
Abstract
Electrolytic ions are known to impact the rates of electrocatalytic reactions, though a molecular understanding of the mechanism of such impact is not well understood. We employ density functional theory to investigate the specific adsorption of potassium and iodine ions on Cu (111), Cu (100) and Cu (211) electrode surfaces under CO2 electroreduction (CO2 ER) conditions and explore their effect on the reaction energetics and binding strength of reaction intermediates. The calculated equilibrium potentials for K* and I* adsorption (* denotes surface-adsorbed species) suggest that a low coverage of adsorbed K* and/or I* is thermodynamically favorable, at different pH, at CO2 ER overpotentials. Co-adsorbed I* serves to primarily weaken CO* binding and may promote CO* desorption or subsequent formation of C-H bonds, phenomena that are typically obstructed by a high surface coverage of CO*. Co-adsorbed K* preferentially increases the binding strength of O-terminated species, including CO* and CHO* relative to COH*. The presence of K* will shift the selectivity and kinetics of CO* reduction towards the CHO* pathway by destabilizing the COH* formation transition state. The impact of solvation in conjunction with the effects of ion specific adsorption on the energetics of CO* reduction are also discussed.
Export citation and abstract BibTeX RIS
The electrolyte composition impacts the catalysis of CO2 electroreduction at the metal/electrolyte interface. Specific adsorption of electrolytic ions to the metal surface can alter the availability of surface sites or the stability of adsorbed reaction intermediates. Subject to the electrolyte composition and the electrode potential, ions may specifically adsorb on to the metal surface.1,2 The specific interaction between an adsorbed ion and the metal surface is not only electrostatic, but is characterized by the formation of a direct bond with some degree of charge transfer. The distribution of ions and solvent molecules near the metal electrode impacts the rate of important electrocatalytic reactions.3,4
Early experiments by Hori and co-workers examined the electroreduction of CO2 and CO on a Cu electrode in hydrogen-carbonate solutions of various alkali-metal cations and concluded that cationic specific adsorption greatly affected the product selectivity of CO2 ER.5 H2 evolution was dominant over CO2 reduction on the Cu electrode with a Li+ containing supporting electrolyte. The selectivity of C2H4/CH4 increased with the cation size: Li+ < Na+ < K+ < Cs+. In another study, Hori et al. rationalized the variation of the electrode potential with solution properties during CO2 ER on a Hg electrode due to the specific adsorption of larger cations, from Li+ to Na+ to K+.6 Anagnostopoulus and co-workers reported increasing faradaic yields of ethylene during CO2 ER on copper electrodes with the cation order: Cs+ ∼ K+ > Li+.7 Ito et al. demonstrated an increase in the current efficiency of CO2 ER on an In electrode in alkali-metal carbonate solutions in the order: Rb+ > K+ > Na+ > Li+.8 Several studies showed that the presence of SO42− and CO32− enhanced the faradaic yield of CO2 ER relative to PO42− anions in solutions using various sp- and d-group metal cathodes.9–12 Zhou and co-workers reported higher faradaic efficiencies up to 95% of formate during CO2 ER on a Sn electrode with Na+ and SO42− supported electrolytes, while HCO3− and K+ containing electrolytes enhanced the production rate of formate over 3.8 μmol min−1 at a faradaic efficiency of 63%.13 These studies substantiate that electrolyte composition impacts the CO2 ER activity, yet highlight the lack of molecular-scale insight into how ions impact these differences. Recently, Strasser and co-workers added 0.3 M potassium halide (KCl, KBr and KI) to KHCO3 electrolyte during CO2 ER on Cu electrodes, and found that the faradaic selectivity and production rate of CO and CH4 can be tuned with the concentration and the nature of added halide (Cl−, Br− and I−). The authors concluded that the modifications in the electrocatalyst performance are mainly attributed to halides adsorbed on the copper surface.14 A mechanistic link between the electrolyte composition, electrode potential, and electrode composition in dictating the CO2 ER activity and selectivity is needed to rationally design the system.
The role of anions, including halides (F−, Cl−, Br− or I−), cyanide, sulfate, perchlorate and phosphate, in affecting surface electrochemical processes has been previously investigated using theoretical and STM, SXS and vibrational spectroscopy (IRAS) techniques.15–29 Single crystal studies using platinum provided clear evidence of anions affecting the rate of electrochemical reactions including oxygen reduction (ORR), hydrogen evolution (HER), and methanol electro-oxidation reactions via specific adsorption.30–34 While anions are known to specifically adsorb onto metal surfaces at high electrode potentials and low pH conditions, cations can specifically adsorb at lower electrode potentials and higher pH. Unlike anion specific adsorption, the phenomenon of specific adsorption of cations has seen limited investigation despite evidence of the cation identity uniquely altering the rate of electrochemical reactions. In previous studies by Markovic and co-workers, the catalytic activity of platinum towards ORR, hydrogen oxidation (HOR) and methanol oxidation in alkaline electrolytes varied with the alkali-metal cation in the order: Li+ << Na+ < K+ < Cs+.35,36 Sitta et al. examined the effect of alkali cation on the oscillatory electro-oxidation of alcohols (CH3OH, C2H4(OH)2, C3H5(OH)3) in alkaline media and reported that the oscillation frequencies followed an inhibition trend in the order of KOH > NaOH > LiOH in which the alkali-metal cations exhibited a site blocking effect due to non-covalent interactions.37,38 While the role of cations in these investigations is rationalized via non-specific interactions, the exact mechanism through which cations exert their influence is not well understood.
Due to the complex nature of the electrochemical interface, the experimental identification of the mechanism by which electrolytic ions affect surface electrochemical processes is highly challenging. Density Functional Theory (DFT) calculations provide a useful tool for examining the phenomenon of ion specific adsorption on metal surfaces. Recently, our group computed the equilibrium potentials for specific adsorption of alkali and alkaline earth metal cations on fcc (111) electrode surfaces and confirmed that the cations could specifically adsorb in the potential ranges of HOR and HER catalysis in alkaline solutions.39 Henson and co-workers examined the role of adsorbed alkali-metal cations (Li+, Na+ and K+) on hydrogen adsorption on Pt (111), Pt (100) and Pt (110) surfaces and demonstrated that cation adsorption was competitive with hydrogen adsorption in alkaline media.40 We previously employed DFT to compute the specific adsorption of aqueous halides (F−, Cl−, Br− and I−) on Cu (111), Cu (100) and Cu (211) surfaces and reported that the adsorption depended weakly on the surface facet relative to the ion identity.41 The adsorption preference, in order of the lowest equilibrium adsorption potential, was calculated as: F− < Cl− < Br− < I−. This study also examined the effect of solvation on the thermodynamic favorability of halide adsorption by explicitly adding water molecules to the copper surface. While these studies provide useful insight on ion surface coverage and the preferred potential range where specific adsorption is favorable; the subsequent effects on CO2 ER have not been addressed previously. Specifically adsorbed ions can impact electrocatalytic rates by site blocking or via complicated interactions that alter the binding strength or coverage of reactive intermediates present on the metal surface.
In this study, we use DFT to investigate whether ions specifically adsorbed to copper electrodes impact the elementary step energetics of CO2 ER. We postulate that, in a similar fashion that anions impact interfacial chemistry via specific adsorption, cations may specifically adsorb onto the metal electrodes at high pH and low potentials. In line with Hori's rationale,6 we hypothesize that the mechanistic modifications of CO2 ER and differences in selectivity in the presence of ions can be attributed to the ion specific adsorption on the electrode surface. The specifically adsorbed ions can (i) block active sites and affect the total available surface sites for electroreduction, (ii) affect the binding strength of reaction intermediates, and (iii) influence the kinetic barriers of elementary reactions. DFT can decouple these effects with a simplified description of the electrochemical environment. This work focuses on the impact of specifically adsorbed ions on the electroreduction of CO2 at different copper surface facets. The binding strength of the reaction intermediates and key reaction energetics are computed with and without ion specific adsorption.
