
Abstract
Platinum is ubiquitous in electrochemical catalysis owing to its ability to accelerate redox reactions involving surface-bound hydrogen and oxygen. Accordingly, Pt is used as a calibration standard and activity benchmark against which novel electrocatalysts are compared. These measurements are often executed in unpurified, reagent grade electrolytes where Pt is also susceptible to deactivation by several routes. This constitutes a challenge where the ease of making measurements with Pt-based catalysts must be balanced against the difficulty of obtaining accurate and consistent results. We report herein a synthetic procedure for unsupported Pt nanoparticles that uses readily available reagents and laboratory apparatus, with the goal of making high-quality control experiments in electrocatalysis as accessible as possible. We also identified conditions under which these particles deactivate in unpurified aqueous acid and base and differentiated between mechanisms involving catalyst poisoning, which dominates at more negative applied potentials, and particle growth, which dominates at positive potentials where Pt-oxide species are produced. Finally, we demonstrated that unsupported Pt nanoparticle films can be used to good effect for reference electrode calibration and benchmarking of hydrogen evolution/oxidation electrocatalysts, even in unpurified electrolytes, provided steps are taken to minimize the impact of deactivation.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: permissions@ioppublishing.org.
Platinum is one of the most widely studied transition metals for applications in thermal and electrochemical catalysis. 1–4 The many thermocatalytic applications of Pt include automotive exhaust treatment 5–8 as well as the production of nitric acid, 9,10 silicones, 11,12 and petrochemical feedstocks. 13,14 Pt is also used ubiquitously in analytical and industrial electrochemistry, and many of these applications revolve around its high catalytic activity toward reactions involving hydrogen. 15–24 For example, carbon-supported Pt catalysts (Pt/C) are used for the hydrogen evolution reaction (HER) in proton-exchange membrane electrolyzers and the hydrogen oxidation reaction (HOR) in low and intermediate temperature fuel cells. 25 Despite its very high catalytic activity, Pt is susceptible to deactivation under electrochemical conditions; thus, the dynamics and mechanisms of Pt deactivation have also been studied extensively. 26–31 The most prevalent deactivation processes generally fall in one of several categories: catalyst poisoning, particle growth, particle detachment, and dissolution. Ultimately, the dynamics of these deactivation processes dictate the practical lifetime of Pt catalysts. Thus, a major research focus on Pt has been on evaluating and minimizing the impact of catalyst deactivation in analytical and practical applications. 32–35
Owning to its very high catalytic activity for hydrogen evolution/oxidation electrocatalysis, Pt is used as a benchmark against which novel HER and HOR catalysts are measured. It is also used to calibrate voltage measurements in aqueous electrocatalysis via the determination of the reversible hydrogen electrode (RHE) potential in the electrolyte of interest. The propensity of Pt electrocatalysts to deactivate under reaction conditions makes these key measurements considerably more challenging. 36–38 Whereas prior studies have generally addressed this challenge by maintaining scrupulously clean experimental conditions, 30,32,33 routine measurements of novel electrocatalysts (and associated comparisons to Pt) are often carried out in unpurified, reagent-grade electrolytes. This motivates the question of whether control experiments using Pt electrodes can be properly executed under these conditions. To address this question, we describe herein a straightforward preparation of unsupported Pt nanoparticles (NPs) as well as their use as a laboratory standard for hydrogen evolution/oxidation studies.
We have focused on experimental conditions that are not scrupulously clean but are nevertheless representative of the methods and materials used for routine measurements in applied electrocatalysis laboratories. Accordingly, we also studied the deactivation of Pt NPs, as reflected in the progressive loss of electrochemically accessible surface area (ECSA). When cycled continuously between oxidizing and reducing conditions in unpurified aqueous acidic and alkaline electrolytes, films of Pt NPs lost 25%–40% of their active surface area over tens of minutes. We also deployed identical location transmission electron microscopy (IL-TEM) 39,40 to differentiate deactivation processes involving changes in catalyst morphology from those that do not. At positive potential limits below 1 V vs RHE, we observed minimal changes in particle size/shape, implying the dominant mode of deactivation is catalyst poisoning rather than particle dissolution or growth. These observations were found to be consistent across acidic and alkaline conditions, but the relative rate of deactivation were modestly slower in base than in acid. By contrast, when the positive potential limit was increased to 1.5 V vs RHE, the Pt NPs were found to grow in size, leading to irreversible loss of surface area. Together, these studies are instructive for the development of analytical protocols where Pt NPs can be used to good effect for analysis and benchmarking novel catalysts for hydrogen evolution/oxidation and other electrochemical reactions of interest.
