Abstract
Lithium-sulfur batteries are considered to be one of the most promising candidates for next-generation "beyond lithium-ion" energy-storage systems. However, the commercialization of lithium-sulfur batteries has been hindered by fast capacity fade caused by polysulfide shuttling problems, as well as by a limited volumetric energy density due to low sulfur loading. Here, porous Fe3O4 nanospheres have been employed as a sulfur host to address the polysulfide shuttling problem through chemical interactions. We have employed optical microscopy and cryogenic scanning transmission electron microscopy (cryo-STEM) with X-ray energy dispersive spectroscopy (XEDS) elemental mapping to study the inherent microstructure of an Fe3O4/S composite with 85% sulfur content. We observe well-faceted, micrometer-sized sulfur particles embedded in a network of the conductive Fe3O4 nanosphere host particles. Our Fe3O4/S composite shows high capacity and outstanding cycling stability. The initial areal capacity is 2.6 mAh cm−2 and a high areal capacity of 2.2 mAh cm−2 was retained after 150 cycles. We believe that both the embedding of sulfur, in a conductive Fe3O4 network, and the strong chemical interactions between Fe3O4 and polysulfides are responsible for the high capacity and high cycling stability of our Fe3O4 composite cathode.
Export citation and abstract BibTeX RIS
Rechargeable batteries are used in a wide range of applications, such as portable devices, electric vehicles, and grid-scale storage of renewable energy.1–3 In the last two decades, lithium-ion batteries, one of the most widespread energy storage devices, have begun to approach their energy density limits.1 Lithium-ion batteries may struggle to satisfy the increasing demand for large-scale applications, such as electric vehicles, with a long driving range, and grid-level electrical energy storage. Thus, it is necessary to develop new materials for rechargeable batteries with high power and energy densities. In recent years, lithium-sulfur (Li-S) batteries have emerged as one of the most promising candidates for next-generation rechargeable batteries.4–8 As one of the most abundant elements on earth, sulfur can provide a high theoretical specific capacity (1,675 mAh g−1) and energy density of 2600 Wh kg−1; ten times higher than that of Li-ion batteries. Compared to lithium-intercalating transition metal oxides in commercial lithium ion batteries, sulfur has a very low cost (less than $100/ton) and is environmentally benign.4 Despite these advantages, a number of challenges must be overcome in order to develop commercially viable Li-S batteries. These challenges include the insulating nature of sulfur, which dramatically affects the upper limit of active sulfur loading in an electrode, and the highly soluble nature of long-chain lithium polysulfides (LiPSs) that are produced during discharge. LiPSs can easily shuttle between cathode and anode and react at both sides, leading to a reduced active material utilization along with fast capacity fade.8–13
Recently, researchers have devoted a great deal of effort to improving the conductivity of the sulfur electrode and mitigating LiPS dissolution. Combining sulfur with carbonaceous materials, such as porous carbons,14–19 activated carbons,20–22 graphene23–26 and conducting polymer materials,27–30 has been the most popular strategy because carbonaceous materials can provide high conductivity and LiPSs may be confined within the porous structures. Although the cycling stability of Li-S batteries can be enhanced to some extent using these host materials, the capacity decay is still severe after moderate to long-term cycling. This is most likely due to the fact that carbon, being largely non-polar in nature, cannot provide strong adsorption sites to bind highly polar LiPSs in the electrolyte.31 Recently, some metal oxides have been found to have strong chemical interactions with LiPSs, and have shown good cycling performance in Li-S batteries. To the best of our knowledge, the metal oxides and hydroxides that have been studied to date include TiO2,32–34 TiO,35,36 MnO2,37–39 MnO,40 La2O3,41 Ti4O7,42,43 Nb2O5,44 SnO2,45 Fe3O4,46,47 Ni(OH)212 and Co(OH)2.48,49 However, as most metal oxides/hydroxides have poor electrical conductivity, the specific capacity of Li-S batteries made with these metal oxides has, so far, been relatively low/modest. Moreover, even though these metal oxides can provide high affinity, the content of sulfur in the entire electrode has been low (<60%). As Gao et al.50 have shown, the sulfur content must be at or higher than 70% in the whole electrode in order for a Li-S battery to compete with commercial lithium ion batteries (for example LiCoO2) in terms of volumetric energy density. A highly conducting metal oxide-sulfur composite with high sulfur content would be most promising.
An additional challenge to overcome, when studying Li-S batteries, is to ensure accurate and precise characterization of the nanoscale distribution of sulfur. Some researchers have routinely used scanning/transmission electron microscopy (SEM/TEM) under high-vacuum conditions to study the nano-structure of sulfur/host material composites. High vacuum SEM/TEM observations have led researchers to attribute battery performance enhancements to the physical confinement of sulfur in or on the host material.31,32,51–53 However, sulfur is prone to sublimation in high vacuum environments, like that of an electron microscope sample chamber.54,55 Recent studies have begun to make the Li-S battery community aware of the fact that sulfur sublimation, in an electron microscope, can cause loss of sulfur from a sulfur electrode sample, as well as a redistribution of sulfur within the sample, due to the capture of sublimated sulfur gas by porous materials.56,57 Characterization of sulfur battery materials by standard, room temperature electron microscopy may therefore give misleading results, and alternative methods are required.