We first employ DFT to estimate the equilibrium adsorption potential (U0) of the specifically adsorbed ions to determine the favorability of adsorption on the copper surface at CO2 electroreduction conditions. In particular, we consider the specific adsorption of K+, I− and co-adsorbed (K+ + I−) on Cu (111), Cu (100) and Cu (211) surfaces at a low surface coverage (θion = 1/9th ML). Of the alkali metal cations, K+ is selected due to its ability to adsorb more favorably to the metal surface than Li+ and Na+39 and for its preferred usage in CO2 ER experiments.42,43 Previous results show that halide adsorption on the metal surface is more favorable than other anions including sulfate and phosphate.15 While I− is less commonly employed, our previous findings reveal that I− adsorbs more favorably than F−, Cl− and Br− onto the copper surfaces.41 Recent experimental evidence of CO2 ER on copper electrodes in the presence of halides demonstrate that addition of I−, relative to Br− and Cl−, dramatically impacts the selective production of CH4.14
CO* (where * denotes surface-adsorbed species) reduction is the key activity and selectivity determining step in the CO2 electroreduction path on copper electrodes.44–47 The effect of adsorbed K* and I* (where * denotes surface-adsorbed species) is investigated on the binding strength and reaction free energy (G) of three CO2 ER intermediates: CO*, CHO* and COH*. In previous electrokinetic DFT studies, the key selectivity determining step in the production of CH4/CH3OH was determined to be the reduction of adsorbed CO* to form either CHO* or COH*.44,48 The path to form CH4 proceeds via the COH* intermediate which is cleaved to form surface carbon C* and water. The surface carbon C* is successively reduced to form CH4. The alternate path proceeds via the CHO* intermediate, which could yield CH3OH through successive reduction, but is also likely a path that leads to dimerization or C2 formation.49 To examine the effect of the specifically adsorbed ions on the selectivity determining step of CO* reduction, we compare the potential-dependent reaction free energies and activation barriers of two steps: CO*→CHO* and CO*→COH* in the presence of K* and I*.
Methods and Computational Details
Electronic structure calculations were performed using the Vienna ab initio Simulation Package (VASP v. 5.3.3)50–52 with the Projector Augmented Wave53,54 method for core-valence treatment. The Perdew-Burke-Ernzerhof (PBE)55,56 exchange-correlation functional described within the Generalized Gradient Approximation (GGA)57 was employed. The plane-wave basis set cutoff energy was set to 450 eV. A Monkhorst-Pack58 k-point mesh of 5 × 5 × 1 and 5 × 5 × 5 was used to sample the Brillouin zones of all surfaces and bulk structures respectively. The Fermi level was smeared with the Methfessel-Paxton59 scheme using a Gaussian width (σ) of 0.2 eV for surfaces and 0.003 eV for isolated molecules. The ionic convergence limit for unconstrained atoms was set to 0.02 eV/ Å while the self-consistent electronic convergence limit was set to 1 × 10−5 eV. A periodic surface slab model was employed to construct the copper surfaces: Cu (111), Cu (100), Cu (211) using an experimental lattice constant (aCu = 3.62 Å). The surfaces were modeled as a 3 × 3 supercell with a thickness of four metal layers and ∼15 Å of vacuum to exclude pseudo-slab interactions. The bottom two layers of the slab were constrained to imitate the bulk arrangement while the top two layers were relaxed. This surface cell allows for isolation of ion-intermediate interactions at low total surface coverage. Structural optimization of the surface bound species was conducted by initially considering high symmetry configurations (atop, bridge, fcc and hcp) of the adsorbate on Cu (111), Cu (100) and Cu (211) to compute the lowest energy stable configuration. The transition state and the corresponding activation barriers were calculated using the climbing-image Nudged Elastic Band (CI-NEB) method60 with 4–5 intermediate images. Vibrational frequency calculations were performed to confirm transition states. The optimized site preference, final converged configurations of the surface-bound adsorbates and transition states are presented in the Supplementary Information.
Estimation of free energies (G)
The free energy of surface bound species X* (GX*) is calculated using Equation 1 and includes vibrational correction terms that correct the free energy from 0 K to 298 K. For the chemical potential of gas-phase species X0(g), computed at standard conditions of 298 K and 1 atm pressure, all degrees of freedom are included in the enthalpy and entropy corrections as shown in Equation 2. For liquid water, the gas-phase free energy is determined at the room-temperature vapor pressure of water (pH2O = 0.031 atm). The free energy of solid-phase species X(s) (GX(s)) is set equal to their DFT energy as shown in Equation 3:
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0002.gif)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0003.gif)
G represents the free energy of constituent X that may be a surface bound (X*), gas-phase (X(g)) or solid (X(s)) species. EDFT and EZPVE refer to the 0 K DFT and zero-point vibrational energy with U, TS and PV as the entropy, internal energy and pressure-volume corrections, respectively. The free energy of aqueous-phase iodide, I−(aq) ( ), and potassium ion, K+(aq) (
), and potassium ion, K+(aq) ( ), are computed using the standard-state free energy of iodine gas, I2(g) (
), are computed using the standard-state free energy of iodine gas, I2(g) ( ) (assuming ideal gas behavior), and solid potassium, K(s) (
) (assuming ideal gas behavior), and solid potassium, K(s) ( ), respectively, along with the experimental standard reduction potential (U0)61,62 in V-SHE as shown in Equations 4–7. Equations 5 and 7 are valid when the free energy change of iodide and potassium dissolution is zero.
), respectively, along with the experimental standard reduction potential (U0)61,62 in V-SHE as shown in Equations 4–7. Equations 5 and 7 are valid when the free energy change of iodide and potassium dissolution is zero.
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0004.gif)
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0005.gif)
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0006.gif)
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0007.gif)
where e represents the absolute value of the elementary charge. Standard reduction potential values (at 25○C) of U0 I2 = 0.54 V-SHE and U0 K = −2.93 V-SHE are used.
Estimation of equilibrium adsorption potential (U0 NHE) for ion adsorption
The specific adsorption of iodide and potassium ions onto the solvated copper surface is described in Equations 8 and 9 respectively:
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0008.gif)
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0009.gif)
where (H2O)n* represents the copper surface with n H2O molecules explicitly adsorbed, I(H2O)n* and K(H2O)n* represent surface-adsorbed iodide and potassium solvated by n H2O molecules and I−(aq) and K+(aq) are the aqueous-phase ions. Owing to the difficulty in representing a dynamic ensemble of the H2O molecules within the framework of DFT, we instead resort to the use of a static explicit structure of H2O molecules at a local minimum. Final converged structures of solvated K* and I* on the copper surface are presented in the Supplementary Information. Detailed discussions on the adopted approach to model ion solvation was previously reported.39,41 We neglect longer range solvent stabilization of adsorbed ions in this model, which would act only to further stabilize adsorbed species.
