Abstract
Two Fe-N-C catalysts comprising only atomically-dispersed FeNx moieties were prepared, differing only in the fact that the second catalyst (Fe0.5-NH3) was obtained by subjecting the first one (Fe0.5-Ar) to a short pyrolysis in ammonia. While the initial ORR activity in acid medium in rotating disk electrode is similar for both catalysts, the activity in alkaline medium is significantly higher for Fe0.5-NH3. Time-resolved Fe dissolution reveals a circa 10 times enhanced Fe leaching rate in acidic electrolyte for Fe0.5-NH3 relative to Fe0.5-Ar. Furthermore, for the former, the leaching rate is strongly enhanced when the electrochemical potential is in the range 0.75–0.3 V vs. RHE. This may explain the reduced stability of ammonia-pyrolyzed Fe-N-C catalysts in operating PEMFCs. In alkaline medium in contrast, Fe0.5-NH3 is more active and more stable, with minimized Fe leaching during electrochemical operation in load-cycling mode. Operando X-ray absorption spectroscopy measurements in alkaline electrolyte reveals similar trends of the XANES and EXAFS spectra as a function of the electrochemical potential for both catalysts, but the magnitude of change is much less for Fe0.5-NH3, as evidenced by a Δμ analysis. This is interpreted as a lower average oxidation state of FeNx moieties in Fe0.5-NH3 at open circuit potential.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Due to growing concerns related to local and global impacts on the Earth's atmosphere and long-term sustainability of fossil fuels, new ways of producing and using renewable fuels are being explored for both transportation and stationary applications. Hydrogen is a promising fuel that can be produced from water and renewable energy via electrolysis or other means, and back-converted to electric power on demand in high-efficiency fuel cells rather than in low-efficiency combustion engines.1 While different types of fuel cell technologies exist, the proton exchange membrane fuel cell (PEMFC) has key advantages such as low internal resistance, fast start-up/shutdown and high electric power density.2–5 This makes it suitable for small and medium size applications ranging from portable devices to automotive application and distributed electric power production. While high power densities at reasonable energy efficiency can be reached with PEMFC, several drawbacks hold its large-scale commercialization. In particular, the sluggish oxygen reduction reaction (ORR) at the cathode requires the most active catalysts to overcome the kinetic barrier of splitting the double-bond in O2. To date, all catalysts that meet the ORR activity and stability targets in the acidic PEMFC environment are based on platinum.4,5 The low natural abundance in the Earth's crust and geographically-constrained resources of this element raises concerns of cost competitiveness and long-term sustainability of the present PEMFC technology.6
Research on non-noble ORR catalysts for PEMFC started ca 50 years ago, and significantly intensified in the past decade.7–10 Among catalysts free of platinum group metals (PGMs), metal-nitrogen-carbon catalysts have hitherto shown the highest initial activity toward the ORR in acidic media, especially Fe-N-C catalysts.7–12 Compared to Pt-based catalysts, the ORR activity of pyrolyzed Fe-N-C materials is highly dependent on the synthetic path, due to numerous Fe species that may form at high temperature, ranging from metallic, metal-carbide and metal-nitride Fe particles (often embedded in carbon) to various FeNx moieties featuring single iron-atoms covalently integrated in the N-doped carbon matrix.13–16 Among the preparation methods, the sacrificial metal-organic-framework method has resulted in Fe-N-C catalysts with state-of-the-art activity and power performance in a rotating disk electrode (RDE) and PEMFC, respectively.17–19 Characterization by 57Fe Mössbauer and X-ray Absorption Spectroscopy (XAS) has revealed that the active sites in Fe-N-C catalysts featuring exclusively atomically-dispersed iron have an FeN4 structure that is similar to the iron-porphyrin core.18,20,21 While Fe-N-C catalysts featuring exclusively Fe-carbide particles embedded in N-doped carbon matrix can also show interesting ORR activity at beginning-of-life (BoL), their stability during load-cycling in acidic medium was recently shown to be insufficient.22–24 In contrast, an Fe-N-C catalyst prepared via ramp pyrolysis in inert atmosphere and exclusively comprising FeN4 moieties showed only 25% loss in activity after 30,000 load cycles at 80°C in inert-gas saturated acidic electrolyte, well above the U.S. Department of Energy target of 40% maximum loss in activity after such an accelerated stress test (AST).24 While such AST protocol cannot capture all degradation mechanisms that will occur in a PEMFC, it is useful in identifying which PGM-free catalysts meet the necessary (but not sufficient) criterion of structural stability when cycled in the cathode potential range and pH environment expected in PEMFC. In particular it is useful to detect demetallation that may occur even in the absence of O2.
If the ORR activity and stability in acidic medium in RDE setup (stability to load-cycling AST in inert-gas saturated electrolyte) of at least some atomically-dispersed Fe-N-C catalysts is unquestionable, their stability in operating PEMFC has hitherto still been low.7,9,17,18,25 This is particularly true for Fe-N-C catalysts prepared via pyrolysis in flowing NH3, resulting in circa 20–30 times higher BoL activity at 0.9 V in PEMFC than similarly prepared Fe-N-C catalysts but pyrolyzed in inert atmosphere.17,18 After 15–20 h of operation in PEMFC, the current density at 0.5 V obtained with NH3-pyrolyzed Fe-N-C is however typically halved, while that with Ar-pyrolyzed Fe-N-C undergoes less than 10% decay.18,26 Advanced ex situ spectroscopic comparison between an Ar- and a NH3-pyrolyzed Fe-N-C catalyst revealed no or small difference of Fe coordination, both featuring FeN4 sites with nearly identical XAS and Mössbauer spectroscopy fingerprints.18 The only key differences are i) the higher basicity of the catalyst's surface and ii) higher micropore volume for the NH3-pyrolyzed material. A previous study on another NH3-pyrolyzed Fe-N-C catalyst revealed that a short immersion in acidic medium (sulphuric acid or perchloric acid) decreased its activity by a factor ten (activity/10), and that this deactivation was partially reversible after the acid-washed catalyst had been subjected to a cleaning treatment at 400°C in inert gas (activity/10 × 5).27 The irreversible activity loss (activity/2) was assigned to metal leaching from weakly bound Fe sites (ca 50% loss of metal upon first acid wash). Since that pristine catalyst contained not only FeNx moieties but also a significant content of crystalline Fe particles, the metal leached during first acid-wash may have however originated (at least partially) from metallic Fe particles. The reversible deactivation phenomenon (between activity/10 to activity/2) was explained as a protonation of highly basic N-groups (leading to a high turn-over frequency, TOF, of FeNx moieties) and their charge-neutralization by the electrolyte's counter-anion.27 The latter anion-adsorption event was proposed to decrease the FeNx site's TOF. Strong support for this reversible deactivation mechanism, not related to any Fe-leaching event, was given by the possibility to recover most of the initial activity of the ammonia-treated Fe-N-C catalyst by removing the adsorbed bisulfate anions (as proven by X-ray photoelectron spectroscopy) during the 400°C treatment. The ORR activity increased from ca only 10% of the initial activity after a short acid wash to ca 50% of the initial activity after removal of the bisulfate anions.27
More recently, the reason for the reduced stability of NH3-pyrolyzed Fe-N-C catalysts in PEMFC was re-investigated and debated in three follow-up papers.25,26,28 Dodelet's group first proposed that the instability of NH3-pyrolyzed Fe-N-C catalysts in PEMFC is due to oxidation of the carbon surface in micropores, increasing the hydrophilicity and leading to micropore flooding.25 Iron was first claimed to play no role in the deactivation, although Fe coordination and demetallation during operation or post mortem had not been characterized. Choi et al then focused on this micropore-flooding hypothesis28 and could demonstrate that this mechanism could not explain the rapid loss of performance in PEMFC observed with another NH3-pyrolyzed Fe-N-C catalyst. From polarization curves recorded at different relative humidity and cyclic voltammograms before/after stability test, they concluded that the micropores were partially or completely filled by water already at BoL, and that the rapid performance loss was mainly due to a decrease of the ORR kinetics, and not due to a decay of the mass-transport performance of the cathode layer. While clearly ruling out the micropore flooding hypothesis, their work could not conclusively point what is the mechanism for the rapid decay of the ORR kinetics of NH3-pyrolyzed Fe-N-C catalysts. Following that work, Dodelet's group agreed that micropore flooding was not the cause for rapid ORR activity decay of their NH3-pyrolyzed Fe-N-C catalyst as well and, after examination of the Fe content and speciation with Mössbauer spectroscopy following various short-duration PEMFC tests (in the range of 0–20 h), could establish similar trends between a) the current density at 0.6 V vs. duration of operation and b) the relative fraction of Fe present as FeNx moieties in the cathode as a function of duration of operation in PEMFC, both experiencing a relative decay of ca 50% after 20 h operation.26 The novel hypothesis was that the fast activity decay of NH3-pyrolyzed Fe-N-C catalyst is due to a demetallation of unstable FeNx moieties in this material, such moieties being specifically located in micropores. Despite the above-mentioned trend, it must be noted however that the current density at 0.6 V in PEMFC is inappropriate to track changes in the ORR activity, and that the relative decay in ORR activity at 0.8 V after 20 h operation was much higher than 50%, in fact ca 90% (Fig. 4 in Ref. 26). The demetallation from specific FeNx moieties existing in highly microporous Fe-N-C materials may thus explain part of the initial ORR activity decay (e.g. from 1.0 to 0.5 in normalized activity, 1.0 corresponding to BoL activity), but not necessarily all of the decay. It is also possible that the loss of 50% of FeNx moieties in fact implied that ca 90% of the electrochemically-accessible FeNx moieties had been leached. Since Mössbauer spectroscopy is a bulk technique, the fact that ca 50% of FeNx moieties were still present after PEMFC operation does not necessarily imply that those moieties did contribute to the initial ORR activity, if they were not located on the top surface of the carbon matrix.
