
Abstract
A simple approach for expanding metalloporphyrins as electrocatalytic materials is introduced. This new method involves the direct electropolymerization of an electrically conducting layer of ferriprotoporphyrin followed by a metal-substitution reaction executed by soaking the electrode in an aqueous metal-salt solution. This process exchanges the iron center of the surface-bound protoporphyrin with the metal ion of the salt solution. X-ray photoelectron spectroscopy and electrochemical methods confirm that this new metal is incorporated into the surface-bound porphyrin structure and alters the electrochemical properties of the electrode. This simple electrode modification process may expand the use of metalloporphyrins for electrocatalytic applications.
Export citation and abstract BibTeX RIS
Electrode-supported metalloporphyrins have been used for various electrocatalytic reactions including the oxidation of NO, CO, SO2, alcohols, dopamine and glucose as well as the reduction of CO2, NO3−, NO, ClO3− and various sulfur-containing compounds (e.g., cysteine) for both sensing and industrial applications.1–5 Metalloporphyrins are attractive for such applications due to their ability to vary their oxidation states without altering their molecular structure. The electrochemical activity toward a given reaction is primarily governed by the metal center coordinated to the porphyrin ring as well as, to some extent, the substituents of the porphyrin molecule.
Unfortunately modifying electrode surfaces with these macrocyclic molecules is not always straightforward. Such methods as electrochemistry, adsorption via either dipping the electrode in the porphyrin solution or dropping the porphyrin solution onto the electrode (i.e., dip-dry and drop-dry methods, respectively), spin-coating, incorporation into other polymers and self-assembled monolayers (SAMs) have been some of the modification methods employed previously.1 Electrochemistry remains one of the more attractive techniques for modifying electrode surfaces as it results in more stable and electrocatalytically-active surfaces compared with most other methods.6 For example, Ramírez et al. compared four methods of modifying a glassy carbon (GC) electrode with Fe(III)-tetrakis(p-tetraaminophenyl)porphyrin. They concluded that the electrode prepared via electropolymerization performed superiorly to those modified using either a drop-dry or modified dip-dry method, and comparably to the electrode prepared by first covalently linking 4-aminopyridine to the electrode surface followed by their modified dip-dry method.6
While some porphyrins can electropolymerize quite readily, others unfortunately do not lend themselves to this approach for a variety of reasons. For example, iron (III) protoporphyrin (FePP) is known to coat electrode surfaces through an oxidative polymerization mechanism,5,7–9 however replacing the iron center with other metals impacts the polymerization process.5,7,10,11 Vago et al. compared the electropolymerization of protoporphyrins (PP) containing Fe(III), Ni(II), Cu(II), Co(II) and Zn(II) using electrochemical quartz crystal microgravimetric analysis.11 They concluded that the different metalloprotoporphyrins each followed a different film growth pattern despite each possessing an identical porphyrin ring structure. Our group has specifically tried to modify Pt electrode surfaces with non-metallated PP, FePP, Zn(II)PP, and Rh(II)PP, but only FePP readily polymerized into an electrically-conductive layer. The reason for this is currently unknown. This limitation in surface modification greatly restricts the exploration of certain metalloporphyrins for various electrocatalytic reactions as either an alternative surface modification procedure or a complicated synthesis is required to obtain other metalloporphyrin-modified surfaces.
However, if a metalloporphyrin (e.g., FePP) could be readily electropolymerized onto an electrode surface and then undergo a simple metal substitution reaction, perhaps new research avenues into applying metalloporphyrins for electrocatalytic reactions can be realized. There has been considerable work on substituting metals into solvated porphyrins,12–14 but there has been no detailed work on performing this with surface-bound porphyrins. Macor and Spiro briefly commented that soaking a ZnPP-coated electrode in H2SO4 followed by contact with MnCl2 in refluxing DMF yielded a (MnPP)Cl film, however no experimental details or results were given.7 The work presented here set out to explore the direct metal substitution of electropolymerized FePP as a new, simpler method for developing electrocatalysts. It should be noted that the purpose of this study was primarily to explore the feasibility of this method and not its optimization as variables such as the metal salt compound (including anion) and concentration, ionic strength, solvent and soaking duration would need to be evaluated independently in order to optimize the method for practical applications.