The thermodynamic free energy change (ΔG) of solvated ion adsorption as a function of the electrode potential on an NHE scale (UNHE) is calculated using Equations 10 and 11:
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0010.gif)
![Equation ([11])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0011.gif)
where the free energy of aqueous-phase ions (G(aq)) is calculated using Equations 5 and 7, μ represents the dipole moment in the surface normal direction, UPZC is the potential of zero charge and d represents the Helmholtz layer thickness. The last term serves as a correction to the free energy change of ion adsorption and accounts for the interaction between the surface dipole moment (μX(H2O)n* − μ(H2O)n*, due to charge retention of the adsorbed ion) and the electrode/electrolyte interfacial electric field (UNHE – UPZC)/d. This term effectively allows ion adsorption with partial electron transfer. The formalism for this correction has been previously reported,39,41,63 and we approximate a UPZC of 0 V-RHE and d of 3 Å as used in these previous studies. The equilibrium adsorption potential (U0NHE) for iodide and potassium adsorption is calculated using Equations 10 and 11, when ΔGADS = 0, as shown in Equations 12 and 13:
![Equation ([12])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0012.gif)
![Equation ([13])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0013.gif)
Estimation of binding energies (BE)
The binding energies of all surface-bound CO2 ER intermediates are calculated using Equation 14. Equation 15 is employed to estimate the effect of the co-adsorbed ion on the binding energy of the CO2 intermediate.
![Equation ([14])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0014.gif)
![Equation ([15])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0015.gif)
where * denotes surface-adsorbed species, and  is the gas-phase DFT energy of the CO2 ER intermediates.
is the gas-phase DFT energy of the CO2 ER intermediates.
Estimation of potential-dependent reaction energies
The reaction free energy change (ΔG) of CO2 ER reactions as a function of electrode potential U can be calculated on a reversible hydrogen scale (RHE) using the computational hydrogen electrode (CHE) model.63 The relative free energy of an intermediate COxHy* formed from CO2(g) in the CO2 ER pathway as shown in Equation 16 is calculated using Equation 17. To estimate the effect of a co-adsorbed ion on reaction free energies of CO2 ER, Equation 17 is modified to account for the free energy of the adsorbed ion as shown in Equation 18.
![Equation ([16])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0016.gif)
![Equation ([17])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0017.gif)
![Equation ([18])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0018.gif)
Estimation of potential-dependent activation barriers (ΔGACT(U))
We have previously developed a simple, transferable method to approximate a potential-dependent activation barrier for a surface-mediated elementary electrochemical reaction .44,48,64 DFT models are unable to accurately capture the chemical potential of an aqueous phase proton in bulk electrolyte, within limited unit cells and static H2O structures. We assume that the reduction reaction in Equation 19 can be approximated as an inner shell reaction, with the transition state being local to the surface species (A*) and that electron motion is rapid once the nuclei attain the transition state. Under these assumptions, we can use local models of adsorbed species and the ion being transferred such that the elementary electrochemical reaction (Equation 19) can be recast in the form of an analogous non-electrochemical reaction as shown in Equation 20. The main approximation of this method is that the transition state located for the analogous non-electrochemical reaction (Equation 20) is equivalent to the potential-dependent reaction (Equation 19) at a specific potential U0. The equilibrium potential for the addition of H into the DFT model cell (to form (A*+H*)), denoted by U0 in Equation 21, allows the determination of the electrode potential at which the proper reference reactant state, A* + H+ + e−, and its intermediate state, (A + H)* have the same chemical potential. This allows the energy of the transition state to be referenced back to the chemical potential of the ion in the bulk electrolyte.
![Equation ([19])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0019.gif)
![Equation ([20])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0020.gif)
![Equation ([21])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0021.gif)
The activation barrier corresponding to the transition state of the reaction in Equation 20 is labeled G0act and is the barrier obtained from DFT with ZPVE corrections at U = U0. The barrier can be extrapolated to other potentials using the Butler-Volmer formalism as shown in Equation 2264
![Equation ([22])](https://content.cld.iop.org/journals/1945-7111/163/6/F477/revision1/d0022.gif)
where β denotes an effective reaction symmetry factor, similar to the Brønsted-Evans-Polanyi coefficient65 and typically varies between 0.3 and 0.7.66 In this study, we approximate β = 0.5 for all reactions. While future work will address improved estimation of the symmetry factor for various elementary steps, within the scope of this study, we assume that the relative activation barriers with and without ion co-adsorption will have minor variations in the values of β.
Results and Discussion
Equilibrium adsorption potentials of K* and I* on Cu (111), Cu (100) and Cu (211)
The equilibrium adsorption potentials, U0 (in V-NHE), for K+ and I− adsorption in a low-coverage limit (θ = 1/9th ML) on Cu (111), Cu (100) and Cu (211) surfaces were calculated using Equations 12 and 13 and are summarized in Table S1 of the Supplementary Information. A low-coverage limit is considered based on previous studies that concur that the favorability of adsorption decreases with increasing coverage, most likely due to the steric or Coulombic repulsion between adjacent ions at higher coverages.41,67 The specific adsorption of K+ is favorable at any potential more negative of  , while I− adsorbs favorably at potentials more positive of
, while I− adsorbs favorably at potentials more positive of  . Iodide adsorption is more favorable on the Cu (100) (U0 = −0.89 V-NHE) and Cu (211) (U0 = −0.85 V-NHE) surfaces and least favorable on the Cu (111) surface (U0 = −0.59 V-NHE). The favorability of potassium adsorption on the copper surfaces follows the order: Cu (211) (U0 = −2.04 V-NHE) > Cu (111) (U0 = −2.50 V-NHE) > Cu (100) (U0 = −2.65 V-NHE).
. Iodide adsorption is more favorable on the Cu (100) (U0 = −0.89 V-NHE) and Cu (211) (U0 = −0.85 V-NHE) surfaces and least favorable on the Cu (111) surface (U0 = −0.59 V-NHE). The favorability of potassium adsorption on the copper surfaces follows the order: Cu (211) (U0 = −2.04 V-NHE) > Cu (111) (U0 = −2.50 V-NHE) > Cu (100) (U0 = −2.65 V-NHE).
The effect of surface solvation on the equilibrium potentials of K* and I* adsorption was investigated by explicitly solvating K* and I* with up to 4–6 water molecules. Surface solvation dependence on the equilibrium adsorption potential of ions was calculated by systematically varying the number of explicit water molecules and is reported in Figure S14 of the Supplementary Information. Based on the analyses, we observe that the equilibrium adsorption potential is converged with respect to the number of explicit water molecules included with 6 molecules, albeit with noise that remains significant on the 111 surface. The equilibrium adsorption potentials of the explicitly solvated ions (Table S1) and the final converged configurations are reported in the Supplementary Information. Solvation increases the favorability of specific adsorption of K+ by shifting  to more positive potentials. The shift is greatest on Cu (100) (1.14 V) followed by Cu (211) (0.73 V) and Cu (111) (0.41 V). This significant increase in adsorption favorability when surface solvation is included matches approximately the solvation interactions during alkali cation specific adsorption onto Pt (111).39 Relative to K+, the effect of solvation on I− adsorption across the copper surfaces is minimal with a shift of 0.01–0.02 V-NHE in
to more positive potentials. The shift is greatest on Cu (100) (1.14 V) followed by Cu (211) (0.73 V) and Cu (111) (0.41 V). This significant increase in adsorption favorability when surface solvation is included matches approximately the solvation interactions during alkali cation specific adsorption onto Pt (111).39 Relative to K+, the effect of solvation on I− adsorption across the copper surfaces is minimal with a shift of 0.01–0.02 V-NHE in  values on Cu (111) and Cu (211) surfaces and 0.16 V-NHE on Cu (100). The relative impact of solvation can be rationalized by examining the charge retained by the ion upon adsorption and the resulting surface normal dipole moment (Figures S4 and S5 of the Supplementary Information). The magnitude of the surface dipole moments are significantly larger for adsorbed K* relative to I*, indicative of the higher amount of charge retained by K* than I*. By explicitly solvating K*, the surface dipole moment is lowered while K* retains more charge due to the H2O molecules that stabilize the adsorbate. Conversely, due to the small surface dipole moment and lower charge retention of I*, the effect of solvation on the stability of I* is minimal. This observation concurs with our previous investigations of the effect of solvation on the adsorption favorability of aqueous-phase halides on copper surfaces.41
values on Cu (111) and Cu (211) surfaces and 0.16 V-NHE on Cu (100). The relative impact of solvation can be rationalized by examining the charge retained by the ion upon adsorption and the resulting surface normal dipole moment (Figures S4 and S5 of the Supplementary Information). The magnitude of the surface dipole moments are significantly larger for adsorbed K* relative to I*, indicative of the higher amount of charge retained by K* than I*. By explicitly solvating K*, the surface dipole moment is lowered while K* retains more charge due to the H2O molecules that stabilize the adsorbate. Conversely, due to the small surface dipole moment and lower charge retention of I*, the effect of solvation on the stability of I* is minimal. This observation concurs with our previous investigations of the effect of solvation on the adsorption favorability of aqueous-phase halides on copper surfaces.41
Figure 1 shows the equilibrium adsorption potentials, U0 (in V-NHE), for explicitly solvated K* and I* in a low-coverage limit (θ = 1/9th ML) on Cu (111), Cu (100) and Cu (211) surfaces. This Pourbaix diagram thus indicates potential-pH regions at which specific adsorption of potassium and iodide on the copper surfaces is thermodynamically favorable. The solid black line in Figure 1 shows the thermodynamic equilibrium potential (U0 = 0.17 V-RHE)46 for CO2 reduction to CH4 (CO2 + 8 H+ + 8 e− → CH4 + 2 H2O). Previous experiments, however, have typically reported significant operating overvoltages of more than 0.5 V on copper electrodes to produce appreciable product yields.68,69 Under CO2 ER conditions, we expect a low coverage of iodide to adsorb on all three surface facets, with I* stability extending to larger CO2 reduction overpotentials at low pH and on Cu (211) and Cu (100) facets. A low coverage of K* can adsorb favorably at higher pH values on all three facets, though a significant CO2 ER overpotential is required for K* adsorption on the Cu (111) facet.
