
Abstract
Previously conducted high-throughput ab initio calculations have identified carbonophosphates as a new class of polyanion cathode materials. Li3MnCO3PO4 is the most promising candidate due to its high theoretical capacity and ideal voltage range. However, a major limitation of this material is its poor cyclability and experimentally observed capacity. In this work we synthesize Li3Fe0.2Mn0.8CO3PO4 to combine the high theoretical capacity of Li3MnCO3PO4 with the high cyclability of Li3FeCO3PO4. Li3Fe0.2Mn0.8CO3PO4 outperforms Li3MnCO3PO4 in cyclability, and shows little capacity fade over 25 cycles. The observed reversible capacity of 105 mAh/g is slightly greater than in Li3FeCO3PO4, and occurs at a higher average voltage.
Export citation and abstract BibTeX RIS
The need for lithium-ion batteries with higher energy density than existing materials has led to significant efforts to discover new cathode materials.1–3 High-throughput ab initio computation is an effective approach employed to accelerate the process of materials discovery.4 This has led to the identification of several novel lithium intercalation materials.3,5,6 One class of novel materials that has been predicted to function as intercalation cathodes for Li-ion batteries is the lithium transition metal carbonophosphates.5 The Li3FeCO3PO4 and Li3MnCO3PO4 compounds are of particular interest, as shown in Table I, which shows data first reported by Chen et al.3 Both are predicted to have accessible 2+ to 3+ redox couples, but the 3+ to 4+ couple in the manganese-containing compound is also expected to be active at a voltage compatible with existing electrolytes. As a result, Li3MnCO3PO4 is of the greatest interest because it has a high theoretical capacity of 231 mAh/g and average voltage of 3.7 V. As polyanionic cathodes, lithium metal carbonophosphates could also be preferred over oxide cathode materials since they are generally less likely to release oxygen at high voltages.4
Table I. A comparison of predicted voltages of redox couples and theoretical capacities of Li3[M]CO3PO4 for [M] = {Fe, Mn} calculated in Chen et al.3
| Li3[M]CO3PO4 | Predicted | Theoretical Capacity | |
|---|---|---|---|
| [M] | Redox | Voltage | between 2 V–4.5 V |
| Fe | 2+/3+ | 3.0 V | 115 mAh/g |
| 3+/4+ | 4.6 V | ||
| Mn | 2+/3+ | 3.2 V | 231 mAh/g |
| 3+/4+ | 4.1 V | ||
The synthesis and characterization of both Li3FeCO3PO4 and Li3MnCO3PO4 have been previously reported.3 The lithium-containing carbonophosphates are not thermodynamic ground states, so the compounds are synthesized using ion-exchange techniques from the thermodynamically stable sodium carbonophosphates. As reported previously by Chen et al., Li3FeCO3PO4 has a theoretical capacity of 115 mAh/g and can be easily synthesized using ion exchange methods. This compound cycles reversibly, close to its theoretical limit at a rate of C/5. In contrast, Li3MnCO3PO4 shows a discharge capacity of 135 mAh/g on its first discharge at a rate of C/100, which is only ∼58% of its theoretical capacity. In addition, the capacity of Li3MnCO3PO4 degrades in subsequent cycles. The poor performance in Li3MnCO3PO4 could be due to many factors. However, we believe one major cause is the residual sodium (∼17%) ions sitting on Li sites as a result of an incomplete ion exchange during synthesis. The better-performing Li3FeCO3PO4 shows no residual sodium after synthesis.3
Our approach is to substitute manganese in the Li3MnCO3PO4 with iron to improve its performance by imparting the ion-exchangeability and cycling performance of the Li3FeCO3PO4 on to Li3MnCO3PO4. Similar mixing techniques have been used in previous attempts to improve the performance of α-LiMnPO4.7–9 In this paper we focus specifically on Li3Fe0.2Mn0.8CO3PO4. Since complete Li-Na exchange is much easier to perform in Li3FeCO3PO4 than in Li3MnCO3PO4, we expect this compound to ion-exchange more completely from its sodium-containing parent compound than Li3MnCO3PO4.