Experimental
Electrode modification
Scheme 1 illustrates the general process used in order to obtain a PP layer with a metal center other than Fe. The FePP was initially electropolymerized onto a polished GC macroelectrode (d = 3.0 mm) using cyclic voltammetry as described in detail previously and in the Supplementary Material.15,16 The FePP-modified electrode was then rinsed with distilled water and cycled in a phosphate buffer saline (PBS, pH 7.4) until a stable i-E curve was obtained (at least 10 cycles) signifying that the electrode possessed a stable electrochemically-active porphyrin layer. The electrode was then placed in an aqueous metal-salt solution, consisting of 1 mg/mL of the salt dissolved in 0.1 M NaCl, and allowed to soak overnight. The solvent was limited to aqueous solutions for simplicity, however any solvent that would dissolve a metal-based salt and not the polymerized porphyrin would presumably be possible. Rh(II), Co(II), Mn(II), Pt(II), Pd(II), and Ru(III) were explored as possible metal substitution candidates based on their availability and their attractiveness for various electrocatalytic reactions. The specific reagents used are listed in the Supplementary Material.16 Following soaking, the electrodes were rinsed with DI water and ultra-sonicated in DI water for 10 min to remove any non-incorporated salt species.
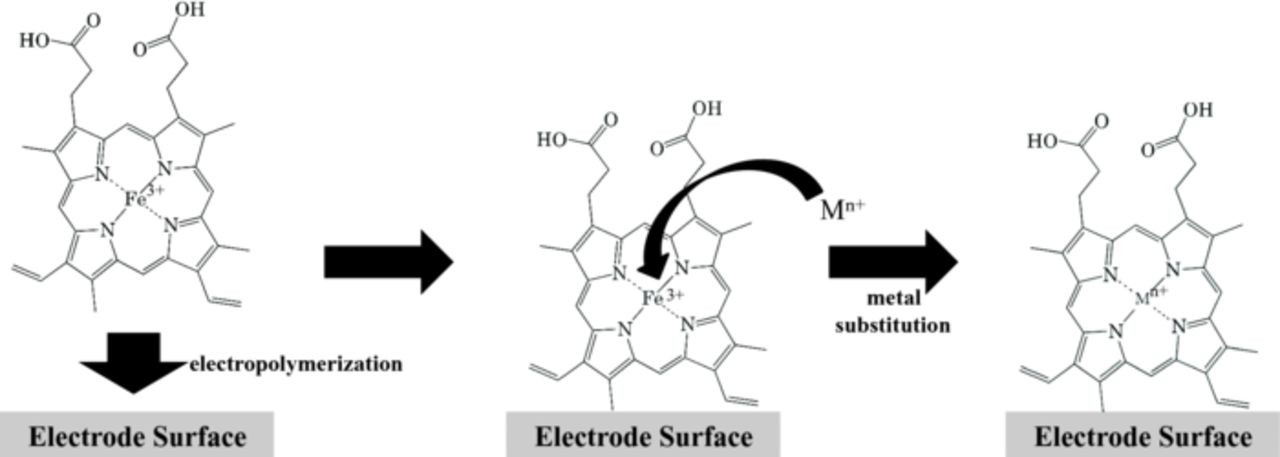
Scheme 1. Overall electrode modification process involving the electrodeposition of FePP followed by soaking in an aqueous metal salt solution, which results in the incorporation of the new metal ion into the surface-bound porphyrin.
Electrode characterization
The porphyrin-modified electrodes were characterized using both X-ray photoelectron spectroscopy (XPS) and electrochemical methods. All electrochemical measurements were performed at room temperature using a single-compartment glass cell and either a CH Instruments model 830B or 842C Series Electrochemical Analyzer (CH Instruments, Austin, TX). A Ag/AgCl (3 M NaCl) reference electrode was used. Nitric oxide and carbon monoxide gas solutions were prepared as described previously.15
XPS measurements were performed by Vince Bojan at the Penn State University Materials Characterization Laboratory (PSUMCL) using a Kratos Ultra XPS with an Al Kα X-ray source. Instrument specifications can be found at: http://www.mri.psu.edu/facilities/mcl/techniques/XPSinstrument.asp.