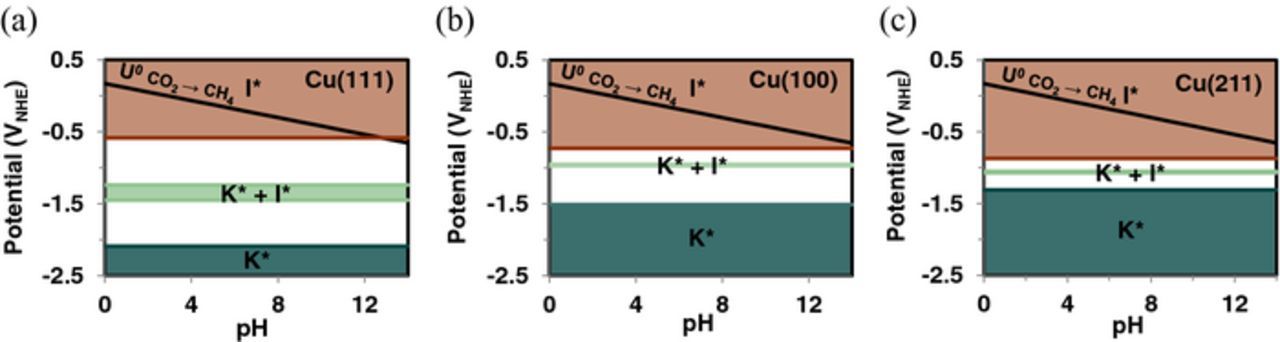
Figure 1. Pourbaix diagram for adsorbed K* (teal), I* (brown) and co-adsorbed (K* + I*) (green) explicitly solvated using six H2O molecules on (a) Cu (111), (b) Cu (100) and (c) Cu (211) surfaces. Solid black line denotes the thermodynamic equilibrium reduction potential for CO2 → CH4.
Though the potential ranges of isolated K* and I* formation do not overlap, the expected attraction between them could create a potential window in which the co-adsorption of K* + I* is favorable. We, therefore, also compute the equilibrium potential range that favors their co-adsorption. Figure 1 also shows the potential window within which the co-adsorption of explicitly solvated K* and I* is favorable. On Cu (111), the  value shifts from −2.09 to −1.24 V-NHE due to I* co-adsorption, while the
value shifts from −2.09 to −1.24 V-NHE due to I* co-adsorption, while the  value shifts from −0.58 to −1.45 V-NHE when K* is co-adsorbed, giving rise to a potential window within which K* and I* can co-adsorb on the Cu (111) surface. Under high pH CO2 ER operating conditions, co-adsorption of K* and I* is only favorable on the Cu (111) surface when explicit solvation effects are considered, and only under a narrow potential range (−1.45 <
value shifts from −0.58 to −1.45 V-NHE when K* is co-adsorbed, giving rise to a potential window within which K* and I* can co-adsorb on the Cu (111) surface. Under high pH CO2 ER operating conditions, co-adsorption of K* and I* is only favorable on the Cu (111) surface when explicit solvation effects are considered, and only under a narrow potential range (−1.45 <  < −1.24 V-NHE). Similar narrow potential ranges for K* + I* co-adsorption are observed for Cu (211) and Cu (100), as shown in Figure 1. Since the co-adsorbed ions are stabilized due to their attractive interaction, as evidenced by a higher charge retention (see Figure S5), it is plausible that higher coverages of co-adsorbed K* + I* could be favorable and expand the co-adsorption window.
< −1.24 V-NHE). Similar narrow potential ranges for K* + I* co-adsorption are observed for Cu (211) and Cu (100), as shown in Figure 1. Since the co-adsorbed ions are stabilized due to their attractive interaction, as evidenced by a higher charge retention (see Figure S5), it is plausible that higher coverages of co-adsorbed K* + I* could be favorable and expand the co-adsorption window.
Effect of adsorbed K* and I* on the binding energies of CO2 intermediates
To calculate the effect of co-adsorbed ions on the binding strength of the reaction intermediates, the ions and the reaction intermediates were adsorbed at their preferred site in a 3 × 3 unit cell (θ = 1/9TH ML/adsorbate). The ions were placed in the preferred site nearest to the reaction intermediate to quantify their interaction. Figures 2–3 illustrate a top view of the configurations used in modeling CO* reduction to CHO* and COH* (with explicit solvation) with and without ion co-adsorption on Cu (111), the results of which are elaborated in greater detail in the subsequent sections. The final converged configurations of the reaction intermediates with co-adsorbed ions on Cu (100) and Cu (211) surfaces are reported in the Supplementary Information. Figure 4 shows the variation in the binding energy of CO2 ER intermediates on three copper facets with ion co-adsorption. The binding energies of the CO2 ER intermediates are calculated using the gas-phase molecular energies as reference. More negative binding energy values correspond to stronger binding of the intermediates with the copper surface. The binding strength of the CO2 ER intermediates on all three copper surfaces varies in the order: COH* > CHO* > CO*. The O-terminated species, including CO* and CHO*, bind slightly stronger to the stepped Cu (211) facet than the close-packed Cu (100) and Cu (111) facets (Figures 4a–4b). This trend is reversed in the case of COH* (Figure 4c) which binds weakest to Cu (211) and strongest to the Cu (100) surface.