With no clear path at the moment on how highly-basic N-groups could be stabilized in acidic medium, our scientific interest was first oriented on the degradation mechanisms of inert-gas-pyrolyzed Fe-N-C catalysts with FeNx moieties as the main active sites. While significantly more durable than NH3-pyrolyzed catalysts, they also suffer from a slow but steady linear decline with duration of operation in PEMFC.26,29 Two main degradation mechanisms of such materials have been identified: (1) irreversible iron leaching from Fe particles imperfectly embedded in carbon or from FeN4 active sites;30 and (2) reversible degradation induced by the hydrogen peroxide by-product formed during ORR in acidic medium;31,32 Mechanism (1) was investigated with online mass-spectrometry and revealed that Fe leaching resulted from carbon corrosion during startup/shutdown AST, while Fe leaching during load-cycling AST mainly originated from Fe particles imperfectly surrounded by a N-C layer. The FeN4 moieties were mostly stable during load-cycling AST in inert-gas saturated acidic medium, in a broad range of temperature and up to 10,000 cycles.24,30,33 The high stability in acidic medium during load-cycling of FeN4 moieties stood in apparent paradox to their poor durability in operating PEMFC. This conundrum was recently clarified by a study showing that the surface modification of Fe-N-C by hydrogen peroxide does not leach FeN4 moieties but decreases their TOF.34 This deactivation originates from the chemical reaction of minute amounts of H2O2 with the FeN4 moieties, leading to the formation of reactive oxygen species. These radicals then react with the carbon surface, introducing a high number of oxygen functionalities on the top-surface. From a combined experimental and theoretical investigation, we showed that this decreases i) the electron density at the Fe center and ii) the O2 binding energy of FeN4 moieties, resulting in a much decreased single-site TOF.34 Interestingly, the controlled ex situ deactivation of Fe-N-C by hydrogen peroxide revealed that this mechanism is highly pH-dependent, the same protocol but applied in 0.1 M solution of KOH instead of HClO4 resulting in no deactivation of an Ar-pyrolyzed Fe-N-C catalyst.34 This makes it promising for the application of Fe-N-C catalysts based on FeN4 moieties in anion exchange membrane fuel cell (AEMFC).35–37 In particular, the AEMFC environment might allow combining the highest ORR activity of NH3-pyrolyzed Fe-N-C catalysts with high durability. Expectation for high durability is supported by i) the stability at high pH of highly-basic N-groups present in NH3-pyrolyzed catalysts and ii) the lack of peroxide-induced deactivation at high pH on a specific Ar-pyrolyzed Fe-N-C catalyst comprising exclusively FeN4 sites.
The aims of this study are to assess the site-structure, activity and stability during load-cycling in alkaline electrolyte of a NH3-pyrolyzed catalyst showing an extremely high ORR activity and exclusively comprising Fe as atomically-dispersed FeNx moieties. A second Fe-N-C catalyst prepared similarly but obtained via a single pyrolysis in inert gas is also studied, allowing the identification of the effect of pyrolysis atmosphere on the properties of Fe-N-C catalysts. Activity and stability during load-cycling AST was also performed in an acidic electrolyte to assess the effect of electrolyte pH. Activity and stability are measured with rotating disk electrode. Fe coordination and Fe leaching were monitored as a function of the electrochemical potential with operando XAS and a flow cell coupled to online mass-spectrometry, respectively, in order to shed light on activity and stability properties.
This study demonstrates that FeNx moieties in a NH3-pyrolyzed Fe-N-C catalyst, while being structurally very similar to those present in the Ar-pyrolyzed Fe-N-C material, show exacerbated demetallation in acidic medium. Online measurement of Fe dissolution rate shows the Fe demetallation from NH3-pyrolyzed Fe-N-C increases when scanning negatively the potential from 1.0 V vs. RHE, to reach a peak of dissolution rate at ca 0.3 V vs. RHE. In alkaline electrolyte, the metal-Nx moieties' stability of Ar- and NH3-pyrolyzed Fe-N-C are comparable, and the very highly ORR-active NH3-pyrolyzed Fe-N-C material is stable in 0.1 M KOH for several thousands of cycles. Operando XAS of the NH3-pyrolyzed Fe-N-C catalyst in alkaline electrolyte shows similar trends as for the Ar-pyrolyzed Fe-N-C catalyst, but with a reduced magnitude of the changes with electrochemical potential. This is interpreted as a lower average oxidation state of the FeNx moieties at open circuit potential in the NH3-pyrolyzed Fe-N-C material compared to the Ar-pyrolyzed Fe-N-C.
Experimental
Synthesis
Two Fe-N-C catalysts are prepared with the sacrificial metal-organic-framework method, using ZIF-8 (Basolite Z1200, Sigma Aldrich) as support, 1,10-phenantroline (≥99%, Sigma Aldrich) as a secondary source of nitrogen and iron (II) acetate (≥99.99%, Sigma Aldrich) as the source of metal. The catalyst precursor is prepared with a weight ratio of 4/1 for ZIF-8/phenanthroline and 0.5 wt% of iron in the complete catalyst precursor. The three precursors are initially mixed using low-energy ball milling at 400 rpm for 2 h 20 min, with 5 minutes pause every 30 minutes of milling. The obtained catalyst precursor is transferred into a quartz boat and inserted in a quartz tube. The first pyrolysis is performed in flash-pyrolysis mode, pre-equilibrating the quartz tube and oven at 1050°C, then pushing the quartz boat and catalyst precursor within 1 min in the heating zone of the furnace with an outer magnet. The pyrolysis duration at 1050°C in flowing Ar is exactly 1 h. The pyrolysis is terminated by opening the split-hinge furnace, removing the quartz tube and letting it cool down at room temperature for 20 minutes. The obtained catalyst is labelled Fe0.5-Ar. To prepare the NH3-pyrolyzed catalyst, Fe0.5-Ar is re-pyrolysed with the same flash-pyrolysis mode, but in flowing pure NH3 and for only 5 minutes at 950°C. The obtained catalyst is labelled Fe0.5-NH3.
Electrochemical measurements and time-resolved Fe dissolution rate measurements
Activity and durability in acidic and alkaline electrolytes are obtained using a RDE set-up (Pine instruments) and either 0.1 M KOH or 0.1 M H2SO4 electrolytes. The three-electrode configuration involves a platinum wire immersed in a H2-saturated electrolyte compartment, separated from the main compartment by a fritted glass, as a reversible hydrogen electrode (RHE) reference; a graphite plate as a counter electrode and a glassy carbon (GC) rotating disk (5 mm diameter, Pine Research) as a support for the active layer forming the working electrode. The ink is prepared by adding in sequence 5 mg catalyst, 54 μL Nafion (5% perfluorinated resin solution), 744 μL ethanol, 92 μL ultrapure water and sonicating for 1 hour in an ice bath. An aliquot of 7 μL of the ink is pipetted on the GC disk and dried at room temperature, resulting in a catalyst loading of 200 μg cm−2. The Initial activity is measured in O2-saturated electrolyte at a scan rate of 1 mV·s−1 (SP-300, BioLogic Potentiostat) and at rotation rate of 1600 rpm. Due to the low scan rate and low catalyst loading, no correction for the capacitive current is needed. To evaluate the durability of the catalysts in RDE set-up, a load-cycling protocol is applied, comprising 5000 triangular cycles performed at a scan rate of 100 mV·s−1 in the potential range of 0.6-1.0 V vs RHE and in N2-saturated electrolyte. The ORR activity after the AST is measured after re-saturating the solution with O2.
