
Abstract
The alkaline stability of poly(arylene ether) backbones in anion exchange polymer electrolyte membranes (AEMs) derivatized with quaternary benzyl N, N- dimethylhexylammmonium (DMH+) and trimethylammonium (TMA+) cation groups were investigated in poly(2,6-dimethyl 1,4-phenylene) oxide (PPO) and Udel polysulfone (PSF) polymers. Previous studies have demonstrated that quaternary ammonium and phosphonium groups trigger backbone degradation in commercially available poly(arylene ether)-based AEMs, despite the base polymers being resilient to alkaline solutions. Herein, we demonstrate that the electron withdrawing or donating character in the poly(arylene ether) backbone ultimately decides whether or not the prepared AEMs will become brittle in alkaline media due to cation triggered backbone degradation. Mitigation of cation triggered backbone degradation was only achieved when electron withdrawing substituents (not including the cation), such as sulfone or bromine groups, were eliminated from the polymer backbone (or, alternately, when electron donating groups were present). Hence, PPO AEMs prepared through chloromethylation, as opposed to free radical bromination, were resistant to backbone hydrolysis in alkaline media because each cation-functionalized repeat unit had two electron donating methyl groups rather than a single methyl group. In summary, this paper presents some design rules for preparing mechanically stable poly(arylene ether) AEMs from low cost, commercially available polymers for alkaline electrochemical devices.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Alkaline fuel cells (AFCs) and solid alkaline electrolyzers using anion exchange polymer electrolyte membrane (AEM) separators have attracted a lot of attention of late because they offer the prospect of low temperature electrochemical devices substantially free of platinum group metals.1 For example, Yan and co-workers2 and Zhuang and co-workers3 have already demonstrated an initial proof-of-concept of a platinum group metal free AEM-AFCs. Another attractive feature of a fuel cell with an AEM configuration is the reduction in methanol crossover when using a liquid methanol fuel stream.4 Because the AEM transports hydroxide ions from the cathode to the anode, there is no electro-osmotic drag of methanol from the anode to the cathode, and the ion flux counters the fuel crossover flux. While much work has been done on AFCs, AEMs are also attractive separators for flow batteries,5,6 water electrolyzers,7 and other electrochemical applications.8 They are also important for bipolar membrane electrolytes for fuel cells, mineral acid and base production, and pH adjustment of food products.9,10
There are two significant concerns when using AEMs for alkaline electrochemical applications. One is their lower ionic conductivity when compared to their cation exchange polymer electrolyte membrane counterparts (e.g., DuPont's Nafion) and the second is their poor stability when exposed to alkaline solutions.11 With respect to alkaline stability, both the polymer backbone and cation group components of the AEM can degrade in the presence of hydroxide ions. Since the 2005 review article written by Slade and Varcoe, significant headway has been made into improving the AEM ionic conductivity and cation group alkaline stability.4 For instance, the hydroxide ion conductivity for AEMs has been reported to be over 100 mS cm−1 in the temperature range of 60 to 100°C in deionized water (DI H2O).12–15 A more recent review by Varcoe and co-authors highlight the recent progress in AEM research for alkaline fuel cells and other electrochemical applications.16 There are reports describing AEMs with exceptional alkaline stability in ≥1 M potassium hydroxide (KOH) (or sodium hydroxide (NaOH)) at ≥60°C.17–21 However, the AEMs with exceptional alkaline stability tend to be of the polyaliphatic type and thus one must compromise on oxidative (and perhaps mechanical) stability when using these materials.
Other AEM systems like polyphenylenes and radiation grafted partially fluorinated backbones have exceptional alkaline stability, but E-beam fabrication of AEMs has not been commercialized, and is still practiced on a batch by batch basis at the laboratory scale. Other AEMs, like polyphenylenes, require the synthesis of many precursors – some of which have low yields.22–24 There are some recent reports of alkaline stable AEMs containing pendant side chains or crosslinked pendant side chains, AEMs prepared from block copolymers and AEMs with the cation group imbedded along the linear polymer backbone.25–29 In the case of AEMs prepared with side chains and from block copolymers, high alkaline stability was obtained and attributed to the phase-separation between the hydrophilic domains and hydrophobic domains, where the fixed cation groups in the hydrophilic domains remained well solvated.25,26,29 Increasing the solvation of the cation group in the presence of the hydroxide ion enhances the stability of the cation.30 Cation groups imbedded along the linear polymer backbones can be sterically hindered, mitigating hydroxide ion attack at these sites.27,28
Poly(arylene ether) backbones are desirable for AEMs because of their high oxidative stability and their simple synthesis from commercially available polymers.31 A major drawback to using poly(arylene ether) backbones (e.g., poly(arylene ether) sulfone (PSF) and poly(2,6-dimethyl 1,4-phenyle oxide (PPO)) for AEMs is that the AEMs become brittle in alkaline solutions over time. This observation was initially made for PSF AEMs by Sata in 199632 and by Hibbs and co-workers in 200923, but neither group was able to determine the root cause of the mechanical degradation of the films. Additionally, this result was contrary to the findings by Zschocke et al. who established that PSF by itself was resilient in alkaline media.33 Recent papers by our group established that adding cation groups to the benzyl position of Udel PSF and PPO caused the polymer backbone to chemically degrade through several different hydrolysis mechanisms in alkaline media, explaining the loss in mechanical properties.34,35 We termed this phenomenon "cation-triggered backbone degradation." More recently, Li et al. showed that changing the cation from quaternary benzyl trimethylammonium (TMA+) to quaternary benzyl N,N-dimethylhexylammonium (DMH+) on a PPO backbone mitigated the backbone hydrolysis degradation mode.36,37 Additionally, crosslinking PPO with quaternary benzyl DMH+ cations further enhanced AEM mechanical stability in alkaline solutions.18 We found these results surprising given that we had shown that changing the electron donating character of cation group at the benzyl position on a PSF backbone did not influence cleavage of the polymer backbone.35,38 It is postulated that the DMH+ cation's long n-hexyl alkyl chain, which exhibits high electron donating character, suppressed the electron withdrawing effect of the affixed cation, thereby minimizing the delocalization of the electronic structure of the polymer backbone, rendering it less susceptible to hydroxide ion attack.
