where lr is the depletion length of the reactant from the bulk to the interface and  is the characteristic time of diffusion. With this approximation, the concentration of the reactant at the interface can be written as
is the characteristic time of diffusion. With this approximation, the concentration of the reactant at the interface can be written as
![Equation ([13])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0013.gif)
where τ is an effective time constant that has units of time and incorporates both the diffusion transport ( ) and embeds any fraction of the e−aq concentration as a result of depletion (β):
) and embeds any fraction of the e−aq concentration as a result of depletion (β):
![Equation ([14])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0014.gif)
When this revised concentration (Equation 13) is plugged into the definition for the faradaic efficiency in Equation 7, a modified expression with transport-limited reactant depletion is derived,
![Equation ([15])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0015.gif)
This expression contains the derived constant τ, and it should be clear that if this value goes to zero (indicating infinitely fast transport of the reactant to the interface), then Equation 7 is recovered. Although in this model, this time constant stems from diffusion of the reactant, it can be modified to reflect the characteristic transport time for other transport processes such as ionic migration via drift or convection.
While the value of τ is unknown a priori, its order of magnitude can be estimated using the diffusion time scale l2r/ , where the diffusivities of ClCH2CO2−35 or Fe(CN)63−36 are known, and approximating the fractional electron depletion β using models for the electron transport.29 Given the extremely high rates of Reactions 3 and 5 and the small interfacial length, the interfacial concentration profiles are expected to reach steady state on the order of microseconds. With this assumption, lr is expected to be constant and can be estimated as
, where the diffusivities of ClCH2CO2−35 or Fe(CN)63−36 are known, and approximating the fractional electron depletion β using models for the electron transport.29 Given the extremely high rates of Reactions 3 and 5 and the small interfacial length, the interfacial concentration profiles are expected to reach steady state on the order of microseconds. With this assumption, lr is expected to be constant and can be estimated as