Experimental
Na3Fe0.2Mn0.8CO3PO4 was synthesized via a hydrothermal synthesis method.3 Two clear solutions were made with FeSO4·7H2O (J. T. Baker) and MnN2O6·4H2O (Sigma Aldrich), and Na2CO3 (Alfa Aesar, 99.5%) and (NH4)2HPO4 (Alfa Aesar, 98%), respectively. The combined molar ratio of transition metal cations was 1:7 to carbonate ions and 1:1 to phosphate ions. The solutions were mixed in a glass bottle, heated in an oil bath to 130 °C and magnetically stirred for 44 hours, all in an Ar-flushed glove box. Next, the reaction products were separated from the ion-exchange solution and washed by centrifuging in the native solution, followed by centrifuging in methanol, distilled water, and methanol again. The wet powder was dried overnight in a vacuum oven at 50 °C.
Li3Fe0.2Mn0.8CO3PO4 was made from its sodium-containing precursor via a Li-Na ion-exchange method. As-synthesized sodium carbonophosphate was added to 2M LiBr in 1-hexanol.10 The suspension was stirred magnetically for 5 days at 140 °C in an Ar atmosphere. The product was centrifuged to separate it from the exchange solution, and was then washed in methanol, water, and methanol again. The wet powder was dried in a vacuum oven overnight at 50 °C.
The ion-exchange product was then ball-milled with carbon in a Retsch PM200 planetary ball mill to improve the material's electrochemical performance through decreased particle size and carbon coating.11,12 Zirconia-lined milling jars were loaded with Li3Fe0.2Mn0.8CO3PO4 and carbon (Super P) in a weight percent ratio of 85:15 in an Ar-filled glove box. The sample was milled for 6 hours at 500 rpm, and the resultant powder was also unloaded in the Ar-filled glove box to limit air exposure.
Pristine powder samples of the sodium and lithium carbonophosphates were characterized by X-ray diffraction (XRD) using a Bruker D8 Advance diffractometer (Molybdenum Kα, λ = 0.7017 Å). Elemental composition of the lithium carbonophosphate material was verified by Inductively Coupled Plasma atomic emission spectroscopy (ICP) measurements using a Horiba Jobin Yvon, ICP-AES (ACTIVA-S). The sample powders were dissolved in 2% nitric acid, which itself was prepared from concentrated nitric acid and ASTM grade I deionized water. The diluted nitric acid was also used as a blank. Calibration curves were constructed using five calibration solutions of different concentrations for each measured element in 2% nitric acid. All measurements for each element were repeated three times and had a relative standard deviation of less than one percent.
To electrochemically characterize the Li3Fe0.2Mn0.8CO3PO4 powder cathode films were made using the as-prepared sample, which was mixed with polyvinylidene fluoride (PVDF) and carbon (Super P) such that the overall ratio of components is in a 6:1:3 weight percent. The films were made using a doctor-blade method on aluminum foil, with each electrode containing ∼2 mg of active material. CR2016 coin cells were assembled in an Ar glove box with a Li metal foil anode, Celgard 2025 separator (Celgard Inc., U.S.A.), and 1M solution of LiPF6 in ethylene carbonate/dimethyl carbonate (1:1) as electrolyte. Galvanostatic experiments were conducted at a rate of C/50 on an Arbin Instruments (College Station, TX) battery cycler.