Experimental
The synthesis method for Pt nanoparticles was adapted from several prior literature reports. 41–43 A representative procedure is as follows. Aqueous stock solutions of 12 mM H2PtCl6 (99.5% Alfa Aesar) precursor and 100 mM sodium polyacrylate (average molecular weight ∼1200, 45 wt% in water, 99.5%, Alfa Aesar), respectively, were prepared by dissolution with ultrasonic agitation for at least 5 min. These solutions were prepared in bulk quantities and used for several synthetic runs. A fresh solution of 100 mM L-ascorbic acid reducing agent was prepared immediately before the synthesis by adding 0.0195 g of anhydrous L-C6H8O6 (99.5%, Alfa Aesar) to 1 ml deionized water and mixed by sonication.
In a 20 ml scintiallation vial, 0.833 g of 12 mM H2PtCl6 and 0.1 g of 100 mM sodium polyacrylate were added to 1 ml ascorbic acid solution. Deionized (DI) water (≥18 MΩ cm resistivity, Millipore) was then added to increase the solution volume to 10 ml. The final concentrations of the reagents in the precursor solution were 1 mM each of H2PtCl6 and sodium polyacrylate and 10 mM ascorbic acid. The precursor mixture was initially transparent with a slight yellow color.
The vial was placed in a preheated bath of silicone oil (Fluke 5010, Type 200.05) at 90 °C. The contents were stirred at 600 rpm for 60 min using a magnetic stir bar. The temperature was monitored using an alcohol thermometer to ensure that the variation was within ±3 °C. Within 15 min, the solution became black and opaque. The reaction was allowed to continue for 60 min, and successive additions of DI water were made every 20 min to maintain a constant total volume of solution. The vial was then removed from the bath and allowed to cool at room temperature. After cooling, 0.4 g NaOH (98%, Alfa Aesar) was added to the suspension to alkalize the solution to pH∼14. The vial was then capped and left to stand at room temperature for 14 days, during which the black suspended solids settled to the bottom of the vial. Note that this settling process was sometimes found to proceed over a shorter timescale, but a 14-day incubation period was always found to be sufficient for particles to settle.
The clear solution above the settled layer was decanted using a pipette and the particles were extracted by centrifugation at 6000 rpm for 15 min. The supernatant was again decanted and the particles were re-suspended and centrifuged using 1 M aqueous NaOH solution a total of 3 times. Finally, the particles were suspended in 1.5 ml of deoinized water; this suspension was found to remain stable for several days. The colloidal solution was either used as-is or further diluted with pure deionized water as appropriate prior to subsequent analysis.
ICP-OES was used to measure the yield and mass concentration of Pt NPs. After the synthesis, the Pt nanoparticles were re-suspended in DI water via ultrasonication for 10 min to ensure a homogeneous particle dispersion. From the 1.5 ml total volume, 1 ml was removed using a repeating pipettor (Thermo Scientific F1 ClipTip) and centrifuged as described above to collect the solid particles. The particles were then dissolved in a mixture of three parts concentrated HCl(aq) to one part HNO3(aq) (aqua regia). The total mass of this mixture was noted to calculate the acid fraction and the metal fractions. Dissolution was allowed to proceed in a fume hood at room temperature over a period of 4-5 days. The sample was then diluted with pure deionized water to target final concentrations of 2, 5, and 10 ppm for ICP-OES (Agilent 5100 VDV) analysis to determine the platinum content. From these measurements the isolated yield was found to be 55–70% relative to the total Pt content of the synthesis mixture.
The final, purified Pt NPs were characterized with respect to composition and morphology using Fourier-transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), and transmission electron microscopy (TEM). FTIR spectra were collected on Bruker VERTEX-70LS instrument using an attenuated total reflection (ATR) accessory at a resolution of 4 cm−1. Deionized water was used as a blank solution, with aqueous Pt NP suspension as the experimental sample. A Bruker D8 system with Cu Kα radiation (λ = 1.54 Å) was used for XRD analysis. Diffraction patterns were collected from 30° to 90° 2θ with 0.02° steps. The data collection time per step was optimized empirically to yield diffraction peaks that were well resolved from the noise floor.
Transmission electron microscopy (TEM) imaging was executed using a Hitachi H-9500 environmental transmission electron microscope at an accelerating voltage of 300 kV. For IL-TEM analysis, the nanoparticles were deposited on center-marked 400 mesh Au TEM grids (Ted Pella). A perforation was made manually near the edge of the grid using sharp tweezers (Ted Pella). Approximately 5 μL of the Pt nanoparticle suspension was dropcast on the grid and dried under an infrared heat lamp. The dried grid was then immersed in water to remove soluble impurities and poorly adhered NPs. Finally, a hooked gold wire (0.5 mm, 99.999%, Fisher Scientific) was inserted through the perforation to make electrical contact for electrochemical measurements.