Resolving the backbone stability issue in AEMs is important because mechanical degradation of the film leads to fuel and oxidant crossover and poor membrane-electrode contact, both of which result eventually in catastrophic failure. Mustain and co-workers examined several commercially available AEMs and they observed that the AEMs became brittle after exposure to alkaline solutions.39 Thus, the issue extends beyond experimental chemistries, and is seen even in established variants. There is therefore a clear need for AEMs that are mechanically stable in alkaline media.
In this work, we investigate with further rigor the question of whether or not the DMH+ cation mitigates chain scission in PSF and PPO AEMs. The PPO AEMs were prepared through two different methods: i.) free radical bromination or ii.) chloromethylation. As discussed in detail below, our results revealed that incorporation of the DMH+ to the benzyl position of Udel PSF does not mitigate backbone degradation. Also, our work did not initially confirm the findings reported in Li et al.'s work that placing DMH+ at the benzyl position of PPO mitigated backbone degradation. However, our PPO backbone initially deviated slightly from that used by Li and co-workers, in that our variant had some bromine groups at the aryl position - an artifact arising from running the free radical bromination reaction at lower temperatures. Chloromethylating PPO, rather than brominating PPO, with the subsequent addition of DMH+ or TMA+ proved to be a more effective method for preparing mechanically stable PPO AEMs in alkaline solutions. Using the DMH+ cation over the TMA+ cation did not yield any gains in the alkaline (chemical) stability of the chloromethylated PPO AEMs. The findings in this paper suggest a new set of design rules for preparing mechanically resilient poly(arylene ether) AEMs for alkaline applications.
Experimental
Preparation of brominated PPO and chloromethylated PPO and PSF
Our previous works details the preparation of chloromethyalted PSF (CMPSF) and brominated PPO (BrPPO) from commercially available Udel PSF and PPO polymers.34,40 BrPPO was prepared via free radical bromination using azobisisobutyronitrile (AIBN) as the initiator and n-bromosuccinimide (NBS) as the brominating reagent. As shown in our previous studies and by Xu et al.,41 the reaction temperature (100 to 130°C) ultimately determined the relative selectivity of bromination to the benzyl and aryl position of PPO, while the amount of NBS added determined the amount of bromine functionalized to PPO.
Chloromethylation of PSF and PPO was performed using a Friedel-Crafts reaction developed by Avram et al.42 Yan and co-workers adopted this procedure for PPO and they observed that the chloromethylation kinetics in PPO were more facile when compared to PSF.43 The Avram et al. procedure specifies stannic chloride as the Lewis acid catalyst and chlorotrimethylsilane and paraformaldehyde as the chloromethylating reagents. Unlike the free radical bromination reaction, the degree of functionalization (DF) to PSF during the reaction was determined by time and not the amount of chloromethylating reagents added. The electronic supplementary information section (ESI) provides a detailed account of the synthesis methods.
The AEMs were prepared by dissolving the halogenated poly(arylene ethers) in an aprotic solvent (either n-methyl-2-pyrrolidone (NMP) or N,N-dimethylformamide (DMF)) and the addition of a tertiary amine - N,N-dimethylhexylamine (DMH) or trimethylamine (TMA). The Menshutkin reaction proceeded for 12 hours at room temperature for TMA and 60°C for DMH. The AEMs were gathered by drop casting the AEM reaction solutions onto glass plates in an oven and evaporating the solvent for 12 hours at 70°C. The films were removed from the glass plate after immersion in deionized water using a knife. See Scheme 1 for the synthesis of the halogenated poly(arylene ether)s and the subsequent formation of AEMs.