Experimental Results
The sodium-containing sample is tan in color, while the lithium-containing sample is light beige, which is consistent with previously reported results.3 The XRD patterns of synthesized phase-pure Na3Fe0.2Mn0.8CO3PO4 and Li3Fe0.2Mn0.8CO3PO4 samples are shown in Fig. 1. The pre- and post-ion exchange spectra exhibit substantial differences, but are consistent with differences between Li- and Na-containing carbonophosphate phases for other transition metals.3 Substantial peak broadening is seen in the Li3Fe0.2Mn0.8CO3PO4 sample, which implies that either a substantial quantity of defects is present in the sample or that there are very small crystal domains in the ion-exchanged powder. Elemental analysis of the Li3Fe0.2Mn0.8CO3PO4 sample was conducted using ICP analysis, the results of which are displayed in Table II. It is evident that the Li-Na ion exchange proceeded almost entirely (>99%) to completion. The other element ratios are also present in very close to the desired ratio, with the exception of iron, which is in excess. The most likely explanation is that some amorphous Fe-containing phase formed during the ion exchange reaction in 1-hexanol, as has been previously hypothesized.3
Table II. Elemental composition of as-synthesized Li3Fe0.2Mn0.8CO3PO4 obtained using ICP analysis.
| Element | mg/L | Normalized Molarity |
|---|---|---|
| Li | 34.49 | 2.97 |
| Na | 0.89 | 0.02 |
| Fe | 24.30 | 0.26 |
| Mn | 73.18 | 0.80 |
| P | 49.14 | 0.95 |
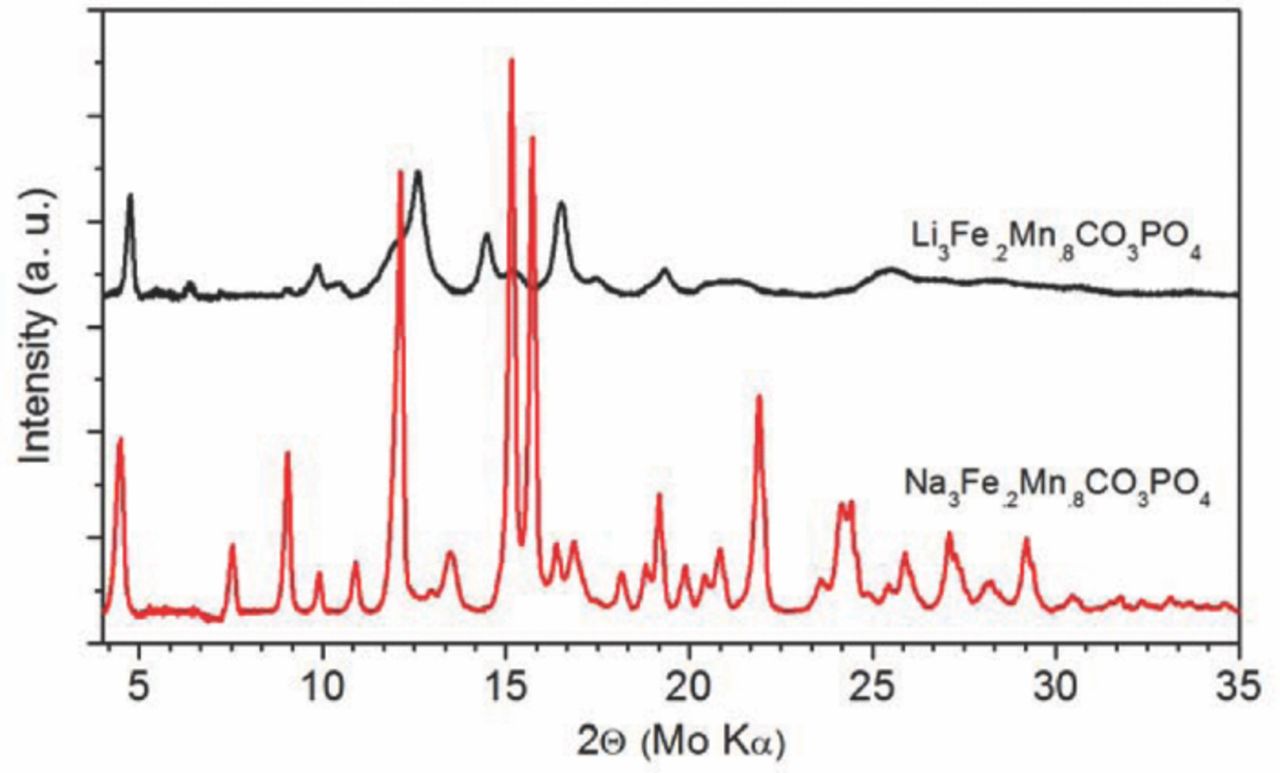
Figure 1. XRD patterns of as-prepared Na3Fe0.2Mn0.8CO3PO4 and Li3Fe0.2Mn0.8CO3PO4 samples.