A Gamry Reference 600+ Potentiostat was used to characterize the electrochemical properties of the Pt nanoparticles. For non-TEM based studies, fixed volumes in the range from 1–10 μL of Pt NP suspension with a known Pt content (based on ICP-OES results) was dropcast onto a 3 mm diameter (0.071 cm2 surface area) glassy carbon electrode and dried under an infrared lamp. The counter electrode was a Pt wire in acidic electrolyte and Ni foil in alkaline conditions; the reference electrode was Ag/AgCl (1 M KCl). The potential from the Ag/AgCl reference electrode was later converted to the reversible hydrogen electrode (RHE) potential by measuring the open circuit potential value of a clean Pt NP film in H2-saturated electrolyte at atmospheric pressure. Electrolytes used for electrochemical analysis were 0.5 M H2SO4 (Certified ACS Plus grade, Fisher Scientific) and 0.5 M KOH (ACS Reagent grade, Sigma Aldrich). Note that the specifications of these reagents allow them to contain transition metal and anionic (e.g. chloride, sulfate, phosphate) impurities in the ppm to tens of ppm range. The electrolytes were used as obtained from the supplier and did not undergo additional purification steps.
To qualitatively assess surface cleanliness and quantitatively estimate the electrochemically active surface area (ECSA), electrolytes were purged with N2 or Ar gas (Zero grade, Matheson Gas). Cyclic voltammograms were then collected at a fixed negative potential limit of 0 V vs RHE while the positive potential limit was varied from 0.5 V to 1.5 V vs RHE at a scan rate of 100 mV s−1.
RHE potential calibration and hydrogen evolution/oxidation catalysis measurements were performed on a rotating disk electrode (RDE) in a sealed teflon cell (Pine Research). For RDE experiments, a known volume of Pt suspension was dropcast on a 5 mm diameter glassy carbon electrode for measurements in acid and on 5 mm Au for analysis in alkaline conditions. The Au substrate in alkaline electrolyte was found to yield improved adhesion, whereas Pt NPs were found to re-suspend when deposited onto glassy carbon. The electrolytes were purged with H2 and the potential was scanned in the range of −0.15 V to +0.1 V vs RHE in acid and −0.2 V to +0.2 V vs RHE in base at 10 mV s−1. Commercial 10 wt% Pt/C (Alfa Aesar) and a polycrystalline Pt disk electrode (Pine Research) were used as controls.
Results and Discussion
Catalyst synthesis and purification
Our synthesis of platinum nanoparticles relies on the reduction of H2PtCl6 using ascorbic acid as the reducing agent and sodium polyacrylate as a capping agent. The synthesis procedure is adopted from prior literature reports, where Pt(IV) salts have been shown to undergo reduction to metallic Pt with two equivalents of ascorbic acid. 44–49 Sodium polyacrylate polymer as a capping agent offers narrow particle size distribution and has been reported previously for Pt synthesis. 50–55 Alkalizing the reaction mixture with sodium hydroxide destabilizes the colloid and facilitates removal of the polyacrylate from the NPs. 56–58 The main advantage of this approach (over the use of commercial Pt/C catalyst, for example) is that it offers a route to synthesize catalytically active Pt NPs that can be deposited on any substrate and analyzed without the confounding effects of supports or polymer binders. Our synthetic approach also makes use of low-cost reagents (other than the Pt precursor itself) and equipment that is readily available in many wet chemistry laboratories.
We executed TEM, XRD, and FTIR measurements to verify the composition and crystallinity of the Pt NPs before and after removal of the polyacrylate capping agent. Figures 1a, 1b show representative TEM images of capped and decapped (based-treated) Pt NPs. The particles in panel a clearly remain encapsulated in a matrix of polymer several nm in diameter. In some cases the polymer encapsulates a single Pt particle and in others several distinct particles are visible, which suggests that the polyacrylate arrests Pt NP growth by encapsulating individual particles, and these core–shell assemblies further aggregate in solution or upon deposition on the TEM grid. By contrast, particles that have been purified with alkaline solution aggregate on the TEM grid. Manual analysis of the particle size distribution (see Supporting Information) shows a normal distribution on the basis of particle number with a mean diameter of 3.1 ± 0.05 nm and circularity index of 0.9. The mass-averaged particle diameter, which is the relevant metric for estimating mass-specific surface area, is 3.4 nm. D-spacings extracted from high resolution TEM imaging (insets of Figs. 1a, 1b) further confirm the particles as face-centered cubic (fcc) Pt.