![Equation ([16])](https://content.cld.iop.org/journals/1945-7111/166/6/E181/revision1/d0016.gif)
where the reactant flux from the bulk toward the interface, Jr, is obtained from the experimental rate of reaction. Given a reactant concentration of ∼1 mM, this would yield a length of lr ∼ 300 nm. Additionally, the interfacial reactant concentration will overlap with approximately β ∼ 5–10% of the peak e−aq concentration at the catholyte surface, due to the sharp decline in e−aq concentration as a function of distance from the interface.29 Taking all of these into account, the value of τ should be close to 10 μs. The corrected theoretical line is shown in Figure 4, where it can be seen that a τ of ∼10 μs provides a good fit to the experimental data for ClCH2CO2− from Figure 2.
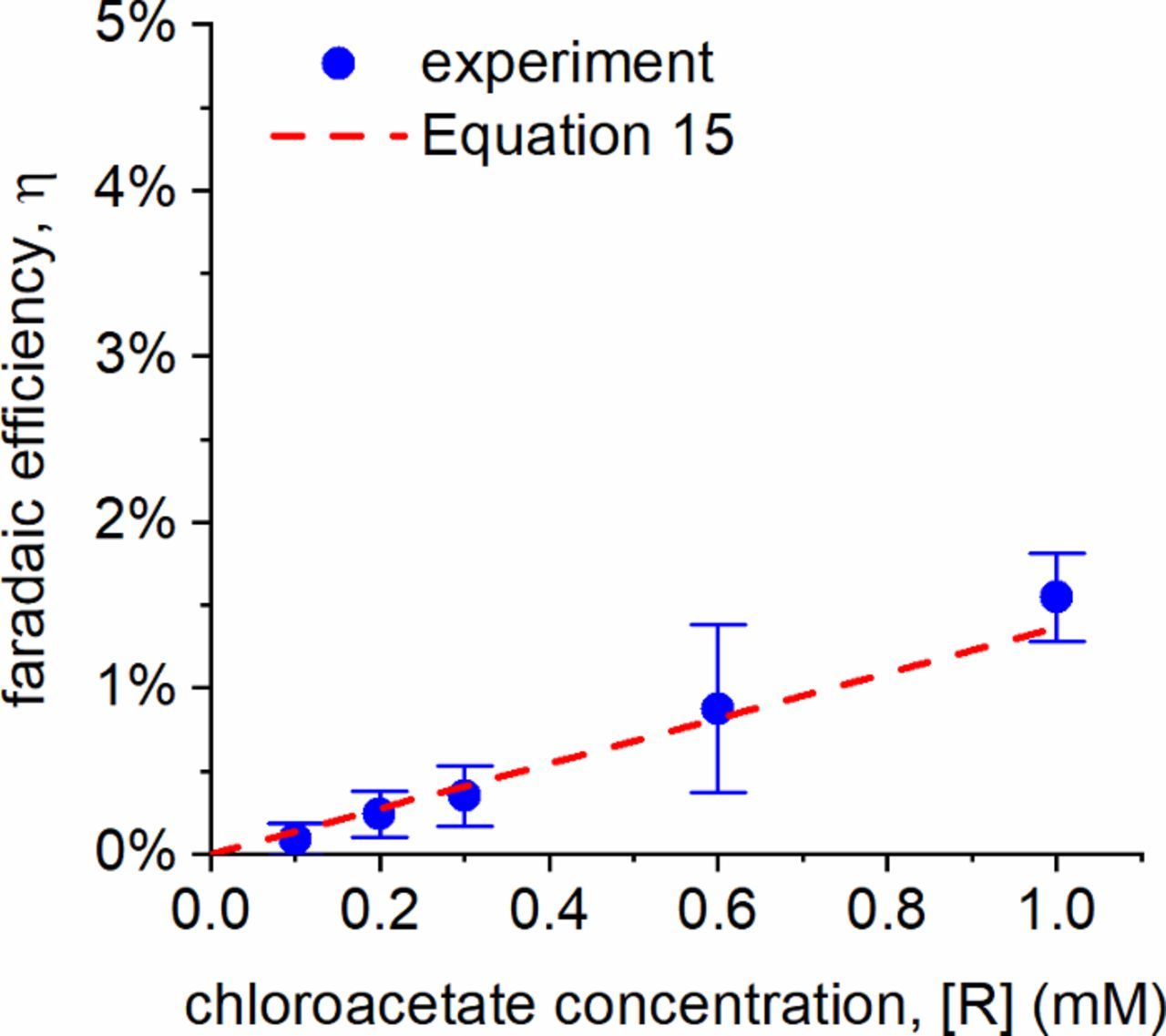
Figure 4. Comparison of experimental faradaic efficiency for ClCH2CO2− with the transport-limited theoretical efficiency in Equation 13 using a τ of 10 μs. Error bars reflect 95% confidence intervals for N ≥ 3 experiments.
These results show that a pure kinetics-based description of the faradaic efficiency in plasma cathode electrolytic systems is insufficient, even when accounting for competing reactions with oxidizing species. Rather, the faradaic efficiency is inherently limited by the transport of the reactant to the interface as well as the depletion of electrons from the liquid surface. It is also worth mentioning that Equation 15 is not meant as an accurate a priori prediction of the faradaic efficiency, but rather an approximate analytical model that is illustrative of how transport negatively affects the faradaic efficiency, with a very clear relationship between the efficiency and important system parameters.
Discussion
Transport-limited reactions are well-known in conventional electrochemical cells, and for this reason constant stirring of the solution is used to enhance convection.37 However, since the depletion layer is so small at the plasma-liquid interface (lr ∼ 100–1000 nm), stirring the solutions during the plasma reaction was ineffective for this system. A comparison for both ClCH2CO2− and Fe(CN)63− with and without stirring is shown in Figure 5, and no measurable effect was observed.