Results and Discussion
Figure 1 compares the X-ray photoelectron spectrum of an FePP-modified GC electrode initially after electropolymerization and that obtained after soaking it in a Rh(II) acetate solution. In order to show the spectral changes due to the individual metal ion, only the range of 200–750 eV are shown. Overall, both spectra are quite similar, in and out of the displayed range, exhibiting peaks characteristic of the porphyrin macrocycle. This indicates that the structure of the surface-bound porphyrin molecule is unaltered by the exchange of metal centers. The two main differences in the spectra are highlighted by the dotted lines. The bottom spectrum (FePP) exhibits two peaks at ca. 710 and 724 eV corresponding to the Fe 2p3/2 and 2p1/2 electrons, respectively. These two peaks are not evident in the RhPP spectrum, however this spectrum shows the emergence of peaks at ca. 310 and 315 eV that were not present in the FePP spectrum. These are attributed to the Rh 3d5/2 and 3d3/2 electrons, respectively. These spectral differences confirm the exchange of the Fe(III) center with Rh(II). It is important to note that all metals investigated showed similar behavior with diminished Fe(III) peaks coinciding with the emergence of peaks associated with the respective metal. The spectra for the other five metals can be found in the Supplementary Material in Figures S1-S5.16 While all electrodes exhibited peaks associated with their new metal center, a complete disappearance of the Fe(III) peaks was not always observed. This is likely a function of the metal salt solution composition (e.g., respective anion for the metal salt, concentration, etc.) and/or soaking time. For example, Rh(II) was the only salt with an acetate anion while the other salts used chloride. The acetate anion may be better at extracting the Fe(III) ion than chloride. As mentioned above, optimizing these conditions would require further research before applying this method to practical applications.
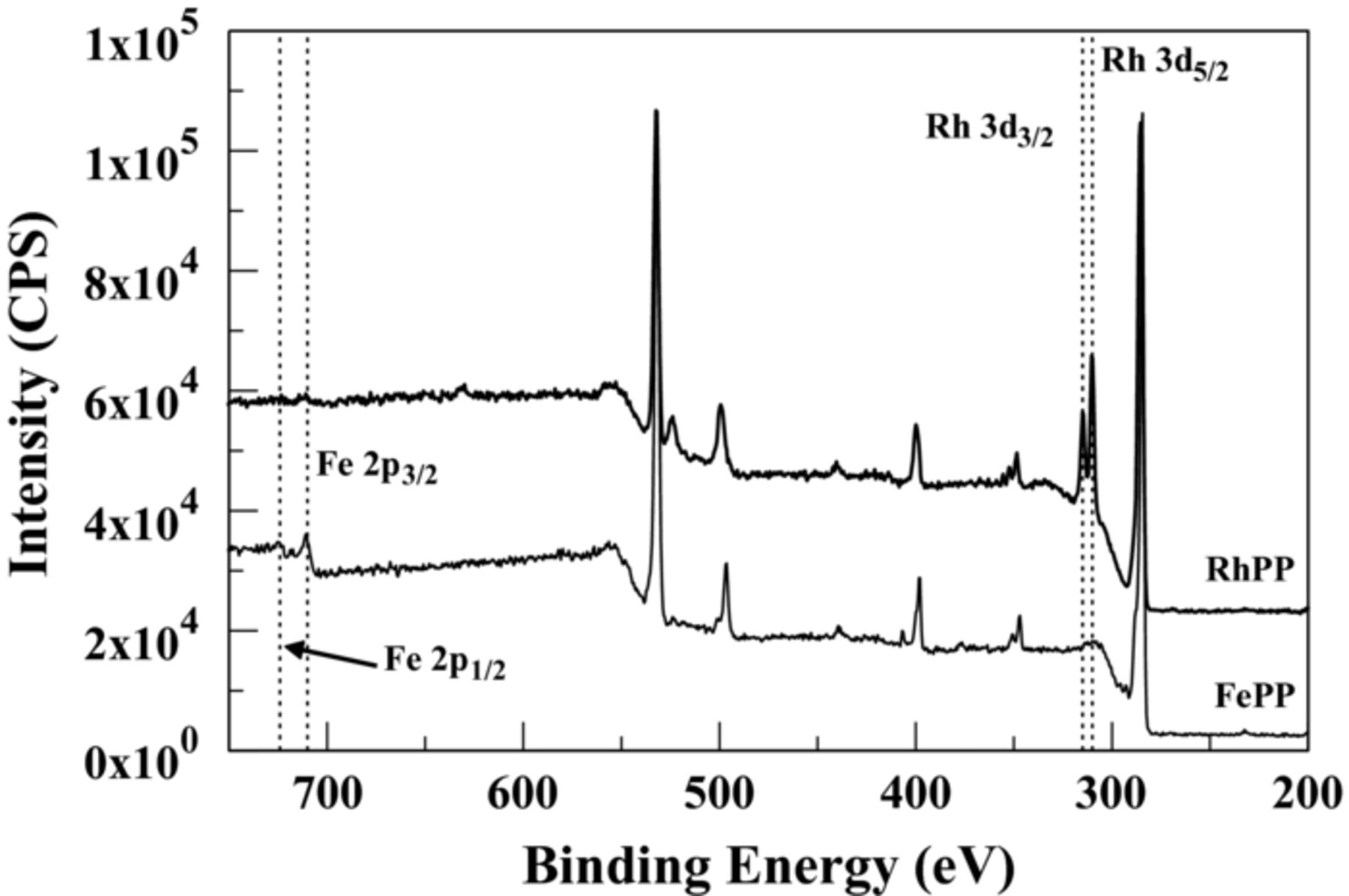
Figure 1. X-ray photoelectron spectra of a PP-modified electrode initially after FePP electrodeposition and another after soaking in a Rh(II) acetate solution.