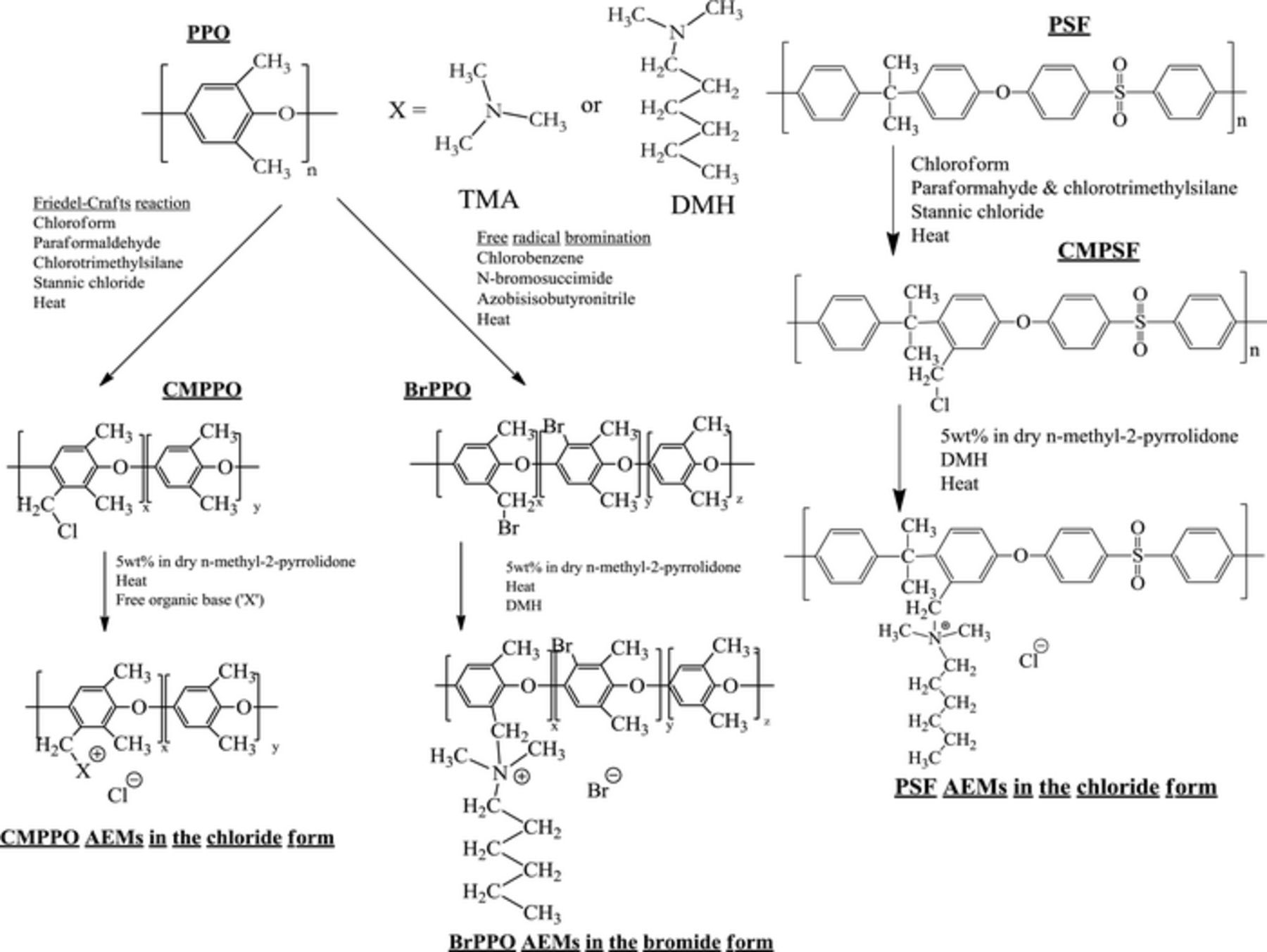
Scheme 1. Preparation of chloromethylated PPO, brominated PPO, and chloromethylated PSF followed by reaction of the halogenated polymers with a tertiary amine to prepare AEMs.
Membranes to be characterized in the hydroxide (OH−) or chloride (Cl−) counter-ion form were ion-exchanged immediately prior to characterization. See our previous work for the procedure.34 The prepared AEMs (before and after exposure to alkaline solutions) were characterized by one-dimensional and two-dimensional NMR methods and FTIR spectroscopy to confirm the structure. Integration of 1H NMR spectra was used to estimate the theoretical ion-exchange capacity (IEC), while the Vohlard titration was used to measure the experimental IEC. Electrochemical impedance spectroscopy was used to determine the in-plane ion conductivity in deionized water (∼18 MΩ) under temperature control; a 4-point probe was employed for this measurement. Water uptake (WU) was measured gravimetrically for AEMs exposed to humidified water vapor. To evaluate mechanical properties, stress-strain curves were generated using a dynamic mechanical analyzer (DMA). See the ESI and our previous reports for full details of each experimental method and for a description of the alkaline stability tests in 1 M KOH at 60°C for 10 days.34,35,38,40,44
Results
AEM preparation and spectroscopic characterization
BrPPO was synthesized by free radical bromination. Varying the reaction temperature yielded different ratios of bromine at the aryl and benzyl positions of the PPO backbone. In this work, two batches of BrPPO (BrPPO Batch #1 and #2) were obtained using different reaction temperatures: 115°C and 130°C, respectively. The amount of bromine at the aryl position in BrPPO Batch #1 was 15% (i.e., 15% of repeat units had bromine to the aryl position) and the amount of bromine at the aryl position in BrPPO Batch #2 was 4%. As noted by Xu et al.,41 high reaction temperatures dictated selectivity of bromine to the benzyl position over the aryl position. The authors observed that adding more bromine to the aryl position made the polymer more hydrophobic. This allowed the use of higher IECs to achieve higher ionic conductivity while retarding excess water uptake. Arges et al. established that AEMs prepared from BrPPO with bromine at the aryl position achieved hydroxide ion conductivities close to 40 to 50 mS cm−1 over the temperature range of 30°C to 70°C, but that the AEMs became extremely brittle in 1 M and 6 M KOH solutions at 60°C.34
Figure 1a reports the 1H-13C HMQC NMR spectrum of BrPPO-DMH+ from BrPPO Batch #1. The 1H-13C HMQC NMR spectrum of BrPPO-DMH+ from BrPPO Batch #2 resembled the spectrum given in Figure 1a and is presented in the ESI under Figure S1. The signal associated with the dimethyl groups adjacent to the quaternized nitrogen in DMH+ was observed at 3.0 ppm for 1H and 50 ppm for 13C. This signal verified that DMH+ was successfully attached to brominated PPO. Integration of the 1H NMR dimethyl signals in DMH+ for BrPPO-DMH+ Batch #1 demonstrated that 103 ± 1% of bromomethylated groups in BrPPO Batch #1 reacted with DMH. Integration of the 1H NMR for BrPPO-DMH+ Batch #2 yielded 98 ± 2% conversion of the bromomethyl groups to DMH+ cations.