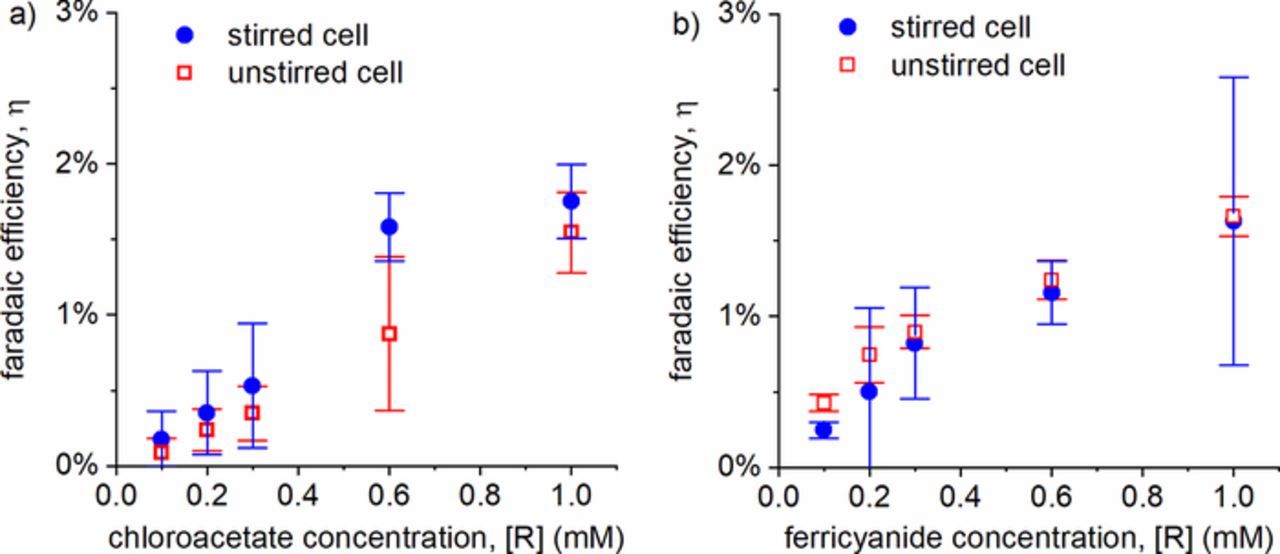
Figure 5. Comparison of experimental faradaic efficiency for (a) ClCH2CO2− and (b) Fe(CN)63− with and without stirring. Error bars reflect 95% confidence intervals for N ≥ 3 experiments.
Because overcoming the effects of transport limitations at the interface is so challenging, Equation 15 indicates that the easiest way to increase the faradaic efficiency is by increasing the reactant concentration. We confirmed this with ClCH2CO2− by measuring the faradaic efficiency at concentrations as high as 1000 mM, as shown in Figure 6, nearly achieving 100% efficiency.
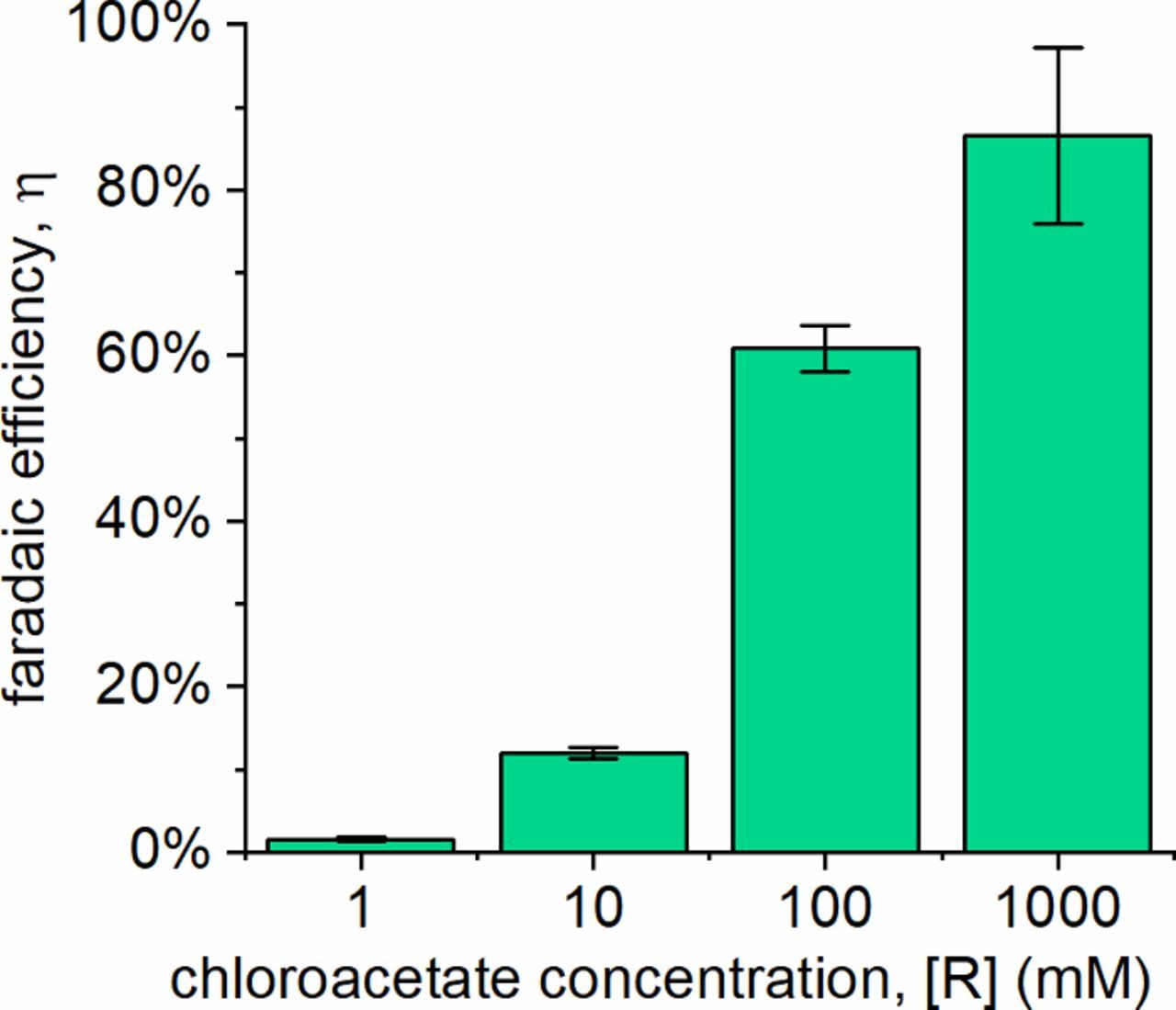
Figure 6. Faradaic efficiency at ClCH2CO2− concentrations of 1 mM, 10 mM, 100 mM, and 1000 mM. Error bars reflect 95% confidence intervals for N ≥ 3 experiments.