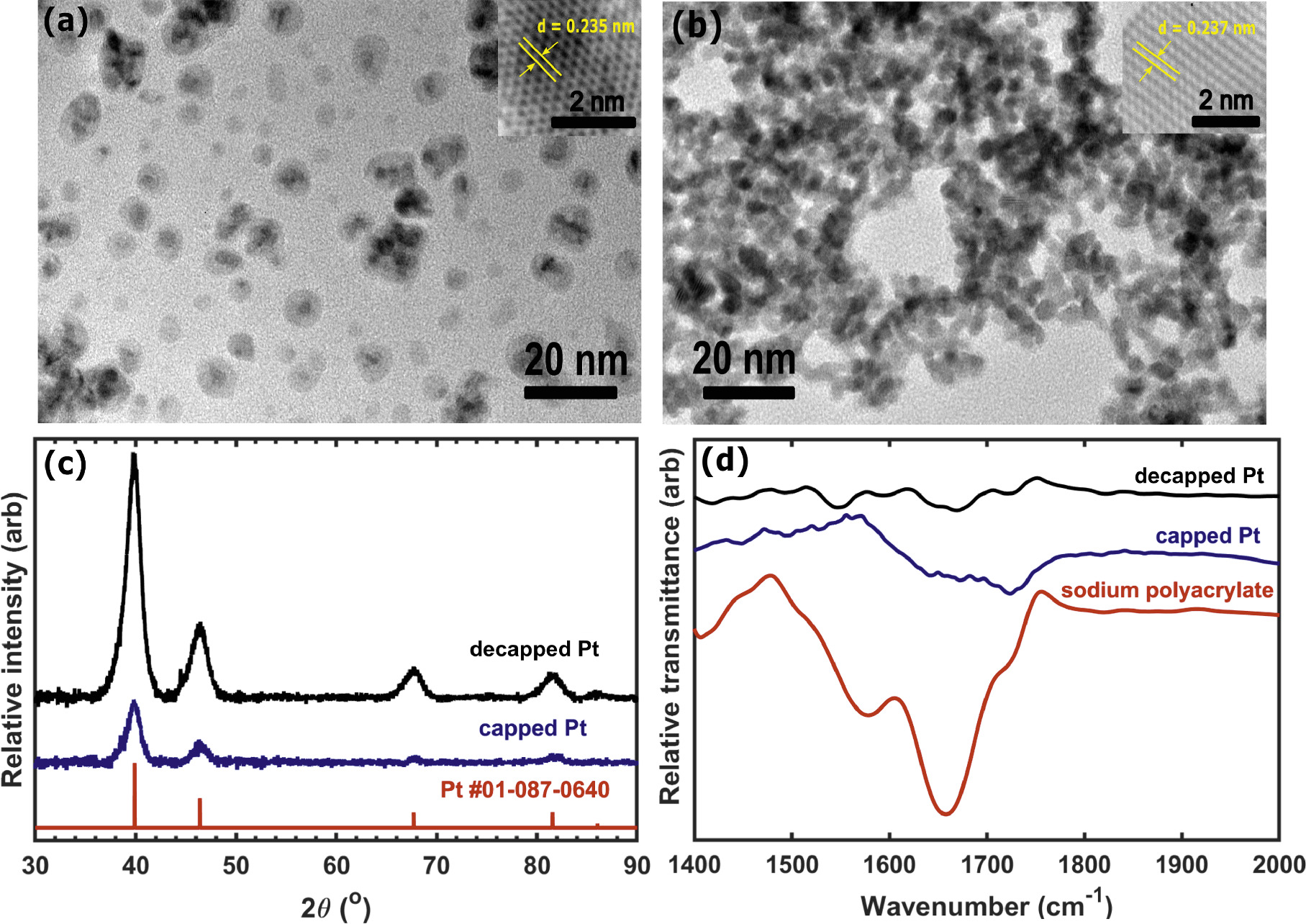
Figure 1. Compiled compositional analysis of Pt nanoparticles via (a), (b) TEM, (c) XRD, and (d) ATR-FTIR. The standard XRD pattern of Pt was obtained from the ICDD database via the numerical index indicated in panel (c).
Download figure:
Standard image High-resolution imageFigures 1c, 1d depict representative XRD and ATR-FTIR data for capped and decapped Pt NPs. XRD results further confirm fcc Pt as the only crystalline component in the reaction mixture before and after purification. ATR-FTIR data from capped Pt exhibit absorbance features at 1640 cm−1 and 1723 cm−1, which we attribute to carboxylic and/or alcoholic peaks that are blue-shifted relative to the aqueous sodium polyacrylate control. This shift has been previously attributed to the interaction between the particle and the polymer. 59–61 The decapped Pt NPs also exhibited absorbance features in this range, but at a considerably lower intensity and without clear evidence for a blueshift relative to polyacrylate in aqueous solution. This further suggests that the base treatment substantially removes polyacrylate from the NPs, but we cannot rule out the possibility that some polymer remains in solution or weakly bound to Pt.
Quantifying initial ECSA
To further characterize the decapped NPs, we used surface voltammetry to assess the accessibility of Pt surface sites via hydrogen underpotential deposition (HUPD) in aqueous H2SO4. These measurements yield one estimate of the electrochemically accessible surface area (ECSA) via integration of the charge associated with hydrogen adsorption–desorption. 18 Figure 2 shows a representative surface voltammogram for a film containing decapped Pt nanoparticles in 0.5 M H2SO4 at 100 mV s−1 and a loading of 130 μg cm−2.