The leaching of Fe during short electrochemical cycling, performed either before or after the AST (AST performed in Ar-saturated electrolyte), is investigated in an on-line electrochemical scanning flow cell (SFC) directly connected to an inductively-coupled plasma mass spectrometer (ICP–MS), previously developed by us.38–40 It is however challenging to continuously measure Fe leaching over the length of the AST, due to drift of the ICP-MS with time and need for constant recalibration. To measure 56Fe, the ICP–MS (Perkin Elmer, NexION 350) is operated in dynamic-reaction-cell mode, using methane as the reaction gas. The cell is calibrated to both an acidic and alkaline standard solution of iron to ensure maximized detection of 56Fe. Daily calibration of the ICP–MS is done by a four-point calibration curve (0, 0.5, 1.0, 5.0 μg·L−1) of standard iron solutions prepared from Merck Centripur ICP standards (Fe(NO3)3, 1000 mg·L−1, in 2–3% HNO3). As an internal standard, we use 58Co (Merck Centripur, Co(NO3)2, 1000 mg·L−1, in 2–3% HNO3) diluted to 50 μg·L−1 in HNO3 (0.15 mol·L−1) to ensure full acidification of the electrolyte in a y- connector before its introduction in the ICP–MS. The SFC consists of a three-electrode setup using a Ag/AgCl (Metrohm, 3 M KCl) reference electrode, a graphite rod counter electrode and a GC RDE as a working electrode, on which the catalyst is drop cast. A positioning stage (Physik Instrumente, M-403.6 DG) is used to approach individual catalyst spots on the working electrode. Stability measurements are conducted in alkaline (99.99%, Suprapur, NaOH, 0.05 mol·L−1) as well as in acidic (Suprapur, 0.05 mol·L−1 H2SO4) electrolyte. The potentiostat (Gamry, Reference 600) as well as purging gases and the positioning stage is controlled by a custom LabVIEW software. The catalyst ink is prepared from the Fe-N-C catalyst, Nafion (5% perfluorinated resin solution) and water, with a mass ratio of catalyst/dry-ionomer of 4 and a catalyst concentration of 3.3 g·L−1 in the liquid ink. An aliquot of 2.75 μL is deposited on the GC, resulting in a catalyst loading of 400 μg·cm−2. Such a high loading is necessary to reach a sufficient signal-to-noise ratio in the ICP-MS measurements. This is due to the aforementioned interference of the 40Ar16O dimers and high background noise of iron in alkaline solution. For Fe-leaching measurement before and after the AST, the latter is conducted in a separate Teflon RDE-cell containing 100 mL electrolyte, and the RDE tip is then quickly transferred from the Teflon cell to the SFC set-up, with the catalyst still wetted by electrolyte. The RDE cell used for the AST consists of four individual compartments, one each for the three electrodes and for the purging tube. The counter and reference electrodes are the same as in the SFC setup.
Physico-chemical characterization
The pristine catalysts are characterized with 57Fe Mössbauer spectroscopy at room temperature. To this end, 57Fe-enriched catalysts are used, prepared identically as the ones otherwise investigated in this study, except for the use of 57Fe acetate during their synthesis. Mössbauer spectra are measured at room temperature with a 57Co:Rh source. The measurements are carried out in triangular velocity waveform using NaI scintillation detector for γ-rays. The velocity calibration is done with an α-Fe foil. A mass of 30 mg of 57Fe-enriched Fe0.5-Ar and Fe0.5-NH3 powders are necessary for a proper signal-to-noise resolution.
The pristine catalysts are also characterized with XAS in both ex situ and operando conditions. The XAS spectra are collected at SAMBA beamline (synchrotron SOLEIL) at the Fe K-edge using a double crystal Si 220 monochromator and a Canberra 35-elements germanium detector for operando acquisition in fluorescence mode. The catalyst ink (10 mg catalyst, 100 μL 5% Nafion solution and 50 μL ultrapure H2O) is prepared via ultrasonication, and 50 μL is deposited on circa 3 cm2 circular area of a larger conductive carbon foil, resulting in a catalyst loading of circa 1 mg cm−2.41 The carbon foil is then inserted in a three-electrode cell, 0.1 M KOH electrolyte is added and the three electrodes are connected, using Pt-wire counter electrode and a Hg/HgO reference electrode. Note that all potentials are however reported in V vs. RHE in this work. Air is continuously bubbled in the electrolyte during the measurements. The operando XAS spectra are collected at open circuit potential (OCP), 0.2, 0.4, 0.6, 0.8 and 1.0 V vs. RHE. Ex situ spectra were collected in transmission geometry on pellets of 1 mm diameter using Teflon powder as a binder.
To measure the specific surface area of carbon in the catalysts, sorption isotherms of N2 are measured in liquid nitrogen (77 K) with a Micromeritics, ASAP 2020 instrument. The sample is previously cleaned at 200°C for 5 h in flowing nitrogen. The specific surface area is determined by the multipoint Brunauer-Emmett-Teller (BET) method.
Results
Initial ORR activity and ex situ spectroscopic characterization
The ORR activity before and after the AST is shown in Figure 1 in linear- and semi-logarithmic scales, for Fe0.5-Ar (Figs. 1a–1b) and Fe0.5-NH3 (Figs. 1c–1d). In each sub-figure, the measurements performed in alkaline electrolyte are shown as purple curves while those performed in acidic electrolyte are plotted in gray color. The curves before and after AST are identified with filled and open symbols, respectively. Before the AST, Fe0.5-Ar shows a similar activity in both electrolytes (Fig. 1b, curves with filled symbols), with ORR mass activity at 0.9 VRHE of 0.35 and 0.25 A·g−1 in alkaline and acidic electrolytes, respectively. A slight difference is visible only at low-potential, with a less defined diffusion-limited current density in acidic medium (Figures 1a, filled gray symbols).
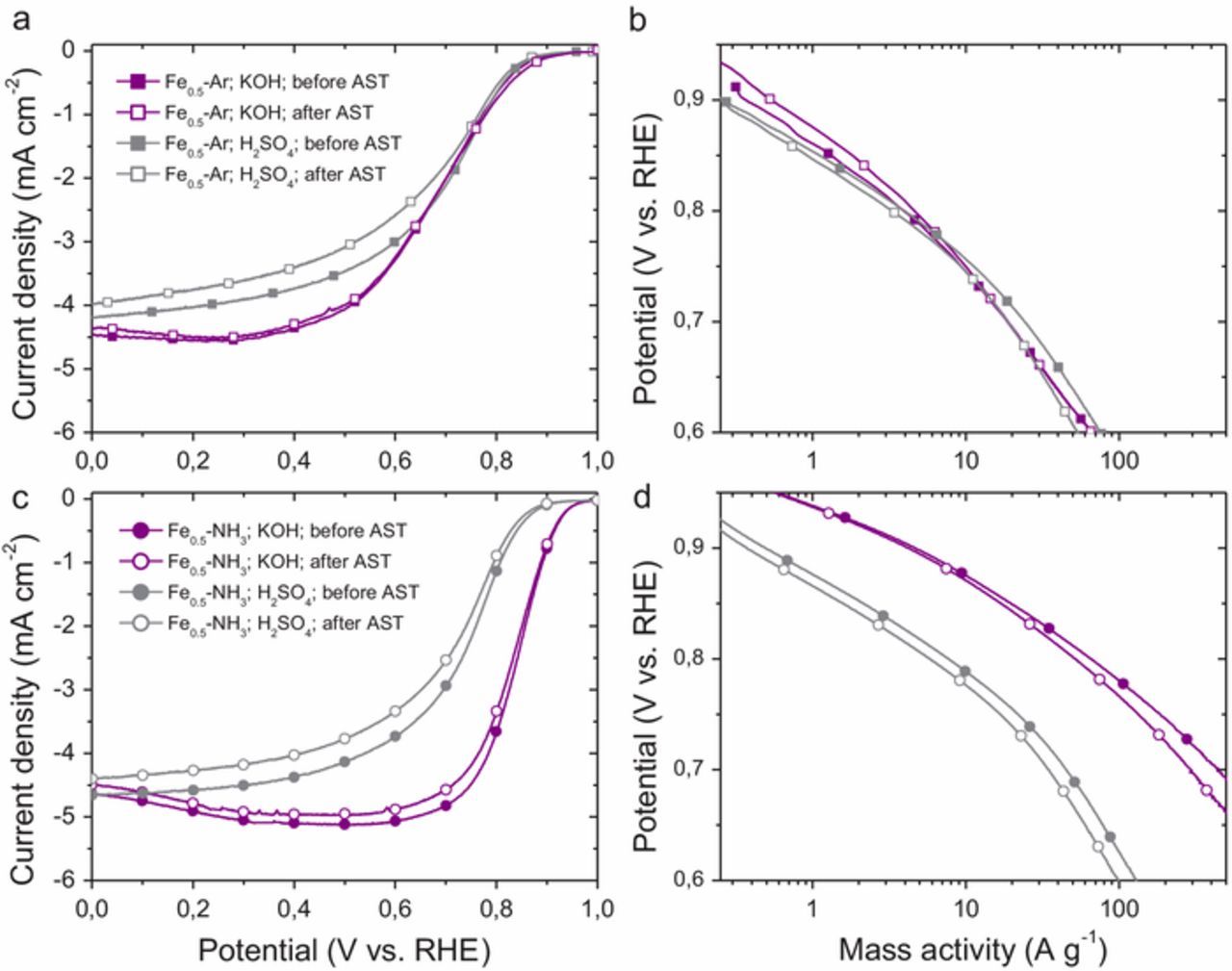
Figure 1. RDE determination of the ORR activity of Fe0.5-Ar (a, b) and Fe0.5-NH3 (c, d) in acidic (gray curves) and alkaline (purple curves) electrolyte before (filled symbols) and after (empty symbols) the load-cycling AST. Polarization curves were measured in O2-saturated electrolyte at a scan rate of 1 mV s−1, a rotation rate of 1600 rpm and a catalyst loading of 200 μg·cm−2. The curves are not corrected for ohmic loss. The AST has been conducted in N2-saturated electrolyte, in a potential range of 0.6–1.0 V vs RHE, with a scan rate of 100 mV s−1 for 5000 cycles. The semi-logarithmic Tafel plots on the left handside have been obtained from the polarization curve by applying the Koutecky-Levich equation, taking the value of diffusion-limited current density as the current density at 0.4 V vs. RHE.