The driving force behind this metal substitution is likely the equilibrium between the two metals. The metal ion in solution, which is much more abundant than the Fe(III) in the surface-bound porphyrin, drives the substitution. It is interesting to note however, that while this would be expected in solution, the metal ion is apparently able to penetrate the polymerized porphyrin layer to remove the Fe(III). The effect of the metal ion size on the reaction is currently uncertain since the metals used in this study all possessed similar sizes. It is likely that most transition metals (within a normal size range) would be capable of being incorporated into the polymerized porphyrin layer.
In addition to XPS analysis, the electrochemical response of these electrodes was monitored as a function of metal center. Figure 2 shows background-subtracted linear sweep i-E curves for the oxidation of 20 μM nitric oxide (NO) in 0.01 M PBS at 100 mV/s. It is clear that the metal center has a profound effect on the oxidation reaction. Both a clean GC surface as well as the FePP-modified GC exhibit a small oxidation peak, located between 0.82–0.90 V vs. Ag/AgCl, which gradually increases to a maximum current of ca. 2 μA. The drawn-out nature of this peak suggests quasi-reversible behavior. However, Mn(II)- and Rh(II)-substituted PP-coated electrodes exhibit sharp oxidation peaks approximately 5.5x higher in current (ca. 11 μA) located at a slightly lower potential (ca. 0.80 V) compared to the clean or FePP-modified GC electrodes. The voltammetric peak shape on these electrodes suggests a much faster oxidation compared with that on the clean or FePP-modified GC surfaces. The dramatic difference between the FePP and both the RhPP and MnPP electrodes further shows that the metal centers are inserted into the porphyrin ring and are in electronic communication, as both catalyze the NO oxidation reaction.
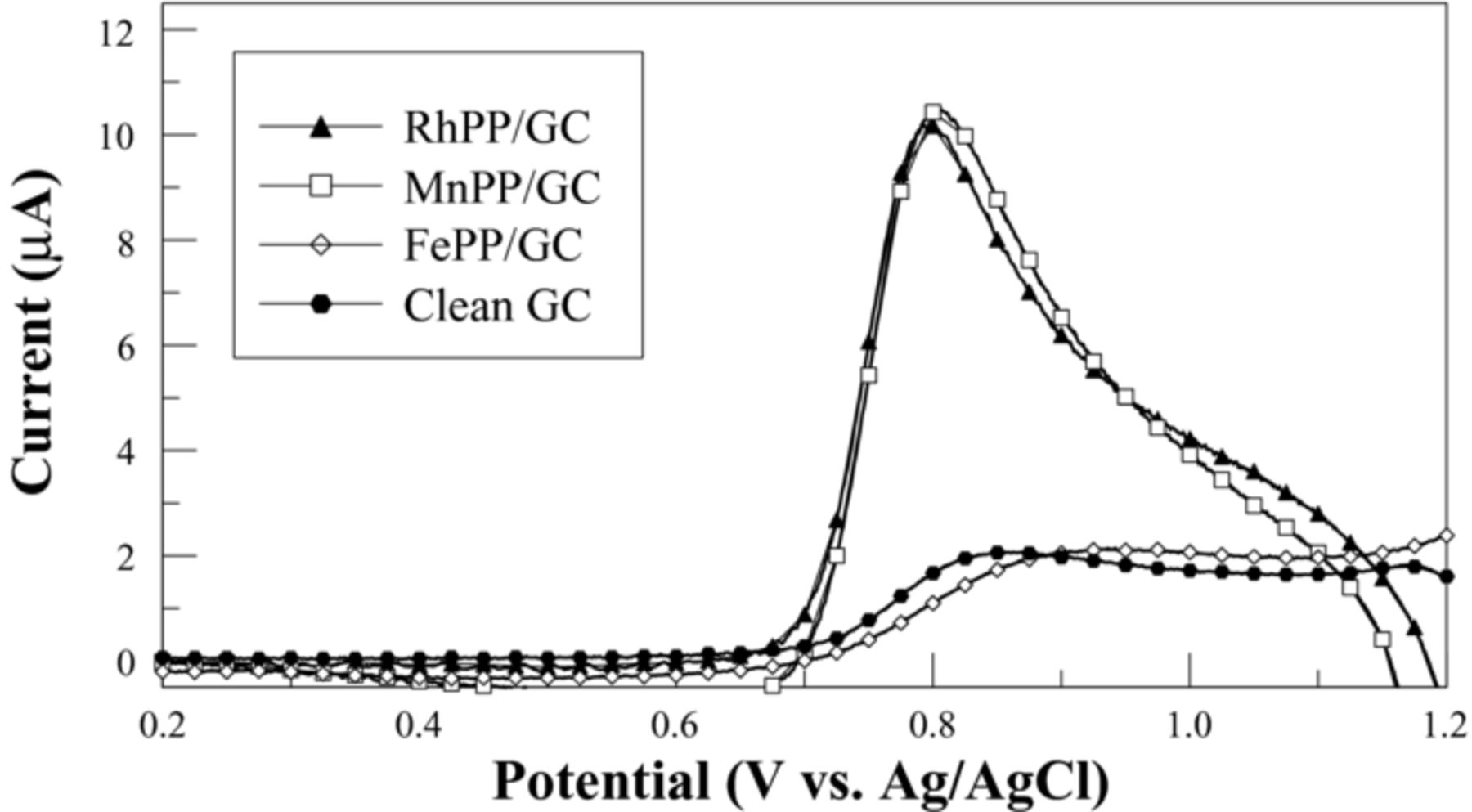
Figure 2. Linear sweep voltammograms for the oxidation of 20 μM nitric oxide at clean GC and different metallized PP-modified GC electrodes in 0.01 M PBS (pH 7.4). Scan rate = 0.1 V/s.