Figure 2. Representative cyclic voltammogram of unsupported Pt NP in 0.5 M H2SO4 under Ar atmosphere at 100 mV s−1 measured with a mass loading of 130 μg/cm2. The shaded region represents the area used for ECSA calculation, lower portion is for proton adsorption while upper portion is for proton desorption.
Download figure:
Standard image High-resolution imageUsing the generally accepted HUPD surface charge density of 210 μC cm−2, the mass-specific ECSA for decapped Pt NPs was found to be 37.13 ± 0.69 m2/g (Error bound is 95% CI, for 29 samples tested) in 0.5 M H2SO4. This is less than half the value of 82 m2/g predicted directly from the TEM-derived particle size as 6/ρ d, where ρ is the bulk density of Pt (21.5 g cm−3) and d is the mass-average particle diameter (3.4 nm). To address this discrepancy, we carried out a series of HUPD measurements as a function of Pt mass loading on a glassy carbon electrode, with the results shown in Fig. 3. These data show the apparent mass-specific ECSA increased with reduced loading to a lower bound in the μg/cm2 range. ECSA values calculated at these very low mass loadings, which correspond to one or a few layers of ∼3 nm particles, were consistent with the value expected from particle geometry alone, albeit with a commensurate increase in statistical uncertainty attributable to the difficulty of obtaining such low mass loadings with high precision via solution-phase deposition. The results of further measurements to rule out contributions from organic contamination are included in the Supplementary Data.
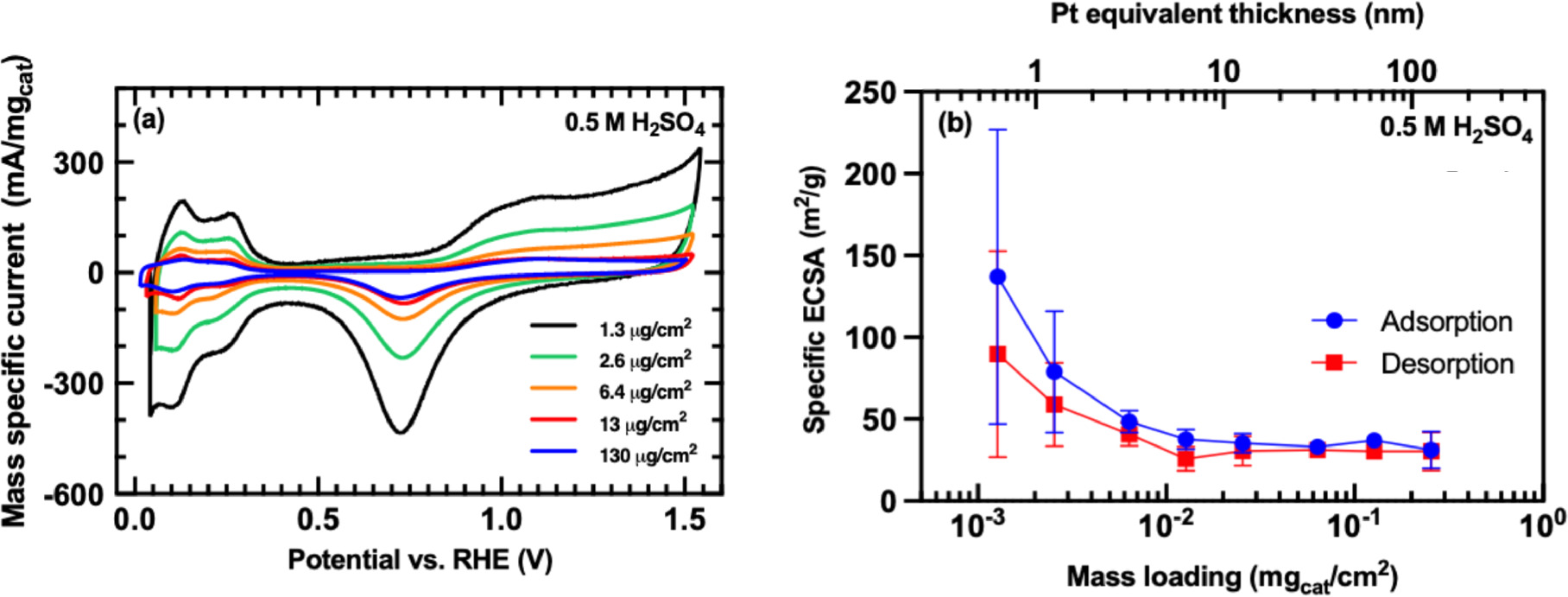
Figure 3. (a) Representative CV data for Pt NP films with a range of mass loadings in 0.5 M H2SO4. The current scale is normalized to catalyst mass to illustrate differences in the apparent mass-specific ECSA. (b) Apparent mass-specific ECSA versus mass loading extracted by HUPD integration of CV data as shown in panel (a). For reference, the upper horizontal axis illustrates mass loading in terms of the equivalent thickness of a dense Pt layer in nm. Error bounds in panel (b) are 95% CI for 3 samples tested at each mass loading.