A distinct behavior appears for Fe0.5-NH3, characterized with a much higher initial activity in alkaline vs. acidic electrolyte (Figures 1c–1d, filled purple vs. filled gray symbols), with ORR mass activity at 0.9 V vs. RHE of 4.6 and 0.5 A·g−1, respectively. The former is among the highest reported value of the ORR activity in alkaline medium for PGM-free catalysts.42–46 Its lower initial activity in acidic than in alkaline electrolyte can mostly be assigned to a fast protonation of highly-basic nitrogen groups followed by anion-adsorption on positively-charged [NH]+ groups, which had been previously shown, on other NH3-pyrolyzed Fe-N-C catalysts, to divide the activity by a factor 5 to 10.27,45,47 High initial activity of NH3-pyrolyzed Fe-N-C catalysts in acidic liquid-electrolyte could previously be achieved only with high catalyst loading and optimized Nafion/catalyst ratio.47 It is anticipated that these two parameters allow a short time protection of the highly basic N-groups from the liquid acidic electrolyte. With the present experimental conditions (low scan rate and low catalyst loading), the high ORR activity of Fe0.5-NH3 cannot be captured in acidic liquid-electrolyte because the highly basic N-groups were likely protonated and charge-neutralized even before the first polarization curve was recorded. The surface state of Fe0.5-NH3 in acidic medium is then similar to that of Fe0.5-Ar, explaining similar initial ORR activities in acid (compare filled gray symbols in Fig. 1b and Fig. 1d). The slightly higher initial ORR mass activity in acid of Fe0.5-NH3 vs. Fe0.5-Ar (0.50 vs 0.25 A·g−1 at 0.9 V vs. RHE, see Table I) can be explained by its higher BET specific area, 970 vs. 635 m2g−1.
Table I. Mass activity at 0.9 V vs. RHE before and after the AST for Fe0.5-Ar and Fe0.5-NH3 in acidic and alkaline electrolytes. The AST comprises 5000 cycles between 0.6 V and 1.0 V vs. RHE, in N2-saturated 0.1 M KOH at room temperature. The catalyst loading was 200 μg·cm−2.
| Mass activity / A g−1 | ||||
|---|---|---|---|---|
| Acidic electrolyte | Alkaline Electrolyte | |||
| Catalyst ↓ | Before AST | After AST | Before AST | After AST |
| Fe0.5-Ar | 0.25 | 0.25 (−0%) | 0.35 | 0.55 (+ 57%) |
| Fe0.5-NH3 | 0.50 | 0.35 (−30%) | 4.60 | 4.15 (−10%) |
A second possibility to explain the lower initial activity of Fe0.5-NH3 in acid vs. alkaline electrolyte is that (at least some) FeNx moieties after NH3 pyrolysis are intrinsically different from those after Ar-pyrolysis and, while being more active for ORR (regardless of which pH), would also be less stable in acidic medium. It might be that such highly-active FeNx moieties were leached in acidic medium even before completing the acquisition of the first polarization curve. Evidence for increased Fe leaching with the Fe0.5-NH3 catalyst in acidic medium is reported in the section entitled electrochemical stability and operando iron leaching study. We also note that the contribution of N-groups (not binding Fe) to the overall initial ORR activity of those two catalysts can be neglected. Two reference materials prepared similarly as Fe0.5-Ar and Fe0.5-NH3 but without addition of Fe acetate were studied in RDE and their initial ORR activity in 0.1 M KOH at 0.9 V vs. RHE is 0.03 and 0.57 A·g−1, respectively. This is circa 10 times lower than the activity of corresponding Fe-NC materials, 0.35 and 4.60 A·g−1 (Table I). The difference would be even larger in acidic medium.
In order to characterize the active-site structure in Fe0.5-NH3 and Fe0.5-Ar, we first resorted to ex situ spectroscopic characterization. The ex situ 57Fe Mössbauer spectra could not reveal any significant difference between the two catalysts (Figures 2a–2b and supporting Table S1). They were fitted with two doublets D1 and D2, each one having similar Mössbauer parameters for both catalysts and being present in similar ratio. These doublets are assigned to atomically-dispersed FeNx moieties.18 Similar Mössbauer spectra imply that the local Fe coordination and site geometry, up to two coordination spheres, are similar in both catalysts. Further identification of the active-site structure was performed using XAS. Figure 2c shows the ex situ X-ray absorption near-edge structure (XANES) spectra of Fe0.5-NH3 (circle symbols) and Fe0.5-Ar (solid curve), that are characteristic for Fe-N-C catalysts free of metallic particles.18 The absence of a strong signal at ca 2.2 Å (Fe-Fe bond distance in metallic and metal-nitride particles, uncorrected for phase shift) in the Fourier transform (FT) of the EXAFS spectra of both Fe0.5-Ar and Fe0.5-NH3 indicates that both catalysts are free of Fe-based particles, or that the amount of particles is below the XAS detection limit, which is approximately 3% relative to the total amount of Fe in the catalysts (Figure 2d). The absence in the XANES spectra of the pre-edge peak at 7118 eV, characteristic for unpyrolyzed Fe(II) phthalocyanine (FeN4 square-planar structure with identical Fe-N bond distances), reveals a broken D4h symmetry of the FeNx moieties. This may be due to structural disorder or existence of ferric moieties with an axial ligand such as O2. The latter case is particularly possible when recording ex situ spectra of catalysts in their resting state in air environment. The white line intensity is slightly stronger for Fe0.5-Ar than Fe0.5-NH3. This may be interpreted as a higher coordination number in the first coordination sphere surrounding Fe (either N, C or O atoms). This hypothesis is supported by the stronger signal of the first peak (at ca 1.4 Å) in the FT-EXAFS spectra for Fe0.5-Ar vs. Fe0.5-NH3 (Figure 2d). Such ex situ changes may be assigned to a stronger FeN4-O2 interaction ex situ for Fe0.5-Ar, possibly due to a higher average oxidation state of Fe in Fe0.5-Ar vs. Fe0.5-NH3. In the latter, a lower oxidation state of Fe may be expected due to the presence of Lewis-base (highly basic) nitrogen groups, if some of those groups are directly involved in Fe cations ligation.