Electrochemical testing of the Li3Fe0.2Mn0.8CO3PO4 cathodes was conducted at a rate of C/50 at room temperature in a 2.00–4.75 V window. The voltage versus capacity data for one of these tests is shown in Figure 2a. As seen in the figure, the first-cycle charge starts at greater than three volts, and exhibits a charge capacity of 284 mAh/g, which exceeds the material's theoretical capacity of 231 mAh/g. The high starting voltage and the differing behavior in subsequent cycles indicate that the Fe2+ in the prepared sample was oxidized during processing. The high charge capacity on the 1st cycle may stem from the irreversible Li+-extraction from an amorphous impurity phase left on the sample surface from synthesis, which is also indicated by the excess of iron seen in the ICP analysis of the ion-exchanged sample. The 1st discharge shows a capacity of 147 mAh/g, but the capacity of the sample reduces over the course of the first 4 cycles to a reversible value of around 105 mAh/g, or 0.9 Li+ per formula unit. This decrease is not believed to be the result of traditional cathodic capacity fade, but instead the result of irreversible cycling of amorphous iron-containing impurity phases between 2.5 V and 2.0 V. These impurities are likely iron-containing hydroxides or oxyhydroxides, which have been previously been shown to intercalate Li+ at voltages in this region.13 As shown in Figure 2a, the difference in discharge capacity between the 1st, 2nd, and 18th cycles stems primarily from decreased capacity in this low-voltage region.
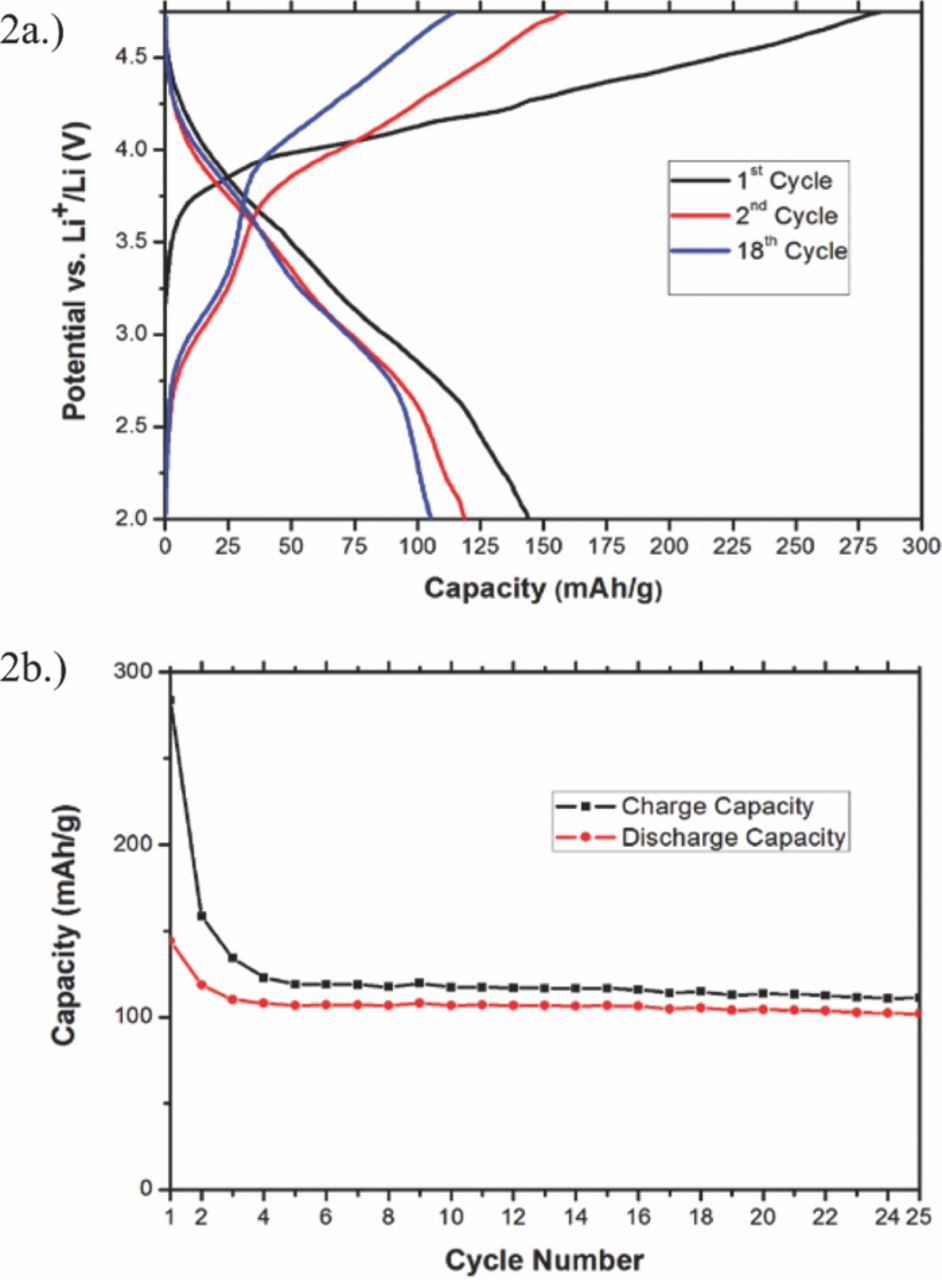
Figure 2. Voltage profile (2a) and capacity retention (2b) of Li3Fe0.2Mn0.8CO3PO4 cycled between 2 Volts and 4.75 Volts. The tests were performed at room temperature at a rate of C/50.