In this work, we have found that Fe3O4 can significantly improve the cycling stability of sulfur electrodes in Li-S batteries. Fe3O4 is one of the most inexpensive metal oxides with high electronic conductivity (2 × 104 S m−1) and low toxicity.58 Porous Fe3O4 nanoparticles were synthesized using a facile hydrothermal reaction, and an Fe3O4/S composite was obtained via a melt diffusion method, which is suitable for large scale production. Our Fe3O4/S composite, with 85% sulfur content, exhibited high capacity and outstanding cycling stability. The initial areal capacity was 2.6 mAh cm−2 and a high areal capacity of 2.2 mAh cm−2 was achieved and maintained after 150 cycles. In order to study the structure and distribution of sulfur in our Fe3O4/S composite, without sulfur sublimation artifacts, we employed a combination of optical microscopy and cryogenic scanning transmission electron microscopy (cryo-STEM). Cryo-STEM has previously been shown to suppress sulfur sublimation in vacuum and preserve the inherent distribution of sulfur in sulfur battery samples in an electron microscope.57,59 This combination of cryo-STEM together with simple optical microscopy, provides a more accurate understanding of the structure of our Fe3O4/S composite, and is useful for studying other Li-S battery systems.
Experimental
Preparation of porous Fe3O4
Porous Fe3O4 nanospheres were synthesized via a hydrothermal reaction.60 1.35 g of FeCl3·6H2O were dissolved in 60 mL of ethylene glycol, followed by the addition of 3.85 g of ammonium acetate. After stirring for 90 min, the solution was transferred to a Teflon-lined stainless steel autoclave with a capacity of 100 mL, and kept at 200°C for 20 h. After cooling down, the precipitate was centrifuged and washed with deionized water.
Preparation of Fe3O4/S composites
Fe3O4/S composites were prepared via a simple melt diffusion method. A mixture of porous Fe3O4 nanospheres and sulfur powder (15:85 weight ratio) was heated under an ambient atmosphere at 155°C for 12 h.
Preparation of Li2S6
A Li2S6 solution was prepared by dissolving stoichiometric amounts of Li2S and elemental S into 1,2-dimethoxyethane and 1,3-dioxolane (DME/DOL, 1:1 in volume) at 60°C overnight in an argon glove box.
Structural characterization
X-ray diffraction (XRD) patterns were recorded from samples of Fe3O4 and the Fe3O4/S composite using a Rigaku Ultima VI diffractometer with a Cu Kα source. Diffraction patterns were collected at a scan rate of 5° min−1 and with an increment of 0.02°.
A white light reflectance optical microscope (Olympus) was used to acquire extended depth of field images of samples of Fe3O4/S composite particles, which were dispersed in ethanol, and transferred onto a glass slide. Fe3O4/S composite particles dispersed in ethanol were transferred onto copper transmission electron microscope (TEM) grids with a lacey carbon film (Electron Microscopy Sciences) for characterization by TEM. TEM grids containing the sample were loaded into a Gatan model 914 cryo-holder under nitrogen gas, near liquid nitrogen temperature. The holder kept the sample at a stable temperature of about −180°C. STEM images were acquired using an FEI Tecnai F-20 microscope operated at 200 keV. XEDS elemental mapping was performed using an Oxford X-Max detector.
Electrochemical measurements
Electrodes with < 1.5 mg cm−2 sulfur loading were prepared for the basic cycling tests by mixing the Fe3O4/sulfur composite described above with Super P carbon and polyvinylidene fluoride (PVdF) binder in N-methyl-2-pyrrolidone (NMP) in a weight ratio of 75:15:10. The resulting slurry was doctor-blade coated onto carbon coated Al foil current collectors to prepare the cathodes. For the areal capacity measurements, the preparation of high sulfur loading electrodes (∼70 wt% of sulfur in the entire electrode composite assembly), Fe3O4/sulfur composites with Super P and PVdF binder were dispersed in NMP in a weight ratio of 82: 12: 6. The slurry was coated onto carbon paper. The electrodes were dried at 50°C overnight.
2032-type coin cells were assembled in an argon-filled glove box (<0.30 ppm oxygen). Celgard 2300 membrane was used as the separator and Li metal as the anode. The electrolyte was 1.0 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in 1,3-dioxolane and 1,2-dimethoxyethane (DOL/DME, 1:1 by volume) with 0.2M LiNO3 as an additive. The volume of electrolyte injected in the coin cell was 40 μL for electrodes < 1.5 mg cm−2 and 60 μL for higher loading electrodes. Cyclic voltammetry (CV) profiles were recorded over the potential range from 1.5 to 3.0 V (vs. Li+/Li) at a sweep rate of 0.1 mV s−1. Galvanostatic charge/discharge measurements and CV were carried out over a voltage window of 1.5–3.0 V using a BT2000 battery cycler (Arbin instruments).