In practice, however, it is not always possible or desirable to increase the reactant concentration. For example, in the processing of dissolved gases, the gas concentration in the liquid is inherently limited by Henry's law; this is the case in the reduction of CO2, where experiments at atmospheric pressure yield low faradaic efficiencies.15 Or in the case of nanoparticle synthesis, low metal salt concentrations may be used to achieve a desired nanoparticle size, as is the case with silver nanoparticles,14 or they may be a consequence of experimental design, as in the anodic dissolution of copper to form copper oxide nanoparticles.38 In these cases, it may be difficult to achieve high faradaic efficiencies without very thoughtful design of the plasma electrolytic reactor. It is also important to consider that, with ions as reactants, drift may start to play a more important role as a transport mechanism in the presence of stronger electric fields, and this can be accounted for by adjusting the time constant in Equation 15; however, given an estimated exponentially decaying electric field of 104 V/m in the double layer on the liquid side,28 drift is not significant relative to diffusion in this particular configuration.
Depending on the application, the importance of a high faradaic efficiency may vary. Nevertheless, considering the depletion of the reactant at the plasma-liquid interface may help in the study of reduction and oxidation mechanisms, i.e., whether e−aq plays a primary or secondary role, and it may lead to the better design of plasma electrolysis systems. Given that the two chemical systems analyzed here fall in typical ranges for diffusion coefficients in liquids (∼1 × 10−9 m2/s) and reaction rates (1 × 109 – 1 × 1010 M−1 s−1) with e−aq, it is likely that other systems of interest will behave similarly at low concentrations. Finally, given that the transport limitation is exclusively based on reactant depletion close to the localized introduction of highly-reactive species, be it e−aq or •OH, this reduction in efficiency should extend to plasma anode configurations, either for synthesis4,5 or contaminant degradation.39 Likewise, given the similarities in chemistry and interfacial lengths (on the order of ∼100 nm), the analysis of this work should extend beyond plasmas in contact with liquids to plasmas in liquids (contact glow discharge electrolysis) as well.40,41
Conclusions
This work analyzed the reduction efficiency in plasma cathode electrolysis systems, using ClCH2CO2− and Fe(CN)63− as reaction models. A kinetics-based theoretical model for the faradaic efficiency, with and without transport, was derived to compare expected efficiencies with experimental measurements. Both systems indicate that, at low concentrations, the faradaic efficiency is limited by reactant depletion near the plasma-liquid interface. Importantly, competing •OH reactions are largely negligible compared to reactant depletion. Overall, because transport limitation seems to be the most important factor reducing the faradaic efficiency, high reactant concentrations and low current density should be used whenever possible. Future work should also focus on better understanding which systems this limitation applies to, when it applies, as well as possible ways to overcome or circumvent this effect.
Acknowledgments
This work was supported by the U.S. Army Research Office under Award Number W911NF-17-1-0119. DMB is supported by the U.S. Department of Energy Office of Science, Office of Basic Energy Sciences under Award Number DE-FC02- 04ER1553.
ORCID
Hernan E. Delgado 0000-0001-8969-6523
David M. Bartels 0000-0003-0552-3110
Paul Rumbach 0000-0002-0154-3045
David B. Go 0000-0001-8948-1442