This novel simple approach of substituting metal ions into an easily fabricated porphyrin-modified electrode has the potential to open doors for various electrocatalytic applications previously not considered due to the difficult nature of modifying electrode surfaces with various metal-substituted porphyrin molecules. For example, our group is currently interested in possibly using this approach to develop an electrode that can selectively oxidize NO in the presence of hydrogen sulfide (H2S) and carbon monoxide (CO); two gases, in addition to NO, produced endogenously in the body that interfere with in vivo NO detection. In order to fully understand its roles, one must be able to detect it in the presence of the other two gas molecules.
In order to explore the possibility that this metal-substitution procedure may be able to help develop catalytic materials for the detection of NO, the amperometric response of the RhPP electrode toward each of the three gases was determined at an applied potential of 0.8 V vs. Ag/AgCl. Figure S6, in the Supplementary Material, illustrates that the RhPP electrode showed a higher sensitivity toward NO than H2S (by ca. 7x), as well as no activity toward CO.16 The selectivity for NO over H2S is by no means high enough for practical applications, but further shows the change in electrochemical properties of the metal-substituted surface. Perhaps further changes to the Rh(II) center or the use of metals not explored in this study will help to progress toward developing materials that can selectively oxidize this gas of interest as well as open the door to exploring new applications.
In conclusion, a new simple electrode modification method has been introduced, which involves the electropolymerization of an electrically conducting FePP layer followed by a direct metal-exchange reaction via soaking the electrode in an aqueous metal salt solution. After soaking, the electrodes clearly show the incorporation of the new metal ion into the intact surface-bound porphyrin structure and exhibit altered electrochemical properties. This may have important implications in future electrocatalytic research, as metalloporphyrins that previously were difficult to adhere to an electrode surface, may now be able to be explored.
Acknowledgments
The authors acknowledge an award from Research Corporation for Science Advancement, Penn State Erie, The Behrend College, as well as an award from the National Science Foundation (CMI; grant CHE-1305660) for the financial support for this work. The assistance of Vince Bojan at the Penn State University Materials Characterization Laboratory with XPS measurements is greatly appreciated.