Results and Discussion
An XRD pattern acquired from a sample of our hydrothermally synthesized porous Fe3O4 nanospheres showed the typical XRD features of magnetite (Figure 1a). The morphology and size distribution of the Fe3O4 nanospheres are presented in Figures 1b–1d. A high angle annular dark-field (HAADF) STEM image of a typical nanosphere (Figure 1b) shows a variation in contrast within the nanosphere, suggesting that porous channels exist inside the nanoparticle, providing large surface area for the Fe3O4 host material to interact with sulfur and LiPSs. A specific surface area of 16.6 m2 g−1 for the porous Fe3O4 nanospheres was determined by Brunauer–Emmett–Teller (BET) analysis. Measurements of particle sizes from an HAADF-STEM image containing many Fe3O4 nanospheres with a typical size range suggests that a majority of the nanospheres are 80–140 nm in diameter (Figures 1c–1d).
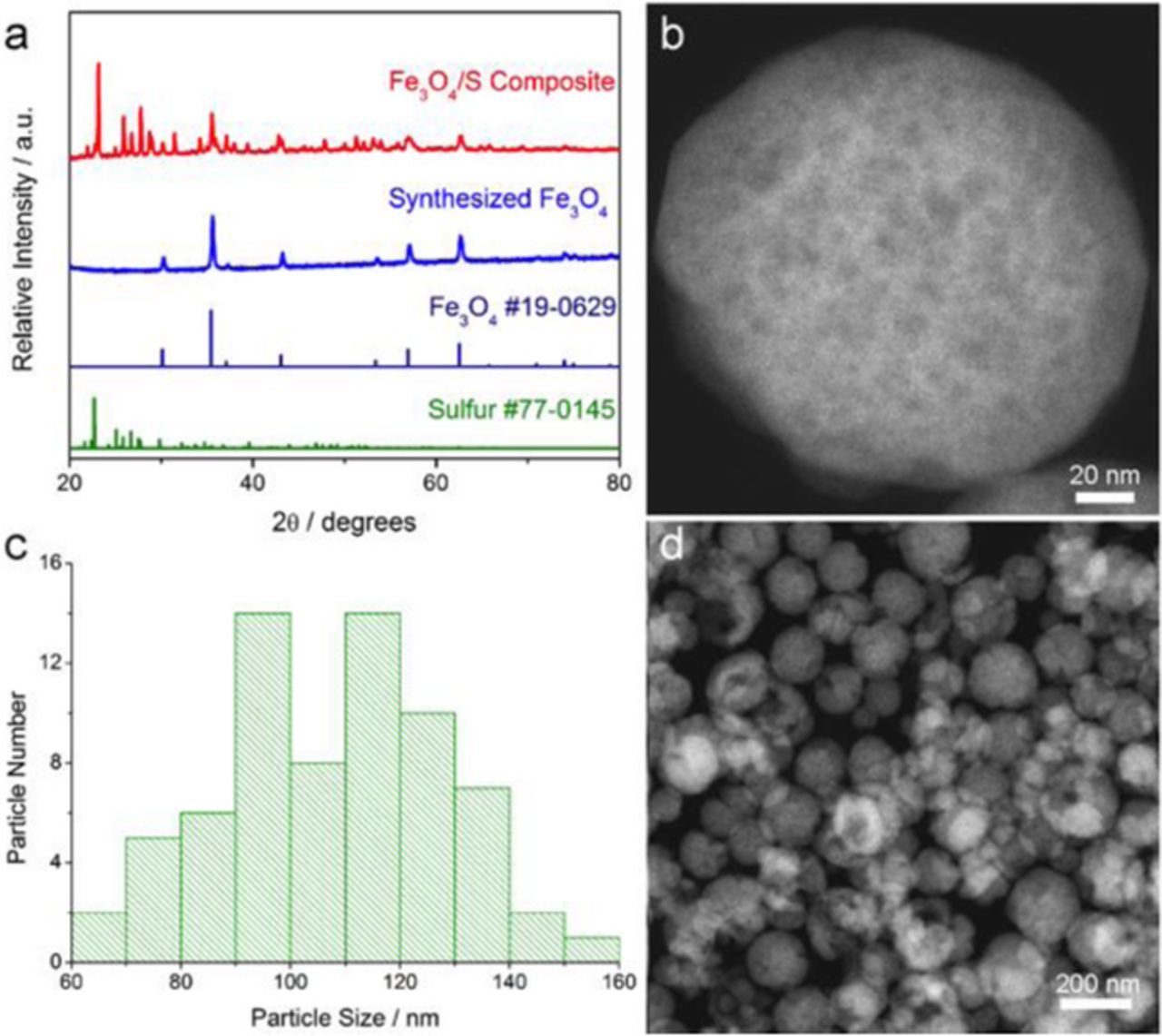
Figure 1. (a) Powder XRD of the as-synthesized Fe3O4 and Fe3O4/S composite compared with the standard XRD of Fe3O4 and elemental sulfur. (b) High-magnification STEM image of a representative Fe3O4 nanosphere with porous inner structure. (c,d) HAADF-STEM image of porous Fe3O4 nanospheres and the corresponding particle distribution.