Figure 1. 1H-13C HMQC spectra of a.) BrPPO-DMH+ Batch #1 and b.) CMPSF-DMH+.
Figure 1b presents the 1H-13C HMQC of PSF-DMH+. Similar to Figure 1a, the dimethyl group adjacent to the quaternized nitrogen in DMH+ was observed at the 1H-13C HMQC coupling of 3.0 ppm for 1H and 50 ppm for 13C. Integrating the 1H NMR for PSF-DMH+ groups revealed that 107 ± 2% of the chloromethylated groups reacted with DMH. The conversion apparently exceeded 100% because of the variability in the determined DF value in CMPSF and the error in the integration of the 1H NMR spectra (which can be up to 10%).45
Note: The assignments of the other signals associated with the PSF and PPO backbones seen in Figures 1a and 1b have been detailed in our previous work and are not repeated here.34,38
Neither CMPPO-DMH+ nor CMPPO-TMA+ could be dissolved into aprotic solvents (DMF, dimethylsulfoxide, and N,N-dimethylacetamide) or chloroform. The lack of solubility prevented us from determining the conversion of chloromethyl groups to quaternary ammonium groups using NMR and from calculating the theoretical IEC. Therefore, FTIR spectroscopy was used to confirm successful preparation of these AEMs. Figures 2a and 2b present the FTIR spectra of pristine CMPPO-DMH+ and CMPPO-TMA+ AEMs. Both spectra show the C-N+ infrared vibration absorption at 1600 to 1610 cm−1.46,47 Additionally, both spectra showed broad absorption peaks in the range of 3300 to 3500 cm−1 corresponding to the O-H vibrational stretching, arising from water solvating the quaternary ammonium cations and chloride anions in the AEM. The absorbance peaks observed in the range 2700 to 3000 cm−1 for both spectra were attributed to the –CH3 stretching from the PPO backbone and the TMA+ and DMH+ cations and the –CH2– groups in DMH+ (in Figure 2b).48 Additionally, the signal for C-Br stretching in bromobenzyl groups, expected at 740 to 750 cm−1, was not observed in the FTIR spectra of the CMPPO-derived AEMs.46,47 Considered together, these observations suggested that the CMPPO AEMs with TMA+ or DMH+ cations were indeed successfully prepared.

Figure 2. FTIR spectra of pristine a) CMPPO-TMA+ AEM and b) CMPPO-DMH+ AEM.
IEC, water uptake, ionic conductivity, and mechanical properties of AEMs
Table I reports the DF values for the halogenated polymer precursors used for preparing the AEMs while Table II provides the theoretical and experimental IEC of the different AEMs prepared. The IEC values given for each AEM material were directly proportional to the DF values of their halogenated polymer precursors. For most samples where both experimental and theoretical IEC values were reported, the values were in agreement, suggesting that both the NMR integration and Vohlard titration methods were accurate and complimentary. However, there was a significant disagreement between the theoretical and experimental IECs of BrPPO-DMH+ AEM Batch #2 – 2.3 mmol g−1 versus 1.5 mmol g−1 respectively. A similar finding was reported by us previously on PSF AEMs with IEC values above 2 mmol g−1,40 where the PSF AEMs with large IECs (over 2 mmol g−1) demonstrated larger theoretical IEC values versus the experimental IECs, while the experimental IEC and theoretical IEC of AEMs below 2 mmol g−1 were in agreement. This trend suggests that the Vohlard titration method may have an upper limit with regard to its accuracy, which would not be unexpected because the accuracy of the method is predicated on complete ion-exchange between the chloride ions in the AEMs with the nitrate ions in the solution. AEMs with larger IEC values may suffer from incomplete ion-exchange. To verify this viewpoint, a two-stage Volhard titration was performed on the BrPPO-DMH+ AEM. The method and results (see Table S1) are given in the ESI section. However, the hypothesis of incomplete ion-exchange seems to be invalid based on the results (i.e., a single stage extraction was sufficient for a near complete ion-exchange). Future work will look to investigate the discrepancy in IEC values obtained by Vohlard titration technique and by integrating the 1H NMR spectrum for AEMs with high IEC.