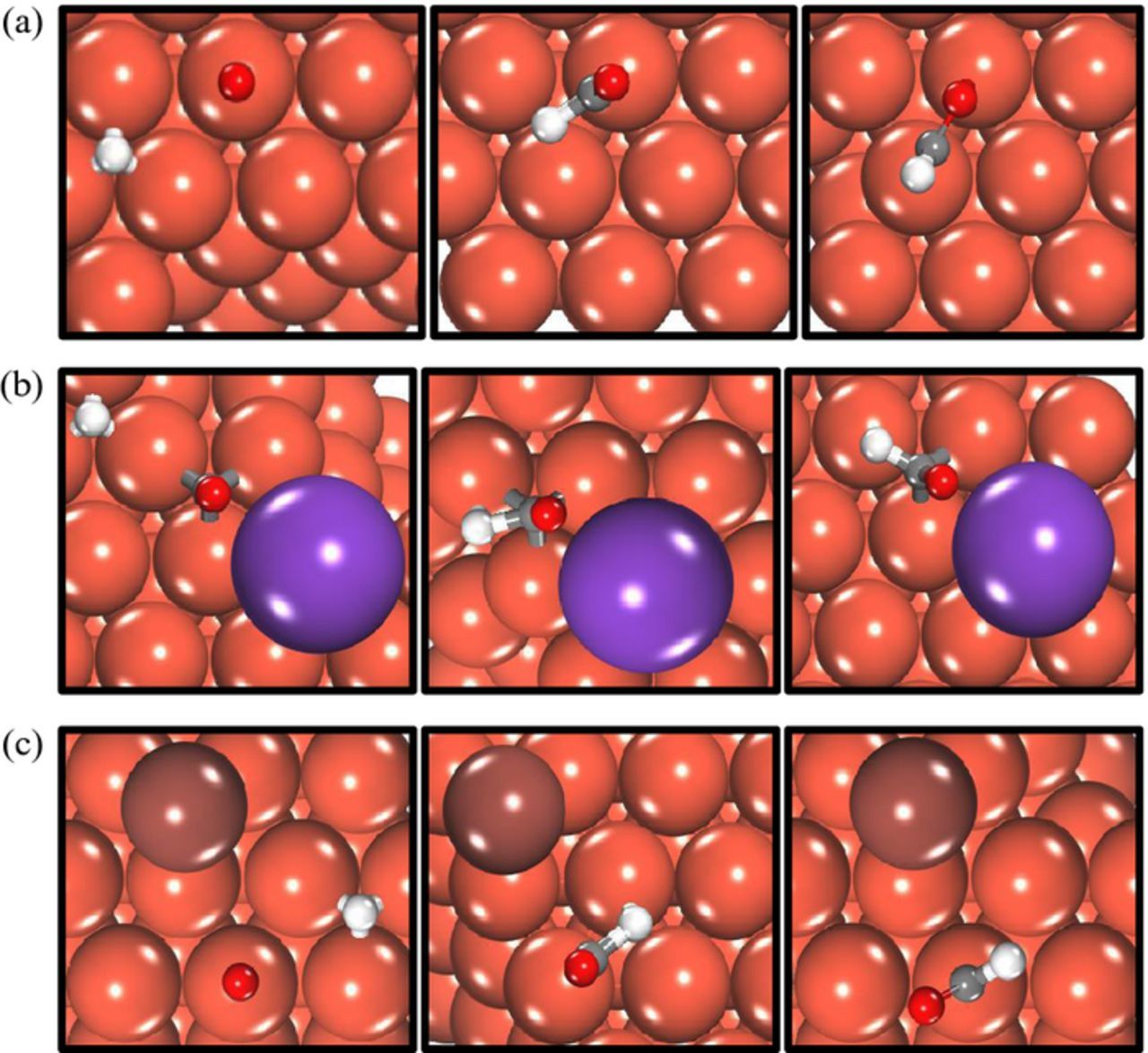
Figure 2. Top view of the initial, transition and final state of CO*→CHO* reduction via direct surface hydrogenation on Cu (111) without ion co-adsorption (a), with co-adsorbed K* (b) and with co-adsorbed I* (c). (Orange = copper, purple = potassium, brown = iodine, gray = carbon, red = oxygen and white = hydrogen).
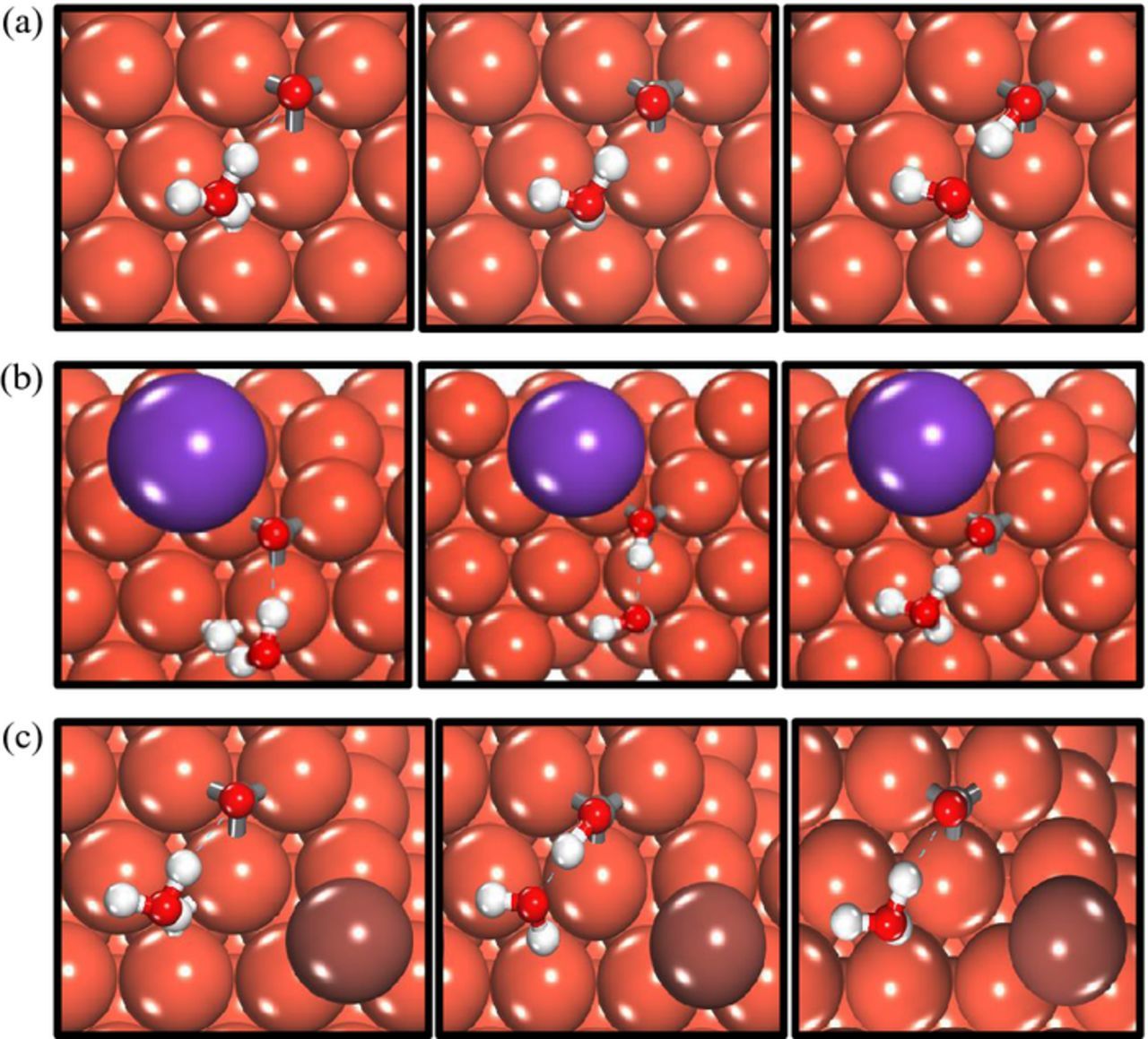
Figure 3. Top view of the initial, transition and final state of CO*→COH* reduction via solvation assisted H-shuttling on Cu (111) without ion co-adsorption (a), with co-adsorbed K* (b) and with co-adsorbed I* (c). (Orange = copper, purple = potassium, brown = iodine, gray = carbon, red = oxygen and white = hydrogen).