An Fe3O4/S composite was prepared by melt-diffusion of sulfur into the Fe3O4 nanospheres. The sulfur content of our Fe3O4/S composite was determined to be 85% by thermogravimetric analysis (TGA) under an argon atmosphere (Figure S1). XRD analysis of the Fe3O4/S composite exhibited the XRD peaks of both magnetite and crystalline orthorhombic elemental sulfur (Figure 1a), indicating that much of the sulfur in the composite is in crystalline form. Fe3O4/S composite particles were further examined by optical microscopy and cryo-STEM equipped with X-ray energy dispersive spectroscopy (XEDS). Optical microscopy is a useful technique for studying materials at the micrometer scale, and screening of samples in an optical microscope, prior to study by electron microscopy, helps to ensure that nanoscale observations made in an electron microscope are truly representative of the bulk material. This is particularly useful and valuable for samples containing elemental sulfur, as samples can be studied in an optical microscope without exposure to vacuum.57 Figure 2a shows an extended depth of field optical image of Fe3O4/S composite particles. The image shows yellow sulfur particles encrusted with black Fe3O4 particles. This observation is consistent with our XRD measurements in Figure 1a, which show the presence of crystalline sulfur and Fe3O4. Optical images from a sample containing only sulfur particles, and a sample containing only Fe3O4 nanoparticle powder are shown for comparison in Figure S2. After screening by optical microscopy, cryo-STEM with XEDS was used to directly image the distribution of sulfur over the Fe3O4 host material. Figure 2b shows an HAADF STEM image of Fe3O4/S composite particles. The XEDS spectrum of these composite particles shows a very strong sulfur peak, which is consistent with the high mass loading (85%) of sulfur in the Fe3O4/S composite (Figure 2c). XEDS elemental maps of these particles in cryo-STEM exhibited well-faceted, micrometer sized, sulfur particles embedded in a network of the Fe3O4 