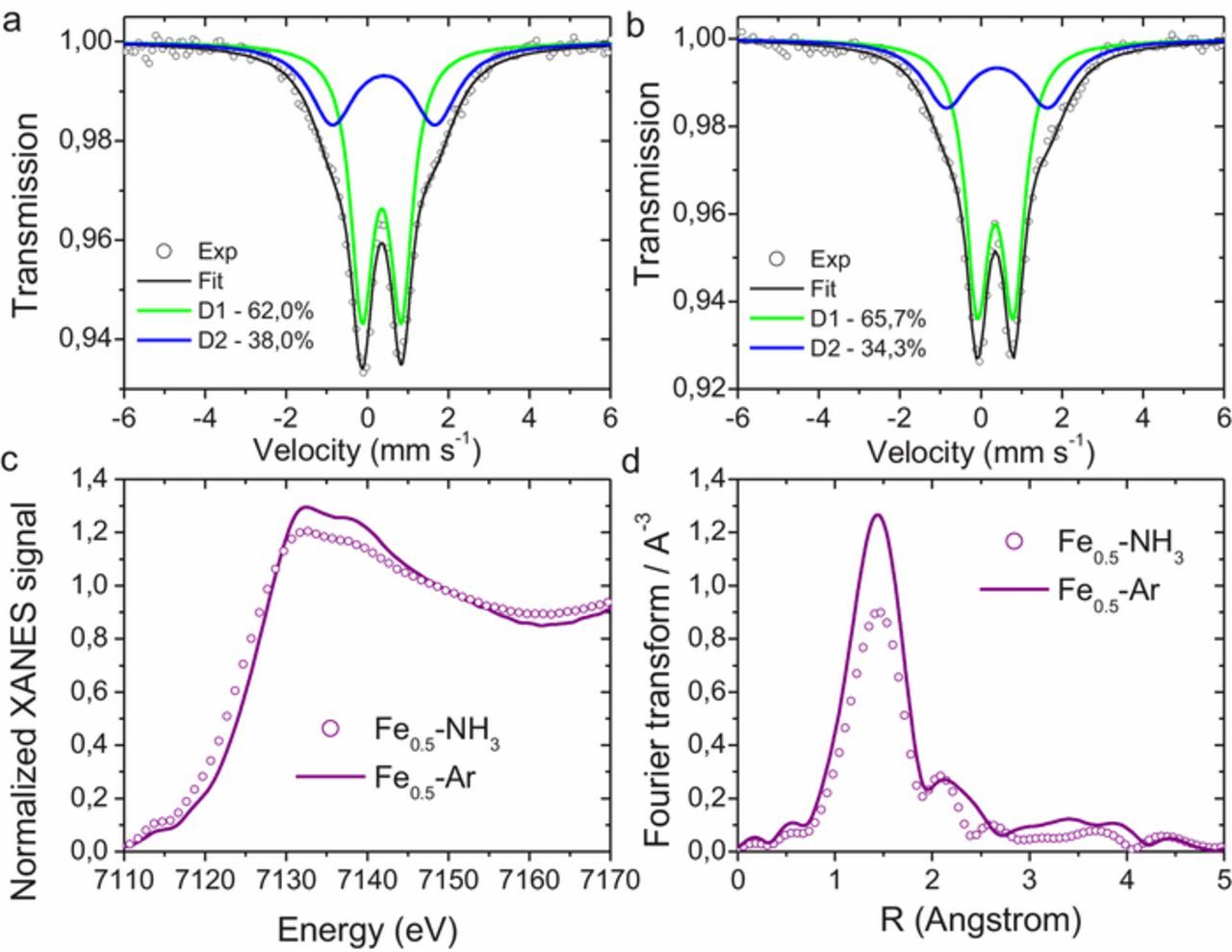
Figure 2. Ex situ spectroscopic characterization of Fe0.5-NH3 and Fe0.5-Ar. 57Fe Mössbauer transmission spectra (top) for a) Fe0.5-Ar and b) Fe0.5-NH3. XAS spectroscopic characterization at the Fe K-edge (bottom) by c) XANES and d) EXAFS. The spectra were recorded in air at room temperature. The distance in the FT-EXAFS spectra is not corrected for phase-shift.
Operando spectroscopic characterization
In order to investigate whether the small differences observed ex situ with XANES and FT-EXAFS spectra of both catalysts remained, disappeared or were exacerbated during the ORR in alkaline medium, we performed operando XAS. The XANES and EXAFS spectra were recorded in alkaline electrolyte between 0.2 and 1.0 V vs. RHE, covering all regions of the RDE polarization curves (Supporting Figure S1). Figure 3 shows the operando XANES spectra (left-handside) and FT-EXAFS spectra (right-handside) for Fe0.5-Ar (top) and Fe0.5-NH3 (bottom). For Fe0.5-Ar, both the operando XANES and FT-EXAFS spectra reveal large changes with the electrochemical potential (Figures 3a–3b). The change of the XANES spectra with potential observed here for Fe0.5-Ar in alkaline medium is similar to that reported by us for the same catalyst in acidic medium.41 The magnitude of change is however ca twice smaller in alkaline vs. acidic medium for Fe0.5-Ar, as can be seen by comparing the Δμ spectra (compare the inset of Fig. 3a in this work with the inset of Fig. 4b in Ref. 41). In the ORR potential region, the FeN4-moieties in Fe0.5-Ar undergo a structural change, as revealed by the complex change and existence of isobestic points at 7130.5 and 7156.4 eV in the set of XANES spectra.
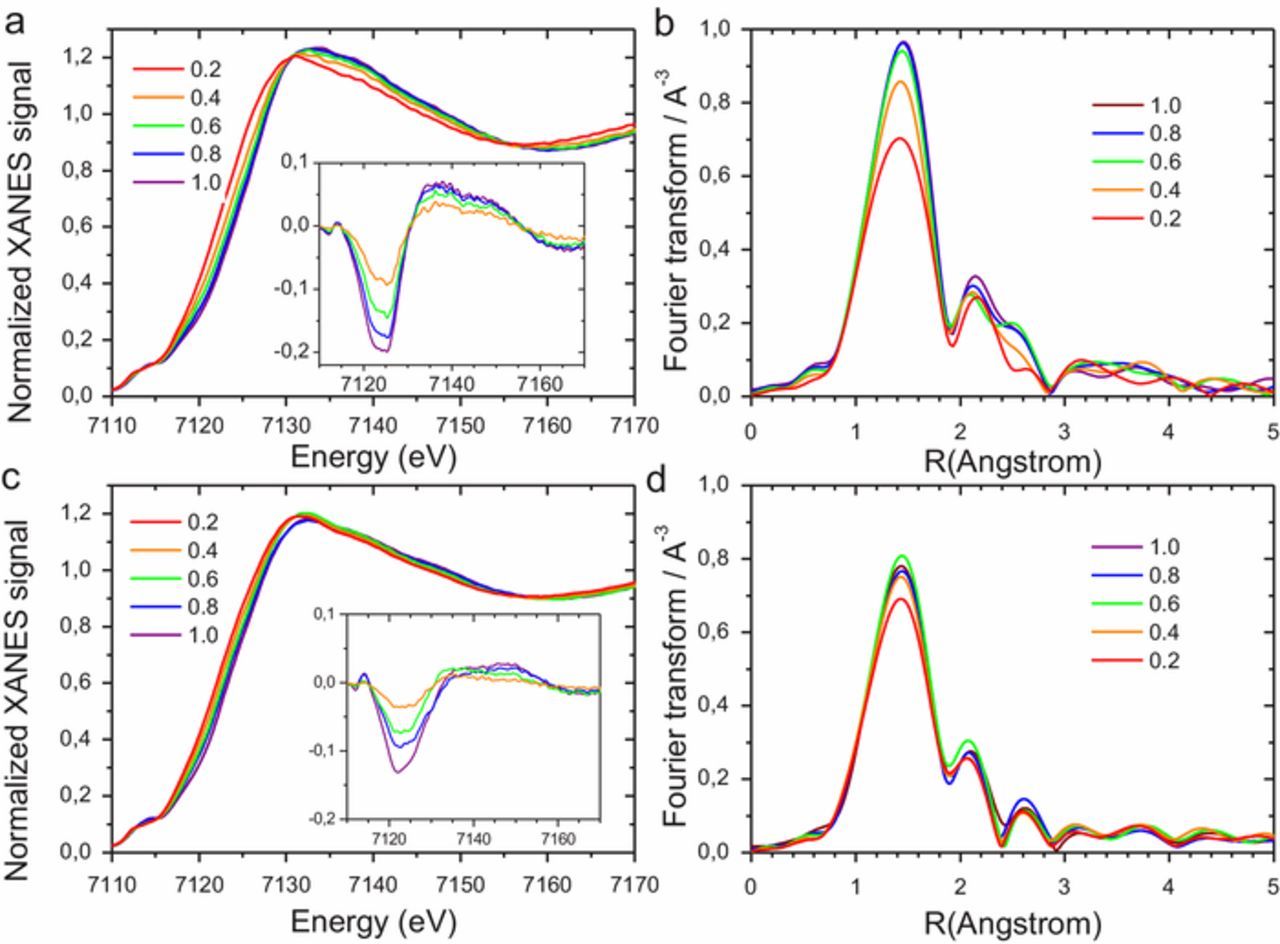
Figure 3. Operando XAS characterization of Fe0.5-Ar (a-b) and Fe0.5-NH3 (c-d) in O2-saturated 0.1 M KOH electrolyte. XANES (left-handside) and FT-EXAFS (right-handside) was recorded between 0.2 and 1.0 V vs. RHE. The legend indicates the potential (in V vs RHE) at which each spectrum was recorded. The insets in subfigures a and c show the Δμ spectra, obtained by subtracting the normalized XANES spectrum at a given potential to the spectrum recorded at 0.2 V vs. RHE.