Download figure:
Standard image High-resolution imageThese results can be interpreted as evidence that adhesion between unsupported Pt NPs is sufficiently weak that a fraction of the particles in the film spontaneously detach from the electrode upon submersion in the electrolyte (note that we used no polymer binders to adhere them to the carbon substrate), whereas films approaching a single particulate layer are more strongly bound. Another possibility is that reduced apparent ECSA for thicker Pt NP films is attributable to mass-transfer limitations, wherein the accessibility of protons to the inner layers of Pt particles is reduced as the total film thickness increases. We speculatively prefer the latter explanation on the basis that a volume occupied by spherical 3.4 nm Pt particles at a volume fraction of 50%, with the interstitial space containing 0.5 M H2SO4, will contain just 1 proton per ∼1000 surface Pt atoms, which illustrates the need for rapid proton diffusion through the dense porous film to accommodate H-adsorption on each Pt surface site. Nonetheless, further experiments were conducted with Pt loadings ≥100 μg cm−2 to comport with common practice for voltammetric studies of Pt NP catalysts.
Quantifying rates of deactivation in unpurified electrolytes
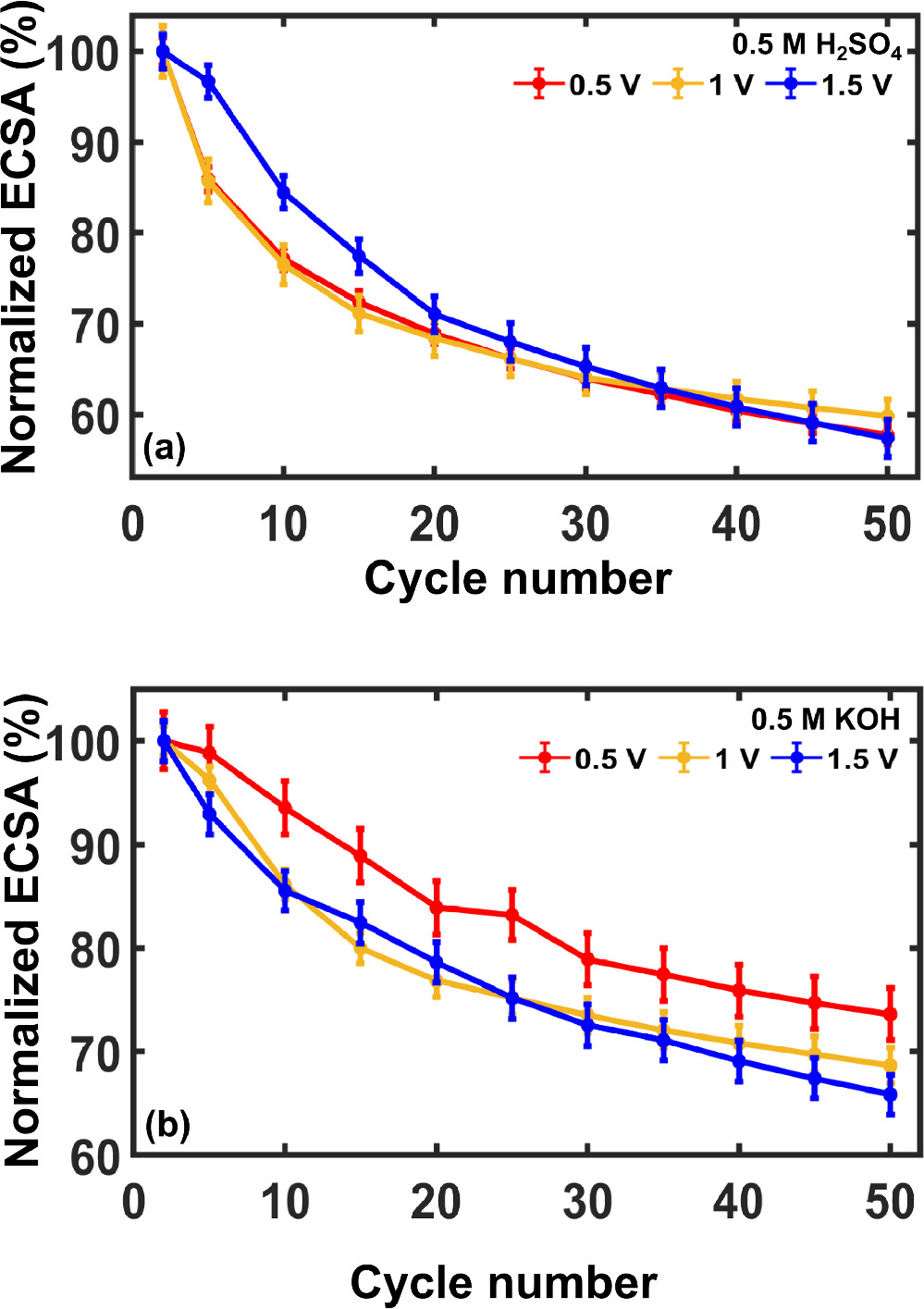
Prior literature reports have emphasized the susceptibility of Pt electrocatalysts to deactivation, which negatively impacts activity and lifetime. 62–64 We were interested in understanding the mechanism or mechanisms by which these unsupported Pt NPs degrade. To do so, we quantified ECSA decay under continuous voltammetric cycling. Figure 4 presents the results as normalized ECSA versus cycle number in acid and alkaline electrolytes. For this analysis, 0.2 mg cm−2 of Pt NPs were swept at 100 mV s−1 over the potential range from 0 V to one of three positive potential limits: 0.5, 1.0, and 1.5 V vs RHE for a total of 50 cycles. A different Pt NP film was used for each potential window and 15 such films obtained from 5 different synthesis batches were tested for the analysis.