On charges following the first cycle, a distinct plateau is present near 3.0 V, which is the predicted voltage of the Fe2+/Fe3+ and near that of the Mn2+/Mn3+ couple.2 The plateau approximately corresponds to the theoretical capacity of iron in Li3Fe0.2Mn0.8CO3PO4 of 23 mAh/g. Additional plateaus, or other distinct features indicative of further redox activity are less apparent, and are discussed later.
The capacity retention is displayed in Figure 2b. Both the charge capacity and discharge capacity drop significantly over the first 4 cycles; from 284 mAh/g to 120 mAh/g and from 143 mAh/g to 108 mAh/g, respectively. After the fourth cycle the charge and discharge capacity remain relatively constant, and drop only ∼5 mAh/g over the next 20 cycles. In addition, significant coulombic inefficiency is observed, with the charge capacity consistently being about 10 mAh/g higher than the discharge capacity after the fourth cycle. This is likely the result of electrolyte decomposition at high voltage.
Discussion
The biggest attraction of Li3MnCO3PO4 is the potential existence of two redox couples in the voltage window of commercially used battery electrolytes. The proposed two-electron activity leads to a high theoretical capacity of 231 mAh/g. To improve the performance of the pure Li3MnCO3PO4, Li3Fe0.2Mn0.8CO3PO4 was synthesized, and its electrochemical performance is shown above. Of the 208 mAh/g theoretical capacity of the material, only about 105 mAh/g can be reversibly cycled for greater than 20 cycles. Despite performance well under theoretical values, this demonstrates a significant improvement over the previously reported performance of Li3MnCO3PO4, which showed much poorer cyclability. In addition, Li3Fe0.2Mn0.8CO3PO4 has a larger energy density than Li3FeCO3PO4. While the reversible discharge capacity for these materials is about the same, the average discharge voltage in Li3Fe0.2Mn0.8CO3PO4 is approximately 0.4 V higher, corresponding to an energy density 40 Wh/kg larger than in Li3Fe0.2Mn0.8CO3PO4.