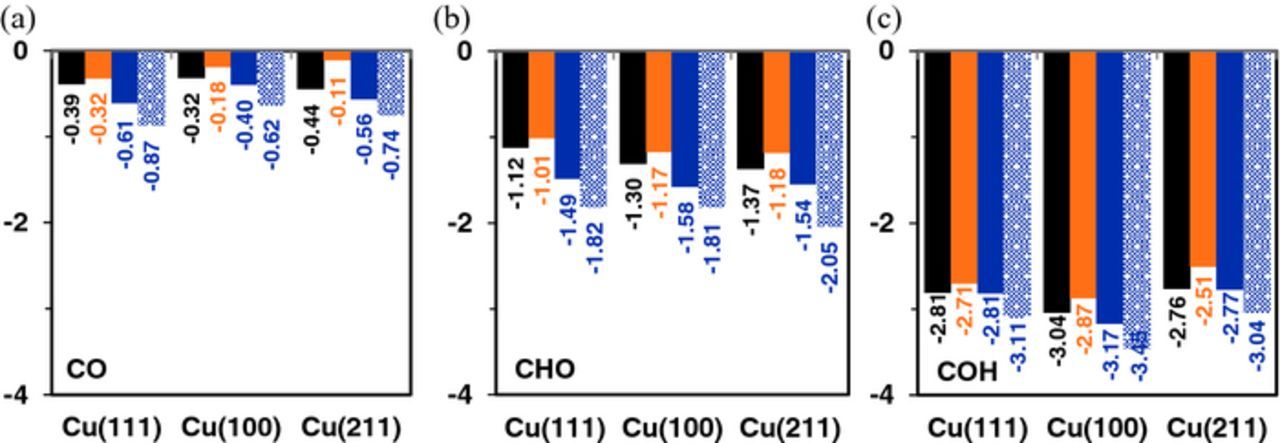
Figure 4. Binding energy BE (eV) of CO2 ER intermediates including (a) CO*, (b) CHO* and (c) COH* on the Cu (111), Cu (100) and Cu (211) surfaces without ion co-adsorption (black) and with co-adsorbed I* (orange), K* (blue), explicitly solvated K* (dotted blue).
When I* is co-adsorbed on the copper surface, the CO2 ER intermediates bind weaker, on average by 0.14 eV. For all CO2 ER intermediates, the weakening effect due to I* co-adsorption is most pronounced on Cu (211) (ΔBE ∼0.26 eV), then Cu (100) (ΔBE ∼0.11 eV) and least on Cu (111) (ΔBE ∼0.08 eV). Conversely, the effect of co-adsorbed K* is opposite but similar in magnitude to co-adsorbed I*. K* co-adsorption strengthens the binding of the CO2 intermediates on the copper surfaces, on average by 0.16 eV. The effect of co-adsorbed K* is more apparent on the binding strength of O-terminated species (CO* and CHO*) than COH*, indicated by the smaller surface dipole moments (Figure S10), with the effect most pronounced on the Cu (111) surface (ΔBE ∼0.20 eV) and the Cu (100) (ΔBE ∼0.16 eV), least on Cu (211) (ΔBE ∼0.01 eV) surfaces. Thus, while co-adsorbed I* weakens the binding of all CO2 ER intermediates, co-adsorbed K* generally promotes stronger binding of the intermediates, more so the O*-terminated species (CO* and CHO*).
Under operating CO2 ER conditions, solvation coupled with co-adsorbed ions likely play a crucial role in affecting the binding strength and stability of the CO2 ER intermediates. Explicitly solvated K* (shown in Figure 4) serves to strengthen the binding of CO2 ER intermediates by 0.29 eV on average relative to co-adsorbed K* alone. Since solvation effects on I* adsorption is minimal, as reported in the previous section, we only explore the impact of explicitly solvated K* on the binding of the CO2 ER intermediates. Solvation alone serves to stabilize the CO2 ER intermediates on the copper surfaces, with a significant effect on COH* (0.58 eV) relative to CHO* (0.35 eV) and CO* (0.15 eV) on the copper surfaces. The preferential stability of COH* with inclusion of explicit solvation can be attributed to a strong H-bond network. When the effects of K* co-adsorption are taken into account, however, the stability of COH* due to explicit solvation is reduced from 0.58 eV to 0.34 eV. Since co-adsorbed K* retains most of its charge, we expect strong cation-water interactions to disrupt the H-bond network and lower the binding stability of COH* on the copper surfaces.
Effect of adsorbed K* and I* on the CO2 ER elementary reaction energies
Figure 5 shows the reaction free energy diagram for CO* reduction on Cu (111), Cu (100) and Cu (211) at U = −0.5 V-RHE with and without ion co-adsorption. Before considering the effects of co-adsorbed ions, we summarize the reaction free energies of CO* formation and reduction without co-adsorbed ions. The relative reaction energy of CO2 gas reduction to CO* formation is downhill in energy on all three copper facets, suggesting that the CO* formation is thermodynamically viable at U = −0.5 V-RHE. The reaction energy of CO* reduction to CHO* on copper is more favorable by 0.34 eV on average than that of CO*→COH*, with reduction being more favorable on Cu (211) > Cu (100) > Cu (111). Previous DFT studies on the reaction mechanism of CO2 ER on Cu (111) report that solvation plays a crucial role in lowering the activation barriers of OH bond formation and preferentially reducing CO*→COH*.44,48 Differences in the reaction energetics and selectivity of CO* reduction due to solvation and explicitly solvated ions are addressed in the latter part of the section.
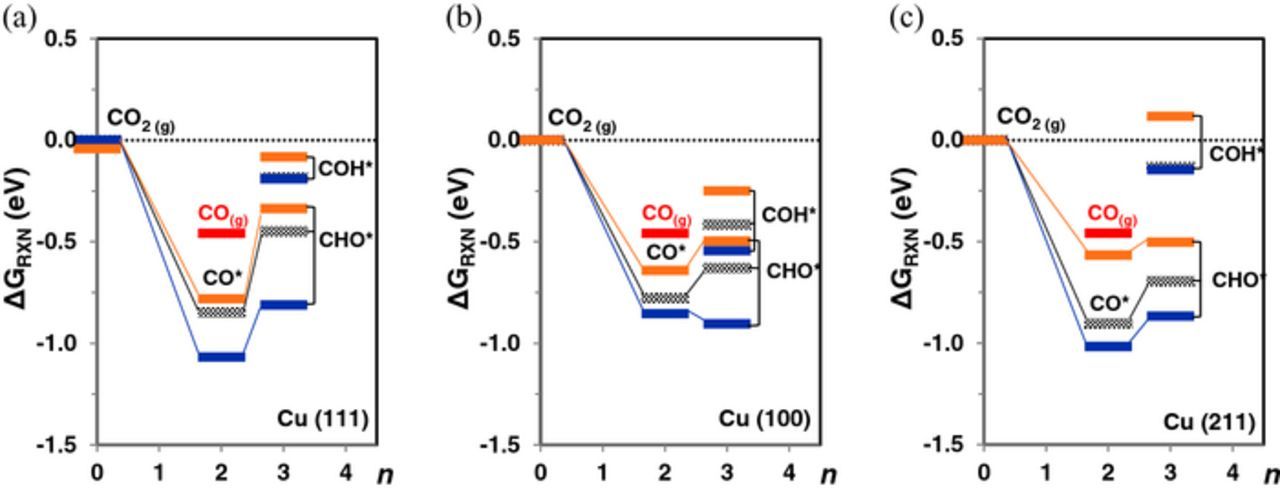
Figure 5. Relative free energy (eV) of CO2*→CO* and CO*→COH*/CHO* reduction at U =−0.5 V-RHE on (a) Cu (111), (b) Cu (100) and (c) Cu (211) surface without ion co-adsorption (gray) and with co-adsorbed I* (orange), K* (blue). Preferred reduction pathway is illustrated by the solid lines. All free energies are referenced to the free energy of gas-phase CO2 (ΔGCO2(g) = 0) at 298 K and 1 atm pressure (black dotted line). Red bar indicates the free energy to form gas-phase CO at standard conditions.