Table I. Degree of functionalization (DF) of halogen groups to the polymer and the conversion of halogenated benzyl groups to quaternary ammonium groups.
| Sample | DF value of Br to aryl position (n = 2) | DF value of halogen to benzyl position (n = 2) | Conversion of halogenated benzyl groups (%) |
|---|---|---|---|
| BrPPO-DMH+ Cl− (Batch #1) | 0.14 | 0.35± 0.02 | 103 ± 1 |
| BrPPO-DMH+ Cl− (Batch #2) | 0.05 | 0.48± 0.06 | 98 ± 2 |
| PSF-DMH+ Cl− | n/aa | 0.84± 0.01 | 107 ± 2 |
| CMPPO-TMA+ Cl− | n/aa | 0.13 | n/ab |
| CMPPO-DMH+ Cl− | n/aa | 0.13 | n/ab |
Note: Error bars are for the range of n = 2 samples. an/a – No functionalization to aryl position because chloromethylation reaction via Friedel-Crafts procedure was performed. bn/a – AEMs were not soluble into NMR solvents.
Table II. Theoretical and experimental IEC and water uptake of prepared AEMs.
| Sample | Theoretical IEC (mmol g−1) | Experimental IEC (mmol g−1) | Water uptake at 25°C at 98% relative humidity (%) |
|---|---|---|---|
| BrPPO-DMH+ Cl− (Batch #1) | 1.8 ± 0.1 | 1.9 ± 0.4 | 6.4 ± 3 |
| BrPPO-DMH+ Cl− (Batch #2) | 2.3 ± 0.1 | 1.5 ± 0.1 | 11.7 ± 5 |
| PSF-DMH+ Cl− | 1.6 ± 0.0 | 1.6 ± 0.1 | 12.8 ± 3 |
| CMPPO-TMA+ Cl− | n/a | 1.0 ± 0.1 | 26.4 ± 4 |
| CMPPO-DMH+ Cl− | n/a | 0.9 ± 0.2 | 22.4 ± 3 |
n/a - AEMs were not soluble into NMR solvents.
Table II also reports the vapor phase water uptake at 98% relative humidity at 25°C for each AEM. BrPPO AEMs Batches #1 and #2 had the lowest water uptake values. The water uptake for the PSF-DMH+ AEM was slightly higher than that of the BrPPO AEMs. CMPPO AEMs had the largest water vapor uptake (22% to 27%) despite having the lowest IEC. The water uptake of AEMs with large IECs can be suppressed through modification of the backbone by introduction of hydrophobic moieties. For instance, BrPPO-DMH+ Batch #1 AEMs had the highest IEC but the lowest water uptake. This was made possible by the incorporation of bromine to the aryl position of the polymer backbone. The vapor phase water uptake for BrPPO-DMH+ Batch #2 AEM at room temperature was 12%, while the vapor phase water uptake for CMPSF-DMH+ was 13%. Both of these AEM materials had similar IEC values (1.5 to 1.6 mmol g−1) suggesting that PSF and brominated PPO Batch #2 were reasonably similar in their hydrophobic characteristics.
Figures 3a and 3b report the in-plane ionic conductivity of the PPO and PSF AEMs in the Cl− form and OH− form in DI water under temperature control. All AEMs, except PSF-DMH+, had a modest increase in their ionic conductivity with increasing temperature. The PSF-DMH+ AEM, on the other hand, demonstrated a relatively larger increase in its ionic conductivity from 30 to 50°C but saw a rapid drop in ionic conductivity at 70°C. The decrease of the ionic conductivity at 70°C resulted from excess swelling in water at the higher temperature, which hindered ion mobility. We observed a similar effect (i.e., excess swelling compromising ionic conductivity) for PSF AEMs with high IECs and in the hydroxide form in our previous reports.40 In future studies, we will seek to determine the appropriate IEC for PSF-DMH+ AEMs that maximize ionic conductivity while simultaneously minimizing water swelling. The hydroxide ion conductivity trend of the AEMs was similar to their chloride ion conductivity trend observed except that the ionic conductivity values were higher for the hydroxide form. The higher ionic conductivity stemmed from the 2.6× higher mobility of the hydroxide ion compared to the chloride ion.8 However, AEMs ion-exchanged to the hydroxide form mostly likely consisted of a mixture of hydroxide, carbonate, and bicarbonate anions despite efforts to purge the water of carbon dioxide by bubbling nitrogen gas through it. Figure S4 in the SI section reports the bicarbonate ion conductivity of one synthesized AEM variant – BrPPO-DMH+ (Batch #1). The bicarbonate ion conductivity was about the same as the hydroxide ion conductivity, thus confirming that the AEMs in the hydroxide form initially suffered from bicarbonate or bicarbonate formation. Hence, comparisons or trends between materials are best defined based on the chloride and bicarbonate ion conductivity.

Figure 3. In-plane a.) Cl− and b.) OH− ion conductivity in deionized water under temperature control before alkaline stability tests. The error bars represent the standard error (n = 3).
The PSF-DMH+ AEM demonstrated substantially higher ionic conductivity than the PPO AEMs (both BrPPO and CMPPO AEMs). In Li et al's work, the PPO AEMs with long alkyl tails (e.g., n = 16 carbons) gave rise to a micro-phase separation in the AEM structure – as revealed by small-angle X-ray scattering (SAXS). The authors also observed a broad peak with the smaller n = 6 alkyl tail in PPO-DMH+ AEMs, but did not observe a scattering pattern in the SAXS pattern of PPO-TMA+. Thus, the micro-phase separation of the ionic domains apart from the hydrophobic n-alky tail domains enabled better ionic conductivity. Because the PSF backbone is more flexible than the PPO backbone, the PSF AEM with DMH+ could display distinct phase separation behavior, which would explain the better ionic conductivity attained.