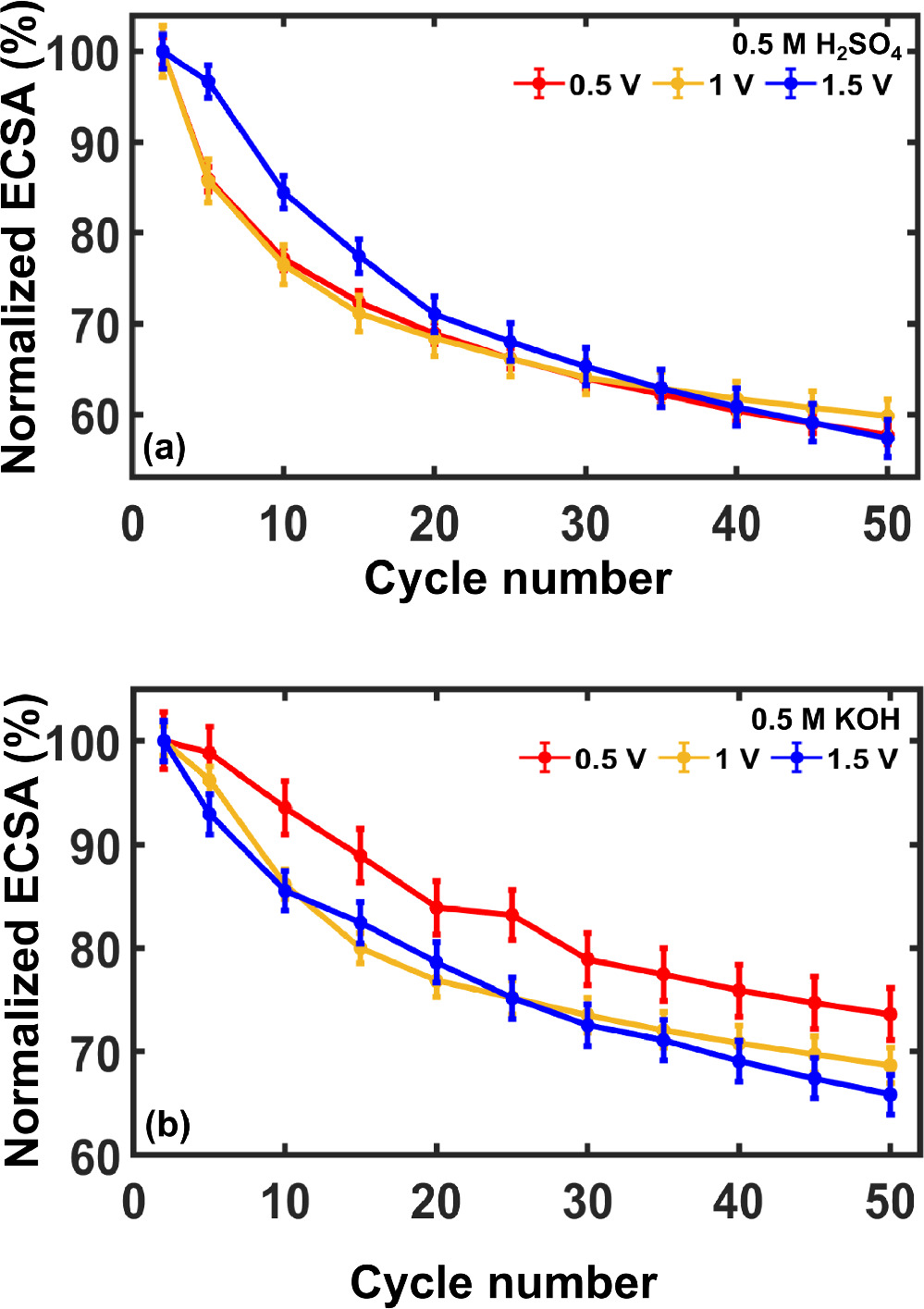
Figure 4. ECSA normalized to the initial value for unsupported Pt NPs as a function of cycle number when cycled between a fixed negative scan limit of 0 V vs RHE and the noted positive scan limits over 50 cycles in (a) 0.5 M H2SO4 and (b) 0.5 M KOH. Experimental conditions were otherwise identical to those in Fig. 2. Error bars represent 95% confidence intervals from 15 distinct films and 5 separate synthetic batches.