The reaction energies of the CO2 ER intermediates are significantly affected by the presence of co-adsorbed K* and I*, as seen in Figure 5. K* promotes CO* formation, whereas co-adsorbed I* weakens the relative energy of CO* formation from CO2 (gas) with the effect most pronounced on Cu (211) (0.34 eV) > Cu (100) (0.14 eV) ∼ Cu (111) (0.067 eV). In our recent study, we confirmed that a tendency to form a high coverage of CO* on transition metals obstructed access of H* to the metal surface sites and prevented subsequent C-H bond formation.70 The presence of co-adsorbed I* could lead to more CO gas phase products by reducing the CO* binding strength. This is evident in Figure 5c, where the reaction energy of CO* formation on Cu (211) at U = −0.5 V-RHE differs by only 0.1 eV from the standard gas-phase CO formation energy.
While co-adsorbed I* weakens the stability of both CHO* and COH* on the copper surface, co-adsorbed K* increases the stability of CHO* on the copper surfaces relative to COH*. On Cu (211), the decreased stability of CO* due to co-adsorbed I* reduces the reaction free energy of CO*→CHO* reduction from 0.21 eV to 0.06 eV and from 0.77 eV to 0.68 eV for the CO*→COH* reduction step. On Cu (100), the relative reaction free energy of CO*→COH* remains largely unaltered with K* co-adsorption, but the reaction free energy of CO*→CHO* is reduced by 0.20 eV, making the pathway exergonic at U = −0.5 V-RHE. Table I summarizes the differences in the relative free energy of COH* and CHO* with and without ion co-adsorption. Positive values reflect GCOH* > GCHO*, with higher stability towards CHO* formation. Co-adsorbed I* weakens the stability of both CHO* and COH* slightly, while co-adsorbed K* increases the relative stability of CHO* over COH* significantly on all copper surfaces. The preferred stability of CHO* over COH* with K* adsorption can be attributed to the higher sensitivity of CHO* BE to co-adsorbed K* than that of COH*.
Table I. Relative stability of COH* versus CHO* formation (in eV) as a function of ion co-adsorption on copper surfaces.
| Cu(111) | Cu(100) | Cu(211) | |
|---|---|---|---|
| Without co-adsorbed ion | 0.27 | 0.21 | 0.56 |
| With co-adsorbed I* | 0.26 | 0.25 | 0.62 |
| With co-adsorbed K* | 0.62 | 0.36 | 0.72 |
| With solvation | 0.03 | −0.12 | 0.46 |
| With co-adsorbed solvated K* | 0.66 | 0.31 | 0.95 |
Table I also compares the relative stability of CHO*/COH* when solvation effects are considered. Solvation preferentially stabilizes COH* with a strong H-bond network, with the effect most pronounced on Cu (100) > Cu (111) ≫ Cu (211). The use of a single explicit H2O molecule in our DFT models to calculate the relative stability of CHO*/COH* is an approximation to the fully solvated electrode/electrolyte interface, with the stability of the adsorbed species being subject to the structure of explicit solvation used. In our previous study, including explicit surface solvation using a single water molecule was found to shift the relative trends in CHO* and COH* binding on Pt, Ni, Co, Cu and Au surfaces by 0.41 eV and 0.60 on average respectively.47 Adding additional water molecules up to a water bilayer has been previously shown to further stabilize the adsorbates by 0.1-0.2 eV,48 though with additional computational demands. The use of a single explicit water molecule serves to capture the major effect of surface solvation on the relative stability of CHO*/COH*. When explicit solvation and K* co-adsorption are considered, the stability of CHO* over COH* is accentuated on the copper surfaces, with the effect most pronounced on Cu (211). Thus, co-adsorbed K* with the inclusion of solvation significantly promotes a CHO* path and generally stronger binding of all three reaction intermediates to the copper surfaces. In contrast, I* does little to affect the selectivity of CHO*/COH* formation, but will promote CO* desorption on the copper surfaces. The differences in binding strength and relative stability of CHO* and COH* species with ion co-adsorption play a crucial role in dictating the CO* reduction selectivity towards CHO*/COH* on the copper surfaces. While these conclusions are arrived at from a thermodynamic standpoint, elementary kinetics and competitive surface coverage effects will impact the formation of CO* and selectivity of CO* reduction to CHO* or COH*. In the next section, we address the impact of co-adsorbed ions on the potential-dependent activation barriers of the CO* reduction to CHO* and COH*.
Kinetic implications of specifically adsorbed ions on CO* reduction
Figure 6 shows the reaction energy diagram and the activation barriers for CO* reduction to CHO* (Figure 6a) and COH* (Figure 6b) on Cu (111) at U = −0.5 V-RHE with and without ion co-adsorption. The relative energies of CO* and CHO* formation shown in Figure 6a are the same as those in Figure 5a, with the corresponding activation barriers now included. Figure 6b shows the relative energies of CO* and COH* formation modeled with using one explicit water molecule. The corresponding initial, transition and final state configurations of both elementary reduction steps are illustrated in Figure 2 and Figure 3. The calculated activation energies (G0act), initial state formation potentials (U0) and activation energies at U = −0.5 V-RHE computed using Equation 22 are reported in Table II. Imaginary frequencies along the reactive modes are also given in Table II.
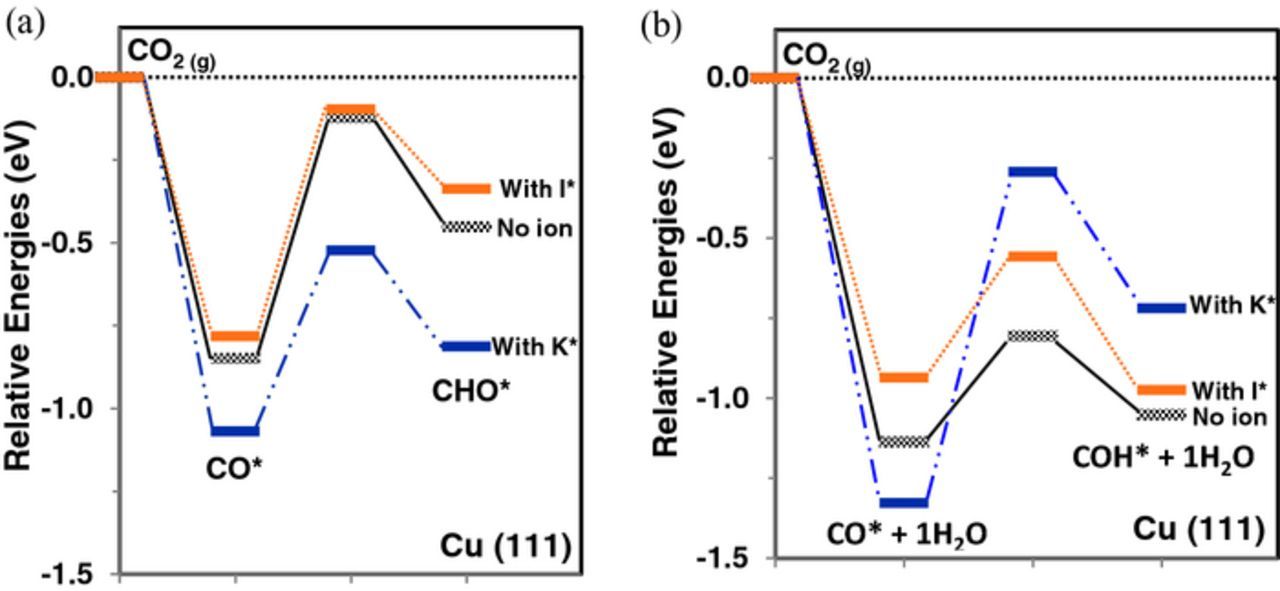
Figure 6. Relative free energy (eV) diagram of (a) CO*→CHO* (via direct surface hydrogenation) and (b) CO*→COH* (via water-assisted H-shuttling) reduction at U = −0.5 V-RHE on the Cu (111) surface without ion co-adsorption (gray) and with co-adsorbed I* (orange), K* (blue). All free energies are referenced to the free energy of gas-phase CO2 (ΔGCO2(g) = 0) at 298 K and 1 atm pressure (black dotted line).