The mechanical properties of the AEMs were investigated using a dynamic mechanical analyzer that collected stress-strain curves. Table III reports the stress and strain at break for each pristine AEM sample in the chloride form. From Table III, it was apparent that the PPO AEMs prepared from chloromethylation rather than free radical bromination had remarkably better mechanical properties. Furthermore, the PSF AEMs had substantially better mechanical properties over the BrPPO AEMs. The poor mechanical properties of BrPPO AEMs were ascribed to the large, bulky bromine atom functionalized to the aryl unit in PPO. The significant difference in mechanical properties of CMPPO AEMs against BrPPO AEMs was attribute to residual, bulky Br present at the aryl position in BrPPO preventing efficient chain packing. Another possible explanation entails that the cation group orthogonal to the methyl group enables better chain packing over the cation attached directly to the methyl group. In this context, we note that the PSF AEMs had substantially better mechanical properties compare to the BrPPO AEMs. The strength of the PSF-DMH+ AEMs was good when compared with a commercially available Nafion 211 membrane (13.9 MPa). The mechanical properties of CMPPO AEMs were slightly better than the mechanical properties of CMPSF AEMs with the same cation. But, both the CMPSF AEMs and CMPPO AEMs demonstrated significantly better mechanical properties over BrPPO AEMs.
Table III. Stress-strain test results of BrPPO, CMPPO and PSF AEMs before and after exposure to 1 M KOH at 60°C for 10 days.
| Sample | Stress (MPa) and strain (%) at break for pristine samples | Stress (MPa) and strain (%) at break for samples exposed to 1 M KOH at 60°C for 10 days |
|---|---|---|
| BrPPO-DMH+ Cl− (Batch #1) | Stress - 7.5 ± 1.7 MPa Strain - 6.7 ± 1.4% | N/A |
| BrPPO-DMH+ Cl− (Batch #2) | Stress - 8.5 ± 4.8 MPa Strain - 2.5 ± 0.6% | N/A |
| PSF-DMH+ Cl− | Stress - 22.3 ± 3 MPa Strain - 28.2 ± 0.8% | N/A |
| CMPPO-TMA+ Cl− | Stress - 28.8 ± 5.8 MPa Strain - 43.4 ± 13% | Stress - 10.3 MPa ± 2.0 MPa Strain - 5.1% |
| CMPPO-DMH+ Cl− | Stress - 32.9 ± 3.1 MPa Strain - 58.1 ± 12% | Stress - 7.0 ± 3.7 MPa Strain - 2.7 ± 0.7% |
*Note: The data for the pristine samples give error bars derived from the calculated standard error for n = 3 measurements. The data for the error bars for the alkaline stability samples were determined by the absolute difference from the average because only n = 2 measurements were made for these samples.n/a - Samples were too brittle to make a measurement.
Alkaline stability
The alkaline stability of the AEMs was evaluated by monitoring the chloride ion conductivity, IEC, mechanical properties, and NMR spectra after exposure 1 M KOH at 60°C for 10 days. Using this combination of functional and spectroscopic tests allowed us to: i.) gauge which materials were resilient to alkaline solutions with a high level of confidence, ii.) determine the modes of hydroxide-ion induced degradation, and iii.) connect different degradation modes to the chemical structure of the AEM. Utilizing a multitude of test methodologies was the best strategy to understanding the diverse and complex degradation modes of AEMs.
The CMPPO-DMH+ and CMPPO-TMA+ AEMs were the only AEMs testable with respect to ionic conductivity after exposure to the 1 M KOH solution for 10 days at 60°C. Despite their mechanical resilience, these AEMs demonstrated significant losses in in-plane chloride ion conductivity. The chloride ionic conductivity of CMPPO-TMA+ dropped from 6.0 ± 0.3 mS cm−1 to 2.1 ± 0.1 mS cm−1 after immersion in 1 M KOH for 10 days at 60°C, while that of CMPPO-DMH+ dropped from 5.0 ± 0.6 mS cm−1 to 1.2 ± 0.4 mS cm−1 under the identical test condition. The BrPPO and PSF AEMs became too brittle to measure ionic conductivity even after 10 days of exposure to 1 M KOH at 60°C.