These isobestic points support the existence of at least two Fe-site geometries, whose fraction switches gradually from 0 to 1 across the potential range of 0.2 to 1.0 V vs. RHE. This observation has also been reported, but only in acidic electrolyte hitherto, for other Fe-N-C materials19,48,49 and interpreted as a change from in-plane FeN4 to out-of-plane FeN4 configuration as a function of potential. The operando FT-EXAFS (Figure 3b) also support a structural change, with a decreasing signal intensity at 1.4 Å with decreasing potential. This can be interpreted as the presence of oxygen adsorbates (O2, OH and H2O) strongly adsorbed on FeN4 sites at high potential, and their absence or elongated Fe-O bond distance at low potential. Those spectroscopic changes were reversible, similar spectra being recorded at a given potential, when scanning down and then up the potential from 1.0 to 0.2 V and then back up to 1.0 V.
The operando XANES spectra of Fe0.5-NH3 in alkaline electrolyte reveal much smaller changes with electrochemical potential, as compared to changes observed for Fe0.5-Ar in the same electrolyte (compare Figure 3c vs. Figure 3a). The Δμ spectra (inset of Fig. 3c) reveals a trend that is however comparable to the Δμ signals observed with Fe0.5-Ar in alkaline electrolyte (inset of Fig. 3a), but with ca twice lower magnitude of change. The smaller spectral changes with electrochemical potential observed for Fe0.5-NH3 vs. Fe0.5-Ar may appear at first counterintuitive, since the former has a higher BET area and expectedly a higher exposure of Fe-sites to the top surface and to the electrolyte. The smaller spectral changes with potential observed for Fe0.5-NH3 must therefore be assigned to a distinct environment of Fe-sites in Fe0.5-NH3 compared to Fe0.5-Ar, in line with their much different ORR activities in alkaline electrolyte (Figure 1).
In line with the operando XANES spectra, the corresponding FT-EXAFS spectra of Fe0.5-NH3 are almost unchanged with potential (Fig. 3d). The trend of the signal intensity at 1.4 Å with electrochemical potential is the same as for Fe0.5-Ar (decreasing signal with decreasing potential), but the magnitude of the change is also much smaller. It is in fact only significant at the lowest potential studied, namely 0.2 V vs. RHE.
Coming back to the initial question whether the small differences observed ex situ with XANES and FT-EXAFS spectra of both catalysts remained or disappeared in operando, the direct comparison of the XANES and EXAFS spectra of both catalysts at low potential (0.2 V vs. RHE, Figure S2) reveals that the spectroscopic signatures are almost identical. Thus, the two catalysts differ in ex situ conditions or in situ at high electrochemical potential, but the differences become smaller as the potential is lowered, and become negligible at 0.2 V vs. RHE. This supports the hypothesis that, in ex situ conditions, Fe is in a higher oxidation state in Fe0.5-Ar than in Fe0.5-NH3, and binds more O2 or oxygen adsorbates. In operando conditions, the difference progressively vanishes as the electrochemical potential is reduced (ferric moieties turning into ferrous moieties in Fe0.5-Ar, becoming then in a similar state as Fe0.5-NH3).
Electrochemical stability and operando iron leaching study
The stability of Fe0.5-Ar and Fe0.5-NH3 catalysts was studied in both acidic and alkaline electrolytes. Table I summarizes the activity observed before and after the 5000 load-cycle AST. Starting with Fe0.5-Ar in alkaline medium, an activity increase from 0.07 to 0.11 mA cm−2 at 0.9 V vs. RHE can be seen after 5000 load-cycles in N2-saturated solution (purple curves in Figs. 1a–1b). Similarly, also Fe0.5-NH3 shows high stability in alkaline electrolyte, with very slight decay in activity at 0.9 V vs. RHE (from 0.92 to 0.83 mA·cm−2, see purple curves in Figs. 1c–1d). In acidic medium, no activity loss was observed for Fe0.5-Ar, but a significant loss observed for Fe0.5-NH3, with a relative decrease of about 24% (Table I).
We attribute this reduced activity following AST in N2-saturated electrolyte to a loss of a fraction of the active centers, i.e. FeN4 or only Fe cations from FeN4 moieties, from the nitrogen-doped carbon network. Since the AST was performed in N2-saturated electrolyte, no ORR occurred during the AST and we can exclude a degradation or deactivation due to hydrogen peroxide or reactive oxygen species formed from peroxide and FeNx sites, which was recently demonstrated to cause a main deactivation of Fe-N-C catalysts in acidic medium.34 To show this loss of iron in situ, we conducted SFC-ICP–MS measurements. Figure 4 summarizes the operando Fe leaching measurements where we applied the same potential-scan protocol (upper plot) to Fe0.5-Ar and Fe0.5-NH3 in oxygen-saturated electrolyte. The electrode was first contacted by the SFC (corresponding time marked with * in the graph, t ∼ 250 s) at open circuit potential (OCP). The applied electrochemical potential protocol is then 20 cyclic voltammograms (CVs) in the range 1.0 to 0.6 V vs. RHE at a scan rate of 100 mV·s−1, a potential range typically occurring during the ORR in fuel cell devices. Another chronoamperometry at 1.0 V was recorded for 200 s, before applying one CV at a low scan rate of 2 mV·s−1 from 1.0 V down to 0.0 V vs. RHE, and back up to 1.0 V vs. RHE to identify in more detail the potential-dependence of Fe dissolution
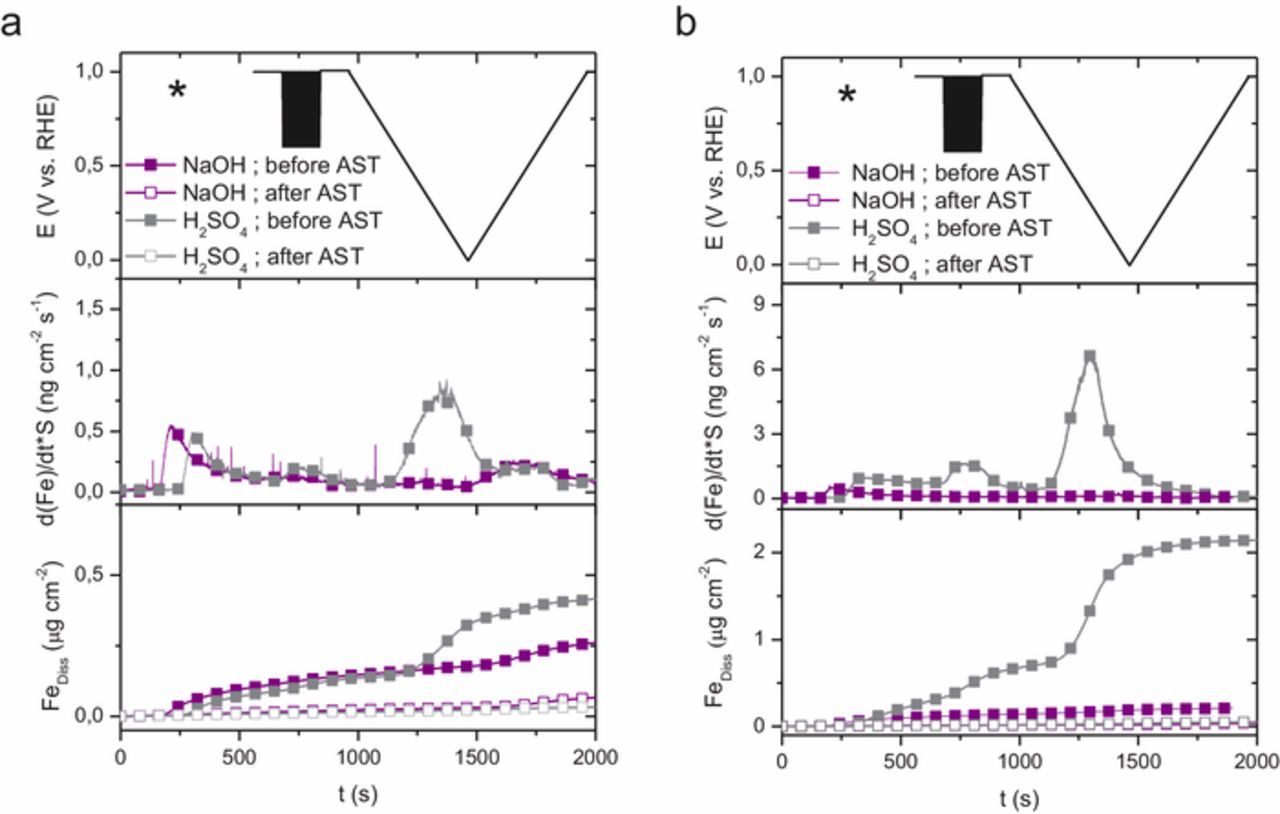
Figure 4. Time-resolved potential (top) dissolution rates (middle) and totally dissolved amount (bottom) of iron from Fe0.5-Ar (a) and Fe0.5-NH3 (b) measured in oxygen saturated electrolyte according to the color scheme of Figure 1. Note the different range of dissolution values in the Y-axis for a) and b). The contact dissolution peak is marked with an asterisk in the upper panels. Dissolution rates after AST in Ar-saturated electrolyte have been omitted for clarity in the middle panels since they are negligible.