host particles (Figures 2d–2f). The XEDS spectrum from some of the Fe3O4 particles exhibits a weak S signal relative to the Fe signal, indicating the relatively weak sulfur infiltration into Fe3O4 nanospheres (Figure S3). Additional images of other composite particles, showing the same structure (Figure S4). From these images, it is clear that a majority of the sulfur particles are in close physical contact with the conductive network formed by Fe3O4 nanoparticle clusters (Figures 2e–2f), which is similar to our recent work on the layered TiS2 as sulfur hosts.61
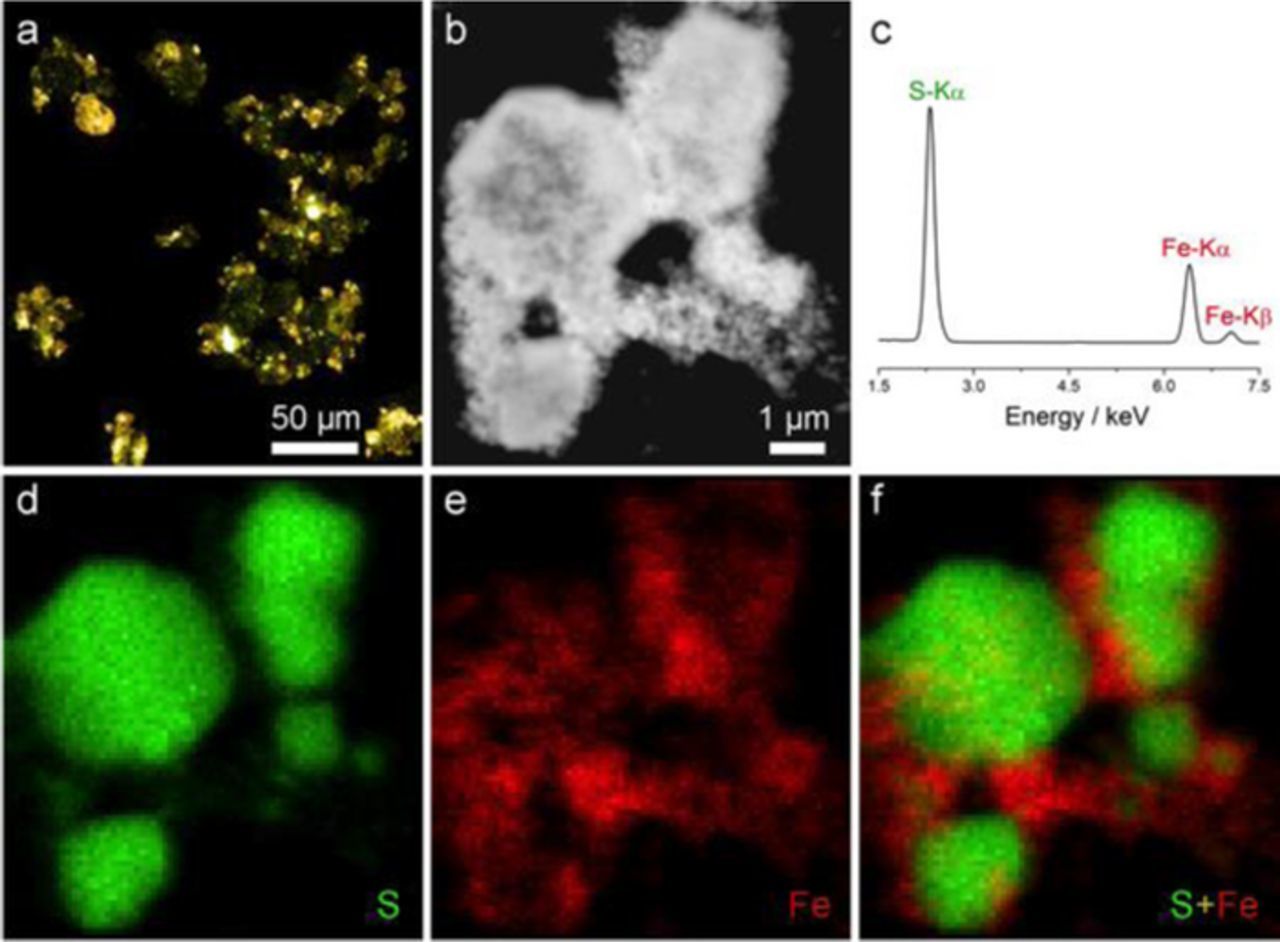
Figure 2. (a) Optical microscopy images, with extended depth of field, for black Fe3O4 with yellow sulfur composite. (b) HAADF-STEM image of Fe3O4/S composite. (c) Part of the XEDS sum spectrum of the Fe3O4/S composite, showing iron and sulfur X-ray peaks. (d-f) XEDS elemental mapping of sulfur, iron and color overlay of sulfur and iron.
The polar nature of both Fe3O4 and LiPSs suggests that LiPSs may adsorb onto Fe3O4 due to chemical interactions during battery cycling. The ability of Fe3O4 to adsorb LiPSs was investigated by adding porous Fe3O4 nanospheres and porous carbon, respectively, to a polysulfide solution of 1 mM Li2S6 in DOL/DME (1:1 by volume). Figure 3a shows that the polysulfide solution becomes colorless after the addition of Fe3O4 nanospheres while the solution remains yellow after the addition of porous carbon. This clearly indicates that there is a strong interaction between the Fe3O4 nanospheres and LiPSs, suggesting that the Fe3O4 nanospheres will interact with LiPSs produced during battery cycling.
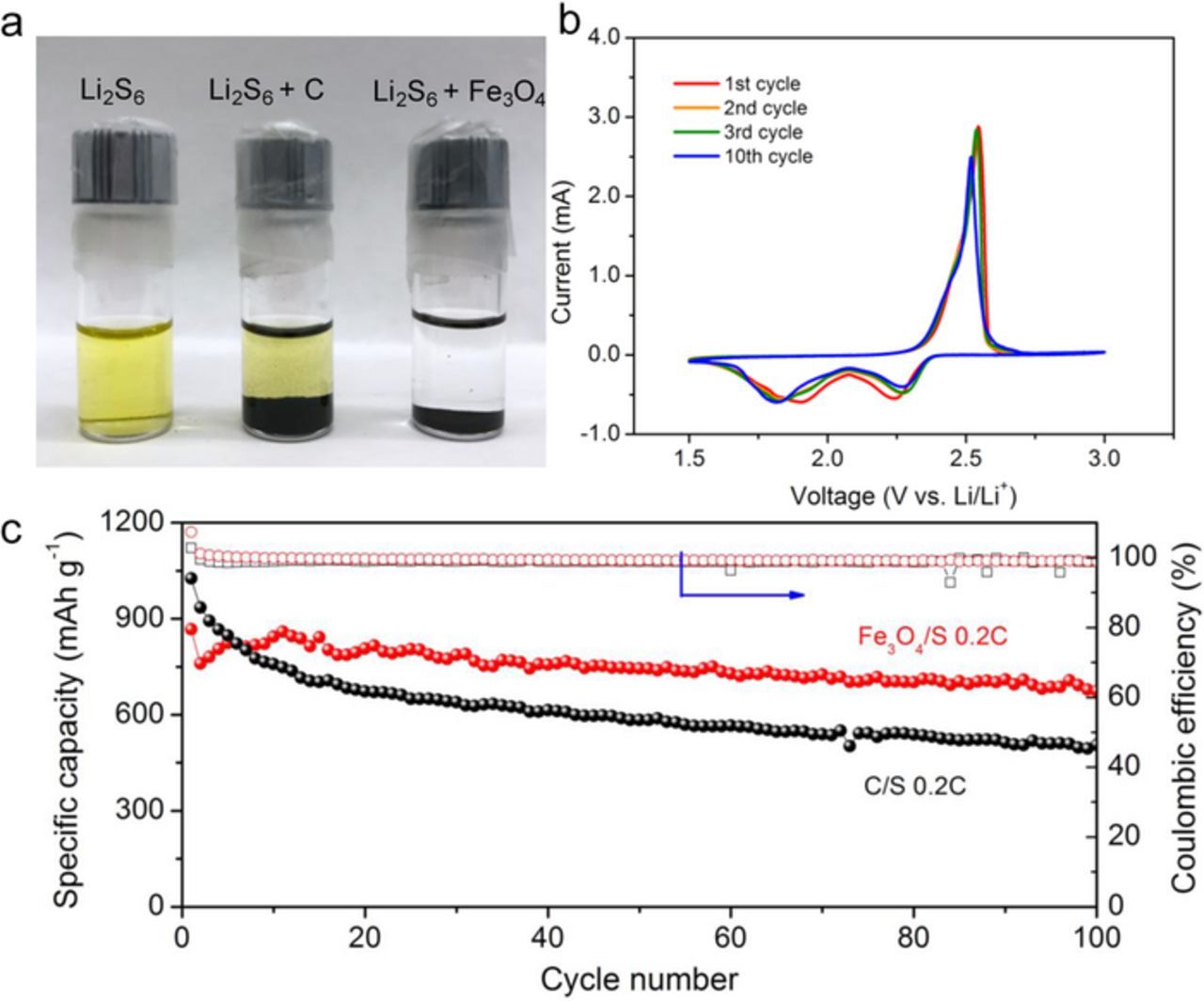
Figure 3. (a) Li2S6 adsorption test. Left to right: Li2S6 solution, Li2S6 with the addition of carbon and Li2S6 with Fe3O4. The solution became colorless after mixing with Fe3O4 but remained yellow after mixing with carbon. (b) Cyclic voltammetry of Fe3O4/S at scan rate of 0.1 mV s−1. (c) Cycling performance of Fe3O4/S composite and C/S composite at 0.2C.