To examine the activity of Fe2+/Fe3+, Mn2+/Mn3+ and Mn3+/Mn4+ redox couples more in depth, which are all predicted to be accessible in the tested voltage window, the differential charge (dQ/dV) capacity is shown in Figure 3 for several cycles. Two charge peaks and two discharge peaks are clearly visible. The first set of peaks around 3.0 V most likely correspond to overlap of the Fe2+/Fe3+ and Mn2+/Mn3+ couples, computed to exist at 3.0 V and 3.2 V, respectively. It has been previously reported that Fe2+/Fe3+ and Mn2+/Mn3+ couples move closer together when mixed in single-phase Li(Fe, Mn)PO4,14 and similar behavior could be present in this system. The second set of peaks around 4.0 V corresponds to the predicted Mn3+/Mn4+ activity in the active material. The slight shift seen in the second set of peaks over the first 12 cycles likely indicates that structural changes may be occurring in the material. In addition, the area of the peaks is noticeably different upon charge and discharge. Specifically, the area under the 3 V charging peak is smaller than the area of the 3 V discharge peak. Finally, the upturn at the high end of the charging curve on Figure 3 indicates that electrolyte oxidation may be occurring. This idea is reinforced since none of this observed charge capacity is returned on discharge, and explains the coulombic inefficiency seen in Figure 2b.
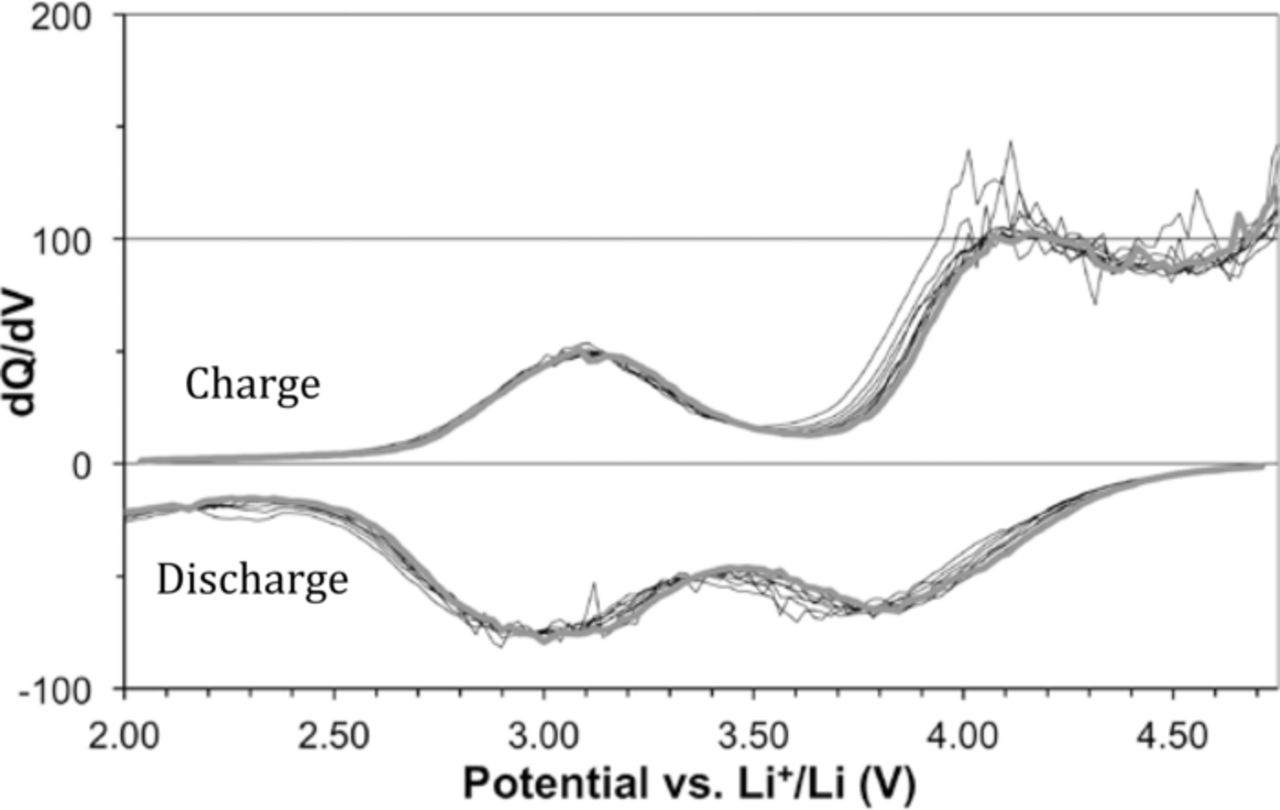
Figure 3. Curves showing the differential charge curves for a Li3Fe0.2Mn0.8CO3PO4 cell for its 3rd through 12th cycles. The reader should focus mainly on the envelope of the curves. The 1st and 2nd cycles are omitted for clarity.
The observed voltage of the Mn2+/Mn3+ redox couple shows one potential advantage carbonophospate cathodes hold over oxide cathodes. The inductive effect of the carbonate and phosphate polyanions causes the Mn2+/Mn3+ redox couple's voltage to be higher (3.2 V) than in oxide materials.15 However, the inductive effect in this mixed-polyanion system is not as large as that in pure phosphates, making the Mn redox couples lie in a useable voltage window, in contrast to LiMnPO4. In order to improve Li3Fe0.2Mn0.8CO3PO4, the capacity fading of the material, and its limited Li+ extraction must be addressed. In addition, the material's ionic and electronic conductivity would also need to be improved.