For each catalyst considered separately, Figure 4 clearly identifies higher Fe dissolution rates in acid than in alkaline electrolyte, especially during the slow CV. We first discuss the transient release of Fe occurring when the SFC contacts the electrode with the electrolyte (time marked with an asterisk), leading to an initial loss of iron. For Fe0.5-Ar, this initial loss is similar in both electrolytes (Fig. 4a, middle panel), while for Fe0.5-NH3 the dissolution rate in acid medium is more than double that in alkaline electrolyte (Fig. 4b, middle panel). Comparing the contact dissolution in acidic medium, the peak dissolution rates are ca 1.0 and 0.4 ngFe·cm−2·s−1 for Fe0.5-NH3 and Fe0.5-Ar, respectively. After 400 s at OCP in acid medium, the Fe dissolution rate became < 0.15 ngFe·cm−2·s−1 for Fe0.5-Ar but remained significant (∼ 1.0 ngFe·cm−2·s−1) and quite constant for Fe0.5-NH3. The cumulative Fe dissolution of Fe0.5-NH3 at that stage is however restricted to ca 0.5 μgFe·cm−2 (Fig. 4b, lower panel), much lower than the total amount of Fe in the catalyst layer (ca 2.5 wt% Fe in Fe0.5-NH3, leading to ca 10 μgFe·cm−2). It is therefore unlikely that the lower activity measured for Fe0.5-NH3 in acid vs. alkaline originates from a very fast dissolution of iron. In alkaline medium, the curves of Fe dissolution rate vs. time of both catalysts are nearly superimposed (from immersion to OCP hold, time 250 to 850 s on x-axis), with a peak value of Fe dissolution rate of ca 0.5 ngFe·cm−2·s−1, quickly decreasing (only ca 0.1 ngFe·cm−2·s−1 before starting the fast CVs). Therefore, a trend is observed that Fe0.5-Ar is more stable than Fe0.5-NH3, and that alkaline environment leads to lower dissolution rate than acidic medium.
The subsequent fast 20 CV scans increased the Fe release rate in acidic medium, especially for Fe0.5-NH3 (increasing to 1.5 ngFe·cm−2·s−1), while it had no impact on the Fe dissolution rate in alkaline medium. Note that, due to the high scan rate used during the 20 CVs, the effect of scanning up or down the potential cannot be distinguished, and only a lump Fe dissolution rate is observed.
The time-resolved Fe dissolution rate during the subsequent slow potential scan then allows us identifying at which potential Fe is dissolved. In sulfuric acid, the onset of Fe dissolution while scanning the potential from 1 V down to 0 V occurs at ca 0.75 V vs. RHE, and the peak of dissolution rate occurs at ca 0.2–0.3 V vs. RHE, for both catalysts. The intensity of the peak of Fe dissolution is however 10 times higher for Fe0.5-NH3 vs. Fe0.5-Ar. At potentials E < 0.2 V vs. RHE, the Fe dissolution rate decreases for both catalysts, and remains very low also during the positive-going scan from 0.0 V to 1.0 V vs. RHE. For Fe0.5-NH3, the cumulative Fe amount leached after the slow CV reaches ca 2 μg·cm−2, representing about 50% of the total Fe content initially present (Fig. 4b, lower panel). Regarding the Fe release rate during the slow CV in alkaline electrolyte, there is no significant effect of the electrochemical potential in the negative-going branch of the scan, while reverting the scan direction from 0.0 V and upwards resulted in increased Fe dissolution rate for Fe0.5-NH3 but unmodified Fe dissolution rate for Fe0.5-Ar. These experiments were repeated multiple times, and showed reproducible trends.
While Figure 4 informs on the electrochemical conditions in which Fe is dissolved, Figure 5 quantitatively shows how much Fe from the catalysts was dissolved as a function of time in the SFC-ICP-MS protocol before the AST. The y-axis shows the %Fe remaining in the catalyst relative to the initial Fe content. The cumulative dissolved Fe content was obtained from the integral of the curves shown in the lower panels of Figure 4 while the total Fe content in each electrode was derived from i) the fixed Fe-N-C catalyst loading value and the exact geometric area investigated by SFC-ICP-MS (verified each time by a microscope) and ii) the knowledge of the initial Fe content in each catalyst. The latter were measured by ICP-MS on the catalyst powders to be 1.45 wt% for Fe0.5-Ar and 1.57 wt% for Fe0.5-NH3. Figure 5 shows that the absolute Fe dissolution is restricted for Fe0.5-Ar (at both pH) and for Fe0.5-NH3 at high pH (5 to 10% relative Fe content is dissolved after 20 fast CVs and a slow scan) while for Fe0.5-NH3 at acidic pH, more than 50% of the initial Fe content present in the active layer was dissolved after the same time.
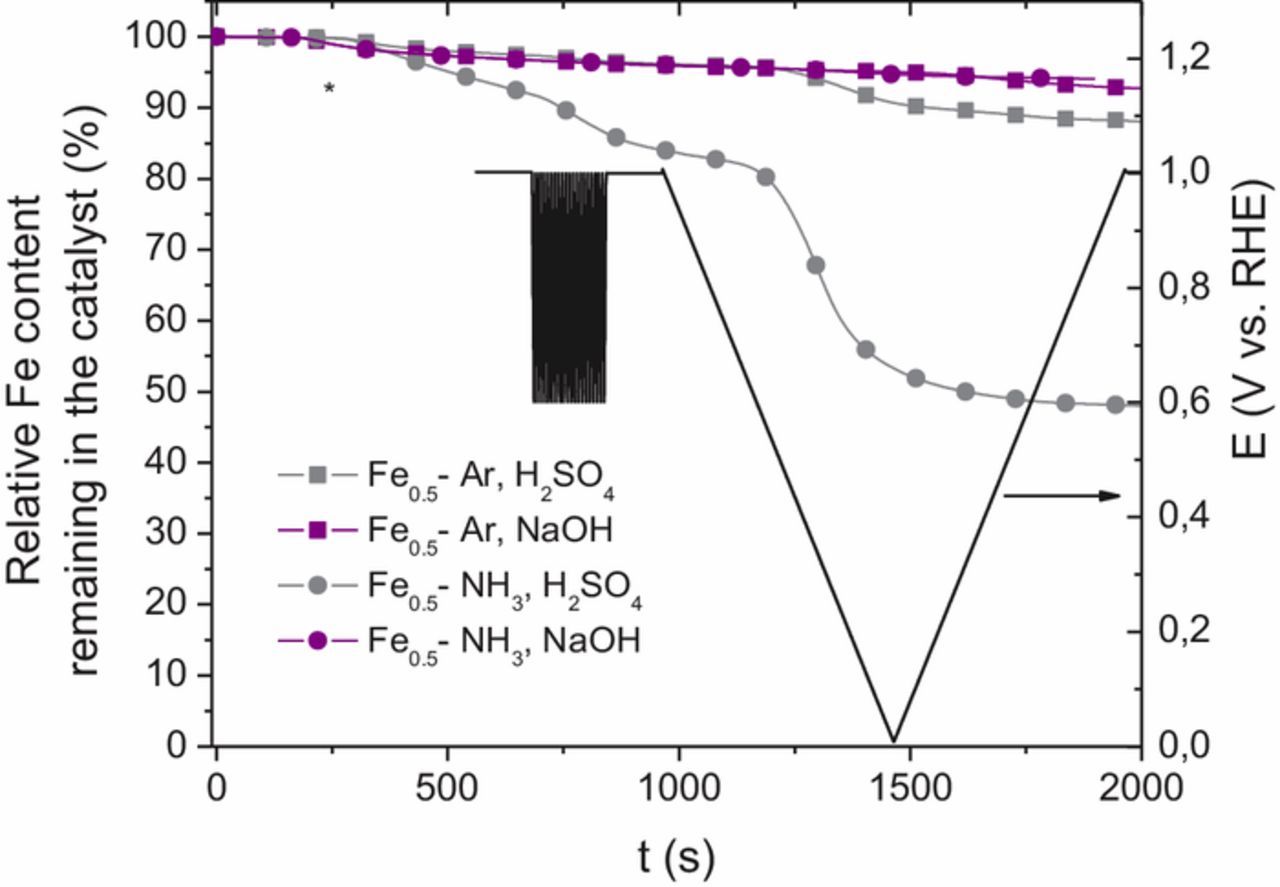
Figure 5. Percentage of initial iron remaining in the catalyst as a function of time. The electrochemical potential applied as a function of time is the same as that shown in Figure 4.