Electrochemical performance testing of the Fe3O4/S composites was carried out in 2032-type coin cells. Cyclic voltammetric (CV) profiles of the Fe3O4/S composite employed as a cathode material in Li-S cells are shown in Figure 3b. In the cathodic scan, two well-defined reduction peaks at 2.23 V and 1.82 V can be observed, corresponding well to the generally accepted multi-step reduction mechanism of S8. The first peak corresponds to the reduction of S8 to higher order polysulfides Li2Sn (4≤n≤8) while the second peak corresponds to the further reduction to the lower polysulfides Li2Sx (x≤2). In the anodic scan, there is one oxidation peak at 2.32 V which corresponds to the oxidation of the lower polysulfides Li2Sx to S8.41 It can be clearly observed that there were no significant changes for either anodic or cathodic peaks after 10 cycles, pointing to the high cycling stability of the composite cathode material.
The cycling performances of a porous Fe3O4/S composite electrode and a C/S electrode at a current rate of 0.2C are presented in Figure 3c. The Fe3O4/S electrode delivered an initial discharge capacity of 867 mAh g−1 while the C/S electrode delivered 1090 mAh g−1. In the first few cycles, we found that the capacity of the C/S cathode was higher than that of the Fe3O4/S electrode, which may be due to the large surface area of carbon having better contact with the sulfur. However, over longer-term cycling, the capacity retention of the Fe3O4/S composite was superior to that of the C/S electrode. After 100 cycles, the Fe3O4/S electrode retained a capacity of 680 mAh g−1, corresponding to a capacity retention of 78%, which is much higher than that of the C/S electrode (49%). A high, stable Coulombic efficiency of >99% was achieved for the Fe3O4/S composite. Long-term cycling tests of Fe3O4/S and C/S were also carried out at a rate of 0.5C (Figure S5). For C/S composites, only 29% of the initial capacity was retained after 500 cycles. In comparison, a capacity retention, as high as 50%, was achieved after 500 cycles for the Fe3O4/S composite, indicating greatly enhanced stability. However, we also acknowledge that this decay rate (0.1% per cycle) is not the best result that has been reported. We believe that this is due, at least in part, to the fact that our Fe3O4/S composite has a very high mass loading of sulfur (85%) and the host cannot provide sufficient contact area with the sulfur and, after long term cycling, some of the active material detaches from the host and diffuses into the electrolyte. We are currently working on metal oxide host materials with larger pore-filling capability and a more effective sulfur infiltration strategy. To rule out any capacity contribution from the Fe3O4, a pure Fe3O4 electrode was also tested over the potential range of 1.5 to 3.0 V. As Figure S6 illustrates, the porous Fe3O4 has negligible capacity within the working voltage window of sulfur.
To evaluate the rate capability and stability, tests were carried out by increasing the C-rates successively from 0.2C to 2C every 10 cycles, and then switched back to 0.2C (Figure S7). Our composite material delivered a high capacity of 450 mAh g−1 at 2C. Upon returning to 0.2C, the capacity was 750 mAh g−1, corresponding to 94% of the original capacity attained at 0.2C, indicating robustness and stability of the composite electrode material.
Due to the importance of high areal sulfur loadings for high volumetric energy density in practical applications, we prepared and tested electrodes with high sulfur mass loadings of 1.9, 2.8 and 3.7 mg cm−2. Figure 4a shows the cycling performances of these high sulfur-loading electrodes. All of the electrodes exhibited excellent stability for over 150 cycles. Galvanostatic charge/discharge profiles are shown in Figure 4b. There are two discharge plateaus and one charge plateau, in good agreement with the CV curves of the Fe3O4/S composite shown in Figure 3b. In addition, it is evident that increasing the areal sulfur loading did not give rise to increased polarization (overpotentials) during charge and discharge cycles, likely due to the fast kinetics of the material. A high areal capacity of 2.2 mAh cm−2 was obtained after 150 cycles.