Ionic conductivity measurement methods cannot discriminate between anion and cation driven conductivity, stability data reported by using ion conductivity as the primary metric can sometimes be misleading.44,49 For instance, if backbone degradation occurs yielding carboxylic acid functionalities but the fixed cation carriers are degraded, the carboxylic acid groups would contribute to conductivity of the sample even though the fixed cation carriers (which is what we are concerned with) were degraded. Hence, the experimental measurement of anion IEC by Volhard titration is one of the most reliable and simplest ways to confirm degradation of the fixed cation groups in an AEM. Table IV reports the IEC of the AEMs before and after exposure to 1M KOH at 60°C for 10 days. This table also reports the IEC data normalized to the AEMs' initial IEC values expressed as a percent. An unexplored consideration is the concentration of fixed ionic carriers (i.e., IEC) on the rate of AEM cation stability when exposed to alkaline solutions. Pan et al. showed that adding less ionic groups per repeat unit in Udel PSF backbone reduced the rate of cation triggered hydrolysis of the PSF backbone in alkaline media.50 A limitation in the analysis of the alkaline stability data reported as the IEC value alone was that the initial IEC of each AEM material differed. Hence, the IEC data expressed as a relative percent provided a more useful comparison. The AEMs prepared from BrPPO-DMH+ Batch #1 retained 79% of their initial IEC after 10 days in 1M KOH at 60°C, while AEMs prepared from BrPPO Batch #2 only retained 13% of their initial IEC after being stored in 1 M KOH at 60°C for 10 days. These results suggested that adding more bromine to the aryl position stabilized the cation from hydroxide ion attack. However, we do recognize that a more systematic study exploring the amount of halogen to the PPO backbone and its influence on cation stability needs to be conducted to test this hypothesis. The PSF-DMH+ AEM retained only 27% of its initial IEC after 10 days after exposure in 1M KOH at 60°C and all the samples broke into pieces after 3 days. The cation groups of the CMPPO AEMs demonstrated better hydroxide ion attack resistance when compared to those of BrPPO AEM Batch #2 and PSF AEMs because their relative IEC drop after 10 days was smaller – the CMPPO AEMs retained 60% of their IEC.
Table IV. IEC of AEMs before and after exposure to 1 M KOH at 60°C for 10 days.
| Sample | Experimental IEC for pristine samples (mmol g−1) | Experimental IEC of AEMs after exposure to 1 M KOH at 60°C for 10 days (mmol g−1) | Normalized IEC data after exposure to 1 M KOH at 60°C for 10 days (i.e., cation groups remaining) (%) |
|---|---|---|---|
| BrPPO-DMH+ Cl− (Batch #1) | 1.9 ± 0.4 | 1.5 ± 0.4 | 79% |
| BrPPO-DMH+ Cl− (Batch #2) | 1.5 ± 0.1 | 0.2 ± 0.1 | 13% |
| PSF-DMH+ Cl− | 1.6 ± 0.1 | 0.3 ± 0.1 | 19% |
| CMPPO-TMA+ Cl− | 1.0 ± 0.1 | 0.7 ± 0.1 | 70% |
| CMPPO-DMH+ Cl− | 0.9 ± 0.2 | 0.6 ± 0.2 | 67% |
Note: The error bars represent the standard error from the average (n = 3 samples were measured).Note: Normalized IEC data was determined by dividing the average IEC after 10 days of exposure to 1 M KOH at 60°C by the initial average IEC of the pristine sample.
Anion IEC and ionic conductivity experiments are practical metrics for assessing whether or not AEM materials are resilient in alkaline solutions. However, these experiments cannot conclusively confirm chemical degradation nor can they provide insight into the degradation products and their corresponding mechanisms. The latter is very important for understanding AEM degradation and for engaging in the rationale design of new AEM materials that are stable in alkaline media. Here, using 1D and 2D NMR spectroscopy, we have confirmed that the DMH+ cation degrades through dealkylation (a direct nucleophiic substitution method) in BrPPO and PSF AEMs and we have confirmed that this cation triggers ether-hydrolysis in PSF AEMs. Figure 4 is the 1H-13C HMQC NMR spectrum of BrPPO-DMH+ Batch #1 after exposure to 1 M KOH at 60°C for 10 days. Figure S2 in the ESI is the 1H-13C HMQC NMR spectrum of BrPPO-DMH+ Batch #2 after exposure to 1 M KOH at 60°C for 10 days. Figure S2 and Figure 4 were qualitatively identical. In both these spectra, a new peak appeared at the cross-section of 2.1 ppm for 1H and 46 ppm for 13C (signal A) when compared to the pristine 1H-13C HMQC NMR spectra of BrPPO-DMH+ Batches #1 and #2 (See Figures 1a and Figure S1 in the ESI). This peak was ascribed to the methyl group(s) in a benzyl tertiary amine moiety. Therefore, the DMH+ cation degraded via dealkylation of one of the methyl groups or the n-hexyl chain. Figure 5 illustrates the dealkylation pathway of degradation of DMH+. These AEMs, as previously stated, were observed to become brittle after exposure to alkali. Arges et al. linked the mechanical (in)stability of BrPPO AEMs to cation triggered chemical degradation of the backbone in alkaline media.34 The peaks associated with the backbone degradation observed for the BrPPO AEMs in Arges et al.'s work could not be detected in Figures 4 and S2 in the ESI because the carbon atoms and protons in the DMH+ cation's n-hexyl alkyl chain had the same chemical shifts (1.3 ppm for 1H and 30 ppm for 13C and 0.9 ppm for 1H and 15 ppm for 13C) as the backbone degradation products observed in Arges et al.'s work. Li et al. demonstrated that this cation - DMH+ - and other variants with long alkyl chains and crosslinked alkyl chains, prevented chain scission in PPO AEMs, whereas PPO-TMA+ AEMs became brittle in alkaline solutions.18,36,37 However, our findings challenge this result. Perhaps the difference lies in the fact that our BrPPO-DMH+ AEMs had residual bromine groups on the PPO backbone that spurred chain scission upon exposing the BrPPO-DMH+ AEMs to 1 M KOH solution at 60°C (Li et al.'s PPO AEMs did not have bromine attached to the PPO aryl position.)