Table II. Parameters in estimating potential-dependent activation barriers for the electrochemical reduction of CO* to CHO* and COH* on Cu (111) assuming β = 0.5.
| Elementary electrochemical step | Model | G0act (eV) | U0 (V-RHE) | TSVIB (cm−1) | Gact at –0.5 VRHE |
|---|---|---|---|---|---|
| CO* → CHO* No ion | No solvation | 1.01 | 0.07 | i 113.9 | 0.73 |
| CO* → CHO* With K* | No solvation | 0.84 | 0.09 | i 239.8 | 0.55 |
| CO* → CHO* With I* | No solvation | 0.97 | 0.07 | i 234.9 | 0.68 |
| CO* → COH* No ion | 1 H2O shuttle | 0.65 | 0.13 | i 814.9 | 0.33 |
| CO* → COH* With K* | 1 H2O shuttle | 1.32 | 0.08 | i 930.4 | 1.03 |
| CO* → COH* With I* | 1 H2O shuttle | 0.59 | −0.07 | i 934.9 | 0.38 |
A direct surface hydrogenation mechanism is employed in calculating the barrier for CO*→CHO* (C-H bond formation in Figure 2). The mechanism transfers H* to surface-adsorbed CO* species. Previous DFT electrokinetic studies revealed that all C-H bond formation reactions proceeded over lower barriers through surface hydrogenation paths, rather than proton shuttling through water.44,48 This conclusion is intuitive given the expected role of the metal in C-H dissociation steps and the principle of microscopic reversibility.
A solvation assisted proton shuttling mechanism is used in calculating the barrier for CO*→COH* (O-H bond formation in Figure 3) based on previous reports wherein the barriers associated with O-H bond formation and C-OH bond dissociation were significantly lowered via the H-shuttling H* transfer mechanism.44,48 Modeling the solvation assisted proton shuttling mechanism using a single water molecule in our work is an approximation. Previous reports indicate that the use of one explicit water molecule can lower the activation barrier of CO2→COOH* on Cu (111) by 0.8 eV and inclusion of a water bilayer can further stabilize the barrier by 0.1–0.2 eV.48 Although the water bilayer generally serves to improve the accuracy of the solvation model and can influence the energetics of CO2 ER, the use of a single water molecule in our model is expected to capture the relative variations in the activation barriers of CO* reduction with and without ion co-adsorption.
Neglecting ion adsorption effects, the presented reaction energetics in Figure 6 and Table II suggest that the CO* reduction to COH* on Cu (111) at U = −0.5 V-RHE has a lower barrier of 0.33 eV compared to a barrier of 0.73 eV for CO*→CHO* reduction. The relative reaction energy for CO*→COH* is also lower by 0.48 eV relative to CO*→CHO*, indicating that solvation plays a crucial role in lowering the activation barriers of O-H bond formation and the preferential selectivity of CO* reduction to COH*.
Co-adsorbed I* slightly weakens the stability of the CO2 intermediates and the activation barriers of CO* reduction to CHO* and COH* on Cu (111) in the presence of co-adsorbed I* are mainly unaltered (∼0.05 eV) relative to without ion co-adsorption. Since co-adsorbed I* loses most of its charge, we expect it to have a low impact on the electron transfer at the transition state for both mechanisms. Co-adsorbed K* preferentially increases the stability of CO* (0.23 eV) and CHO* (0.36 eV) on Cu (111) as shown in Figure 6a, however, decreases the stability of solvated COH* by 0.33 eV (Figure 6b) by disrupting the H-bond network between COH* and the water molecules. The activation barrier for CO*→CHO* reduction is lowered by 0.18 eV (at U = −0.5 V-RHE) in the presence of co-adsorbed K*. The barrier for CO*→COH* reduction, via the water-assisted proton shuttling mechanism, increases by 0.70 eV with co-adsorbed K*, inverting the selectivity of CO* reduction to CHO* when the effects of co-adsorbed K* are taken into account. The preferred selectivity towards CHO* formation with co-adsorbed K* can be rationalized in terms of the higher charge retained by K* that increases the surface dipole moment, thereby facilitating the electron transfer process from the surface to the transition state in a surface hydrogenation mechanism. For the water-assisted proton shuttling mechanism, the extreme 0.70 eV barrier increase occurs due to the difficulty in shuttling a positive charge through a water molecule interacting with a partially positively charged K* species. The high charge on K* (qK = +0.82 e) and the shuttling H* (qH = +1.00 e) in the transition state destabilizes the state and increase the corresponding activation barrier, rendering the CO*→COH* path kinetically unfavorable relative to CO*→CHO*. This specific 0.70 eV impact of K* is reliant on the relative positions of K*, CO*, and H2O, however, the qualitative effect of K* retaining positive charge, slowing H+ transfer to CO* adsorbed in its vicinity, is expected to be robust. Although the solvation model, reaction mechanism and the approximations in estimating potential-dependent activation barriers can give rise to quantitative variations in the activation barriers, the qualitative impact of co-adsorbed K* on the kinetics and selectivity of CO* reduction are emphasized here.
Conclusions
In summary, we have used DFT calculations to explore the impact of specifically adsorbed ions on the reaction energetics of CO2 electroreduction on Cu (111), Cu (100) and Cu (211) surfaces. The calculations show that under different pH conditions for CO2 ER, a low coverage of I* and/or K* can be specifically adsorbed on the copper surfaces. The binding strength of the CO2 ER intermediates is significantly affected by the presence of the specifically adsorbed ions. Co-adsorbed I* generally weakens the binding of CO*, CHO* and COH* intermediates and therefore will impact the surface coverage of the intermediates during CO2 ER. I* has a minor effect of the elementary reaction energetics between the reduction species. Co-adsorbed K* strengthens the binding of all three CO2 ER intermediates and promotes the stability of CO* and CHO* over COH*, thereby making paths through CHO* more favorable. The results suggest that co-adsorbed I* can promote CO(g) formation, and may allow for greater subsequent C-H bond formation by reducing the coverage of CO* and allowing for more surface H*. Co-adsorbed K* may promote paths through the CHO* intermediate that can lead to C2 products. However, more rigorous microkinetic analyses are needed to draw robust product distribution conclusions. By accounting for the presence of specifically adsorbed ions under CO2 ER conditions, this study lays emphasis on the qualitative impact these ions can have on reaction energetics of elementary electrochemical reactions.
Acknowledgments
This work was supported by the National Science Foundation, Grant # CBET – 1264104. I.T. McCrum acknowledges support from The Pennsylvania State University Diefenderfer Graduate Fellowship and NSF NRT #1449785.