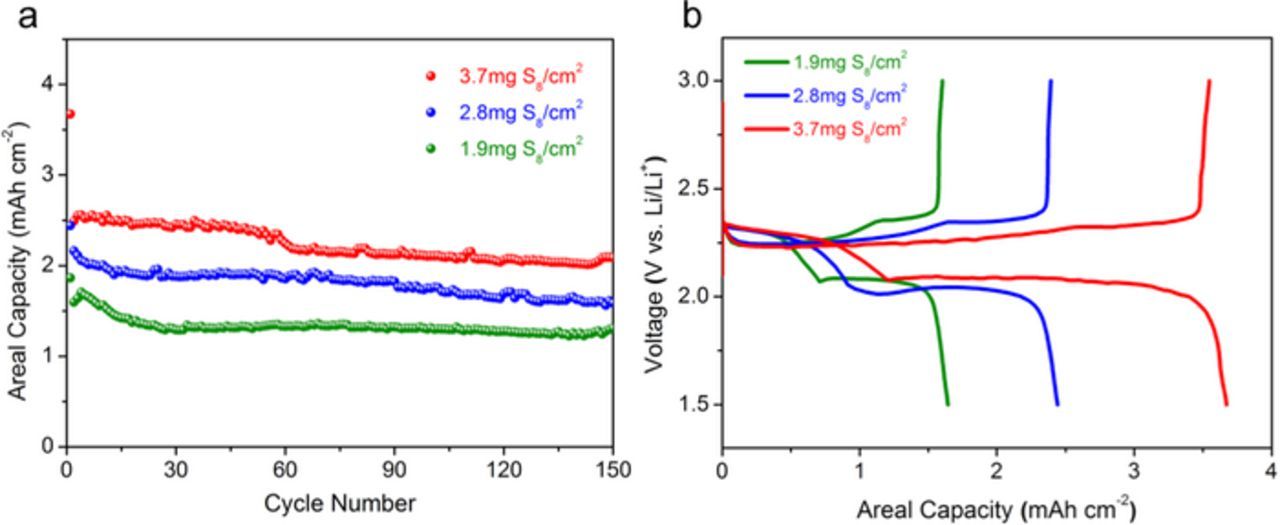
Figure 4. (a) Cycling performance of high-loading Fe3O4/S at a charge/discharge rate of 0.2 C for 150 cycles. (All cells were pre-conditioned at 0.05C for the first cycle.) (b) Initial cycle voltage profile of electrodes with various sulfur mass loadings.
A comparison of sulfur content in the electrode and areal capacity after 150 cycles with reports from the literature that used metal oxides as host material for sulfur is presented in Figure 5. This figure clearly shows that our Fe3O4/S composite electrode has high sulfur content and outstanding areal capacity after 150 cycles when compared to other similar metal oxide/S composites. Our data indicate that our Fe3O4/S composite electrode could potentially be used in practical applications requiring high-energy and long life-time Li-S batteries.
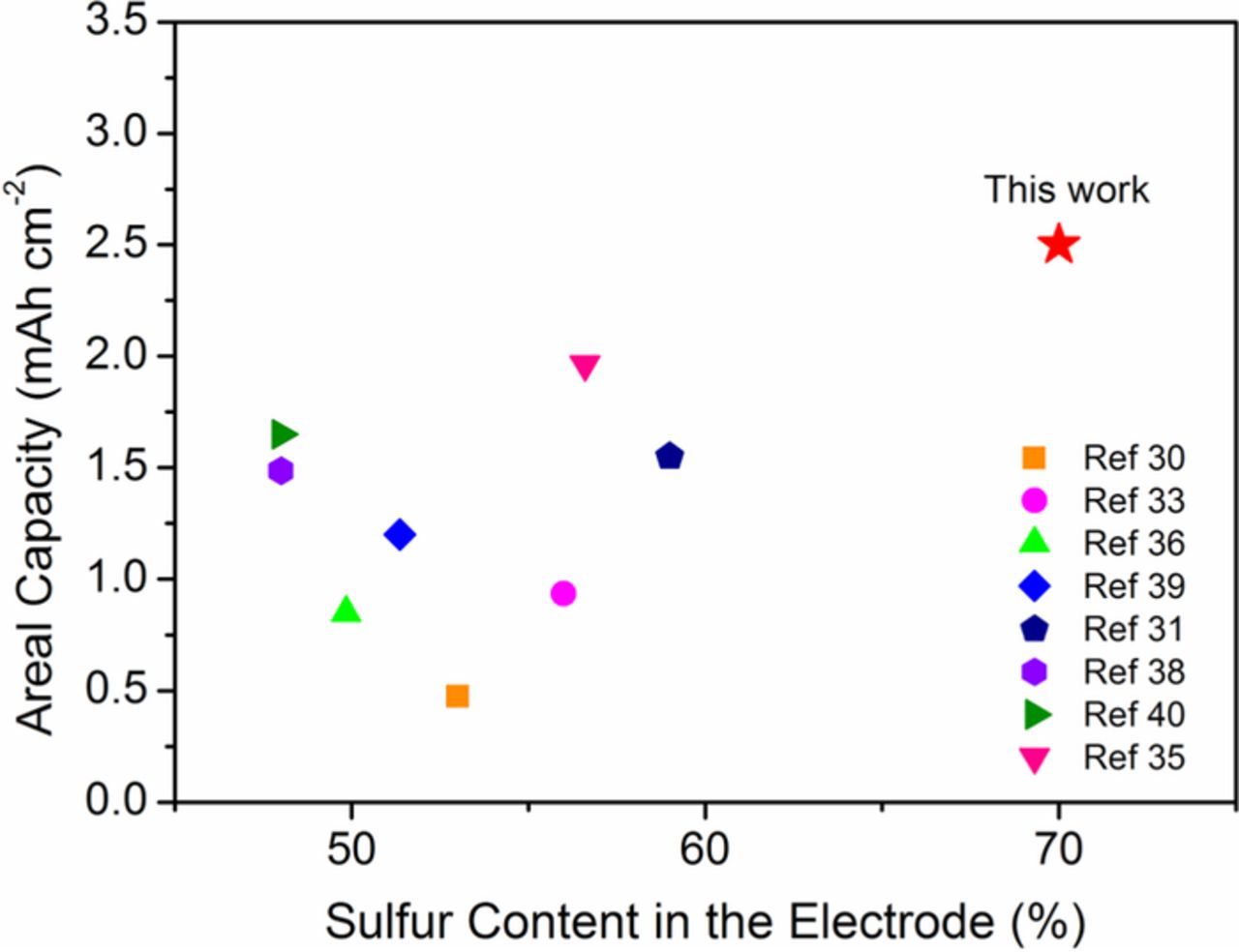
Figure 5. Comparison of sulfur content in the whole electrode and areal capacity of our Fe3O4/S electrode with other electrodes using metal oxides as the host matrix for sulfur, reported in the literature.
The excellent overall electrochemical performance of our Fe3O4/S cathodes can be attributed, at least in part, to the following factors. First, as a host material, Fe3O4 provides high electrical conductivity for electron transfer. We have observed, in optical microscopy and cyro-STEM, that sulfur particles are embedded in a conductive, porous Fe3O4 nanosphere network (Figure 2). This conductive Fe3O4 nanosphere network will facilitate charge transfer during cycling, and the pore structure of the nanospheres observed in Figure 1 may also provide physical entrapment of LiPSs. Second, as we have observed from the color change of polysulfide solutions (Figure 3a), the polar nature of Fe3O4 can provide a strong affinity for polar LiPSs, which could enrich the concentration of polysulfides near the surface of the conductive host, and further entrap LiPSs, mitigating the leaching of sulfur and/or polysulfides from the electrode during battery cycling. The porous structure of the nanospheres observed in Figure 1 provides a large surface area for interactions between Fe3O4 and LiPSs to occur. Chemical adsorption of LiPSs during charging and discharging plays a key role in the cycling stability of our composite.