While this work demonstrates preliminary results of carbonophosphates as Li-ion cathode materials, a number of aspects of this materials performance remain to be understood. One notable unexplained cycling feature is the apparent asymmetric capacity seen in the Mn2+/Mn3+ redox couple on charging and discharging. In addition, the possibility that the increased discharge capacity in early cycles may result from an impurity phase in the material will need to be addressed in future works. Finally, the reason why the inclusion of iron in Li3Fe0.2Mn0.8CO3PO4 significantly improves the material is not yet known.
Conclusions
In this work we reported an improvement over the performance of Li3MnCO3PO4 by doping the compound with iron. We demonstrated the synthesis of Li3Fe0.2Mn0.8CO3PO4 via hydrothermal synthesis of a sodium-containing precursor followed by a Na+-Li+ ion exchange reaction. The Fe doping allows to ion exchange to proceed to near (>99%) completion. Li3Fe0.2Mn0.8CO3PO4 cathodes show a reversible capacity around 105 mAh/g with excellent cyclability over 25 cycles. The observed initial drop in capacity comes from irreversible cycling of impurities formed during synthesis. Three active redox couples, Mn2+/Mn3+, Mn3+/Mn4+, and Fe2+Fe3+ are active in this material. The results open new opportunities to improve the performance of the novel carbonophosphate cathode materials by doping and structural tuning. This is particularly meaningful because as recently reported, besides Fe and Mn, many alkaline earth and transition metals can also form carbonophosphate in pure or doped form.16
Acknowledgments
The authors acknowledge the Robert Bosch Company and Umicore for their financial support. This work also used the shared facilities in the Center for Materials Science and Engineering (CMSE) at MIT. We thank Geoffroy Hautier, Rahul Malik, Nancy Twu, and Jinhyuk Lee for their valuable assistance in this work.