These time-resolved Fe dissolution data reveal that the Fe-based sites in Fe0.5-NH3 are less stable in acidic medium than those present in Fe0.5-Ar, while in alkaline medium the stability of Fe0.5-NH3 is as good, or even better, than that of Fe0.5-Ar. While the data might be interpreted by assuming that a much higher fraction of all FeNx sites are exposed to the electrolyte in Fe0.5-NH3 than in Fe0.5-Ar, this assumption should have resulted in a slightly increased Fe dissolution for Fe0.5-NH3 in alkaline electrolyte compared to that for Fe0.5-Ar in the same electrolyte. This is however not observed. The operando XAS data are also not in support of an increased fraction of FeNx sites being exposed to the electrolyte in Fe0.5-NH3 (smaller magnitude of change for the XANES and EXAFS spectra with potential than for Fe0.5-Ar). Thus, the electrolyte-exposed FeNx sites in Fe0.5-NH3 seem to be intrinsically less stable in acidic medium than those in Fe0.5-Ar.
Discussion
The operando XANES and EXAFS data in alkaline electrolyte reveal that the catalyst Fe0.5-NH3 experiences less change of its site geometry and Fe oxidation state as a function of the electrochemical potential, as compared to Fe0.5-Ar. This is assigned to a lower average oxidation state of Fe cations in FeNx moieties in the resting state for Fe0.5-NH3 than for Fe0.5-Ar. These fine differences between FeN4 sites in Ar-pyrolyzed and NH3-pyrolyzed catalysts are revealed here for the first time by operando XAS, and can explain the higher TOF at high potential for ORR of Fe0.5-NH3 relative to Fe0.5-Ar. The lower average oxidation state of Fe in NH3-pyrolysed catalysts may be a consequence of the presence of nitrogen groups with Lewis basicity. It can be reasonably proposed that the involvement of highly basic nitrogen groups in Fe ligation in Fe0.5-NH3 results in increased electron density at the Fe centers, increased O2 binding and also introduces the possibility to immobilize protons near the Fe centers, which could reduce the energy barrier during the rate determining step of the ORR. However, if highly basic nitrogen groups are directly involved in the coordination of all or some FeNx moieties, it can be expected that such moieties will be stable only in alkaline electrolyte, and not in acidic medium. The operando Fe leaching measurements support this hypothesis, with increased Fe leaching specifically observed for Fe0.5-NH3 in acidic medium. The instability in acidic medium of some FeNx moieties present in Fe0.5-NH3 may thus be assigned to the higher basicity of N-groups that ligate some of the iron cations. Upon their protonation in acidic medium, the covalent bond that previously existed between such Fe cations and nitrogen is broken or weakened, and the iron cations are dissolved in the electrolyte.
It is however unresolved from the dissolution data whether such unstable FeNx moieties in acidic medium account for the vast majority of the ORR activity of pristine Fe0.5-NH3, or both stable and unstable FeNx moieties co-exist in comparable amount. The latter hypothesis is more likely. Due to the disorder of the system formed during high-temperature pyrolysis in NH3, one might expect that two types of moieties coexist, i) FeNx moieties with Fe ligated by at least one highly-basic nitrogen group, and ii) FeNx moieties with Fe ligated only by nitrogen groups with low pKa value (non-protonating in pH 1). The existence of this mixed system of FeNx moieties would explain the irreversible loss of ORR activity experienced by NH3-pyrolyzed Fe-N-C catalysts after an acid-wash but also the fact that the very low ORR activity after acid-wash (activity / initial activity = 0.1) can be recovered to about 0.5 of the initial activity after a mild re-heat-treatment at 300°C (that removes anions and restores the N-groups in a non-protonated state).27 Bringing further complexity, the online Fe dissolution data reveals here that the Fe leaching from Fe0.5-NH3 in acid medium is significantly enhanced when the electrochemical potential is < 0.75 V vs. RHE, and almost peaks at 0.5 V vs. RHE, a potential close to the one often chosen during stability testing of PGM-free cathode catalysts in PEMFC.
Thus, while there is no doubt that the nitrogen protonation and anion-binding phenomenon reduces the high activity of NH3-pyrolysed Fe-N-C catalysts in liquid acid electrolyte in RDE set-up, it is unclear whether this effect is responsible for the fast decay of NH3-pyrolysed Fe-N-C catalysts during the first 10–15 h of operation in PEMFC. The online Fe dissolution data presented here suggest that the Fe dissolution rate of NH3-pyrolysed Fe-N-C catalysts in acid medium may be very fast at cathode potentials of 0.3-0.6 V vs. RHE. The circa 10 x faster Fe leaching rate from Fe0.5-NH3 than from Fe0.5-Ar in liquid acid medium in this potential range is in line with the relative degradation rate of Fe0.5-NH3 vs Fe0.5-Ar in PEMFCs. Further study exploring the potential-dependence and atmosphere-dependence (O2, air or simply N2) of the performance degradation of Fe0.5-NH3 during potentiostatic control of PEMFC cathodes, combined with Fe dissolution measurements may strengthen this hypothesis.
The antagonism between ORR activity and stability of Fe0.5-NH3 revealed here in acid medium does not exist in alkaline electrolyte, where high activity and high stability are simultaneously met. This supports the idea that highly-basic N-groups are at the root of the high ORR activity of FeNx moieties in ammonia-pyrolyzed Fe-N-C catalysts. Such catalysts are therefore proper candidates for replacing Pt-based catalysts in AEMFCs.
Conclusions
Two Fe-N-C catalysts comprising only atomically-dispersed FeNx moieties were prepared, differing only in the fact that the second catalyst (Fe0.5-NH3) was obtained by subjecting the first one (Fe0.5-Ar) to a short pyrolysis in ammonia. While the initial ORR activity in acid medium in RDE setup is similar for both catalysts, the activity in alkaline medium is significantly higher for Fe0.5-NH3. Operando XAS measurements in alkaline electrolyte reveals similar trends of the spectra as a function of the electrochemical potential for both catalysts, but the magnitude of change is much less for Fe0.5-NH3, as evidenced by a Δμ analysis. Accelerated stress tests in alkaline and acidic electrolyte revealed that the ORR activity of both catalysts was very stable in alkaline electrolyte, while some activity decay is observed for both catalysts in acidic electrolyte after 5000 cycles. Time-resolved Fe dissolution combined with previous literature studies point that the lower ORR activity of Fe0.5-NH3 in acid vs. alkaline liquid electrolyte is the outcome of two phenomena, i) the leaching of a fraction of acid-unstable FeNx moieties, and ii) the protonation and charge-neutralization by counter-anions of the electrolyte of highly-basic N-groups. Overall, ammonia pyrolysis of Fe-N-C catalysts is shown to result, in alkaline medium, in high ORR activity of atomically-dispersed FeNx moieties, high ORR durability and minimized Fe leaching during electrochemical operation in load-cycling mode. In acid electrolyte, the ammonia pyrolysis of Fe-N-C catalysts results in circa 10 times enhanced Fe leaching relative to the reference inert-gas pyrolyzed catalyst, with a Fe leaching rate that is strongly enhanced when an electrochemical potential in the range 0.75 to 0.3 V vs. RHE is applied. This may explain the recognized reduced stability of ammonia-pyrolyzed Fe-N-C catalysts in operating PEMFCs.
Acknowledgments
The project CREATE leading to these results has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 721065. We also acknowledge synchrotron SOLEIL (Gif-sur-Yvette, France) for provision of synchrotron radiation facilities at beamline SAMBA (proposal number 20171318) and Andrea Mingers (Max Planck Institute for Iron Research, Düsseldorf, Germany) for Fe bulk content measurement of the catalysts with ICP-MS.
ORCID
Qingying Jia 0000-0002-4005-8894
Sanjeev Mukerjee 0000-0002-2980-7655
Serhiy Cherevko 0000-0002-7188-4857
Frédéric Jaouen 0000-0001-9836-3261