Conclusions
In conclusion, we have demonstrated that Fe3O4 is a promising sulfur host for high-energy and stable Li-S batteries. Porous Fe3O4 nanospheres were synthesized via a facile hydrothermal reaction, and mixed with sulfur via melt-diffusion to form an Fe3O4/S composite. Cryo-STEM equipped with XEDS, together with optical microscopy, enabled a reliable characterization of the distribution of elemental sulfur in our Fe3O4/S composites without the sulfur sublimation artifacts associated with room temperature electron microscopy.56,57 Sulfur particles were found to be embedded in the conductive network of the Fe3O4 host material, which can facilitate charge transfer during cycling. Furthermore, the porous channels within the Fe3O4, and the strong affinity between Fe3O4 and LiPSs, help entrap LiPSs during battery cycling, reducing the loss of sulfur to the electrolyte. Due to the high conductivity and chemical adsorption properties of Fe3O4, the porous Fe3O4/S composite cathode exhibits excellent cycling stability. High sulfur loading electrodes showed superior cycling performance with a high areal capacity of 2.2 mAh cm−2 after 150 cycles. By combining the high conductivity of Fe3O4 with the chemical confinement of LiPSs, the performance of lithium-sulfur batteries has been greatly enhanced. We believe that these findings will be of great interest to the energy community, in general, and to the battery field, in particular.
Acknowledgments
This work was supported as part of the Energy Materials Center at Cornell, an Energy Frontier Research Center funded by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-SC0001086. This work made use of SEM/TEM facilities of the Cornell Center for Materials Research (CCMR) which are supported through the National Science Foundation Materials Research Science and Engineering Center (NSF MRSEC) program (DMR- 1719875). We thank John Grazul for the assistance in the TEM facility.
ORCID
Na Zhang 0000-0003-1978-4469
Héctor D. Abruña 0000-0002-3948-356X