Figure 4. 1H-13C HMQC spectrum of BrPPO-DMH+ (Batch #1) after 10 days exposure in 1 M KOH at 60°C.

Figure 5. The dealkylation degradation mechanism of DMH+ in PPO and PSF AEMs.
Similarly, the 1H-13C HMQC NMR spectrum of PSF-DMH+ AEMs after exposure to 1 M KOH at 60°C for 10 days demonstrated the same tertiary amine signal (2.1 ppm for 1H and 46 ppm for 13C - signal B) from the deakylation of DMH+ as seen in Figures 4 and S2 in the ESI for BrPPO-DMH+ AEMs. See Figure 6. Furthermore, phenyl alcohol was observed at 6.0 ppm for 1H and 120 ppm for 13C (signal C) in the 1H-13C HMQC NMR spectrum of PSF-DMH+ after exposure to alkaline solutions. The presence of signal C indicated the degradation of the PSF backbone in PSF-DMH+ AEMs via ether hydrolysis.35 Hence, while DMH+ mitigated chain scission of the PPO backbone in PPO AEMs (provided there was no residual bromine in the backbone – as in Li et al.'s work), the same effect does not hold true when switching to an alternative poly(arylene ether) backbone like Udel PSF.

Figure 6. 1H -13C HMQC spectrum of PSF-DMH+ after 10 days exposure in 1 M KOH at 60°C.
CMPPO AEMs were examined by FTIR spectroscopy after exposing them to 1 M KOH for 10 days at 60°C. See Figures S3a and S3b in the ESI section. No significant changes were observed in the FTIR spectra. The experimental IEC measurements demonstrated that the cation groups remained in CMPPO AEMs after 10 days of exposure to 1 M KOH for 10 days at 60°C; the C-N+ stretching signal was still apparent at 1600 to 1610 cm−1 in Figures S3a and S3b in the ESI. Cation degradation would result in benzyl alcohol groups or tertiary amines, but existing substituents in the PPO backbone and water would obscure those signals. For example, the 'C-N' stretch corresponding to tertiary amines would arise between 1050 cm−1 to 1100 cm−1. However, the 'C-O' stretching in aryl ethers also appear in this range. Likewise, the 'O-H' stretch in benzyl alcohol, resulting from direct nucleophilic substitution of the quaternary ammonium groups, would overlap with the 'O-H' stretching signal of water solvating remaining cation groups.
Table III also reports stress and strain results at the breaking point for BrPPO, CMPPO and CMPSF AEMs with DMH+ or TMA+ cations at 50°C and 50% RH as a function of exposure to alkaline solutions. After 10 days exposure to 1 M KOH at 60°C, PSF-DMH+ AEMs and BrPPO AEMs were too brittle to measure. Thus, neither of material was suitable for alkaline electrochemical applications at elevated temperatures. Conversely, CMPPO AEMs maintained adequate mechanical properties prior to and after exposure in alkaline media at 60°C for 10 days though their stress and strain values at break were slightly diminished after exposure to alkaline solutions. The mechanical stability of the CMPPO AEMs over the BrPPO AEMs and PSF AEMs stemmed from: a) the absence of electron withdrawing substituents in the CMPPO backbone (like bromine at the aryl position and sulfone), and b) the fact that each repeat unit containing a cation had two electron donating methyl groups rather than a single methyl group.
Conclusions
The strategy used in tethering quaternary ammonium groups to PPO backbones was shown to play a strong role in cation and backbone stability when the resultant AEMs was exposed to alkaline solutions at elevated temperatures. Previous studies have shown that the DMH+ cation, rather than the TMA+ cation, mitigated PPO backbone hydrolysis when exposed to alkaline media. However, this cation demonstrated herein did not have the same affect on PSF backbones because the electron withdrawing sulfone, in addition to the electron withdrawing DMH+ (albeit less electron withdrawing than TMA+), spurred backbone degradation in alkaline solutions. Furthermore, PPO AEMs with bromine at the aryl position in addition to containing the DMH+ cation were not immune to backbone chain scission because of the electron withdrawing bromine substituent. Altering the tethering strategy in PPO AEMs through chloromethylation, rather than free radical bromination, mitigated PPO backbone hydrolysis in alkaline solutions, regardless of the type of quaternary ammonium cation attached. The mitigation was possible because i.) the CMPPO AEMs did not have electron withdrawing substituents in the backbone, such as sulfone or bromine, and 2.) it had two electron donating methyl groups in each repeat unit along the backbone. Although cation triggered backbone degradation of PPO AEMs was mitigated in this work with a commercially available polymer, future efforts will focus on enhancing CMPPO AEM cation stability by experimenting with different cation chemistries or having the cation tethered to polymer backbone with long, n-alkyl chains that provide space between the cation and the polymer backbone.
Acknowledgments
The authors would like to gratefully acknowledge the Office of Naval Research for a grant under the Young Investigator Program (under contract # N00014-10-1-0752; PI: Vijay Ramani) for funding this work. We also thank the Research Resource Center (RRC) at the University of Illinois at Chicago for access to the 360 MHz Bruker NMR, and the University of Chicago's Chemistry Department for access to their FTIR spectrometer. Christopher G. Arges and Lihui Wang contributed equally to this article.