Download figure:
Standard image High-resolution imageError bars representing 95% confidence intervals about the mean of 15 measurements illustrate remarkable consistency in the relationship between relative ECSA and cycle number. A monotonic decrease in ECSA was observed in all cases over 50 cycles. Moreover, we observed that the relative (normalized to initial ECSA) rate of ECSA loss per cycle was nearly identical when Pt nanoparticles were scanned at 0.5 V and 1 V vs RHE, suggesting similarity in their deactivation mechanisms. When scanned to 1.5 V vs RHE, the rate of deactivation was initially slower, but accelerated after several cycles such that the total fractional loss in surface area was ∼40% in all cases.
ECSA values also decreased monotonically with cycle number in alkaline electrolyte across all positive potential limits. The relative rates of deactivation in base were also found to be similar at 1 V and 1.5 V vs RHE, whereas the less positive potential limit resulted in marginally slower ECSA loss. These observations are consistent with the ability of aqueous base to remove residual capping agent from the Pt NP surface (which negates the benefit of positive potential cycling to remove contaminants) and suggests the presence of a second deactivation mechanism that dominates at positive potentials.
We consider it significant that these Pt NPs lost 25%–40% of their initial ECSA over just 50 voltammetric cycles, which required as little as ∼10 min. This extent of ECSA loss is large enough to conclude that the Pt particles are no longer pristine, thereby calling into question their use as experimental controls or activity benchmarks. Hence, it is reasonable to adopt as a general guideline that unsupported Pt NPs of this type should be freshly deposited onto a clean electrode at relatively short time intervals (every few minutes or perhaps between each individual measurement) if they are to be used to good effect in unpurified electrolytes.
Differentiating between modes of deactivation
Several deactivation mechanisms are understood to proceed for Pt catalysts under electrochemical conditions. These include catalyst poisoning via surface adsorption or electrodeposition of impurities; oxidative dissolution, which could be chemically or electrochemically induced; particle growth resulting from Ostwald ripening or similar processes; and particle detachment from the electrode substrate. In acidic electrolyte, unsupported Pt nanoparticles are known to undergo dissolution by repeated cycling across the potential range required to generate and reduce Pt-oxides. 63–68 The dissolution proceeds primarily during the reductive scan when platinum oxide is nominally reduced back to metallic Pt; this reaction proceeds in parallel with dissolution of Pt2+ ions. Under continuous cycling, these ions may remain in the electrolyte or they may be redeposited as the electrode potential is swept further negative. This dissolution-redeposition process may then induce particle growth via electrochemically accelerated Ostwald ripening. 69 Nevertheless, it is difficult to entirely eliminate oxidative cycling in practice, as this treatment is also highly effective at removing organic and/or inorganic contaminants from the Pt surface, either through catalytic oxidation or as a direct result of the dissolution of Pt surface layers. 70
By contrast, the deactivation of Pt nanoparticles has not been as thoroughly characterized in alkaline conditions as in acid. Deposition of transition metal impurities on the electrode surface and analogous dissolution processes have each been implicated as the root causes of reduced catalytic activity. 19 The ECSA variation versus cycle number data in Fig. 4 yield puzzling results where the relative rates of deactivation (20%–40% over 50 cycles) are similar across nearly all conditions studied. These observations leave open the question of whether the dominant mechanism of ECSA loss is similar or different across all potential windows and pH conditions.
Figure 5 compiles the results of an experiment directed at differentiating between reversible catalyst poisoning and irreversible loss of surface area, wherein Pt NP films were first subjected to 50 cycles to a positive limit of 0.5 V vs RHE, followed by 50 cycles to 1.5 V vs RHE, and then a third set of 50 cycles to 0.5 V. The ECSA decay on scanning at 0.5 V vs RHE for the first 50 cycles in acid and alkaline solutions again resulted in a 20%–40% loss in ECSA. When the positive potential was shifted to 1.5 V vs RHE, ECSA values in acid fully recovered over 5–10 cycles and in some cases even exceeded the initial values. This indicates that the Pt surface was renewed after scanning at oxidizing potential, which strongly suggests catalyst poisoning dominates at the positive limit of 0.5 V vs RHE. Nonetheless, after the initial recovery at 1.5 V vs RHE, the ECSA decayed again by nearly the same amount as in the initial set of 50 cycles. Returning the potential window to a 0.5 V positive limit had little further impact on the ECSA, which continued to diminish at a reduced rate through a final set of 50 cycles. These data suggest the mechanism by which ECSA diminishes when cycling to a more positive potential limit is irreversible—dissolution, particle growth, or detachment.
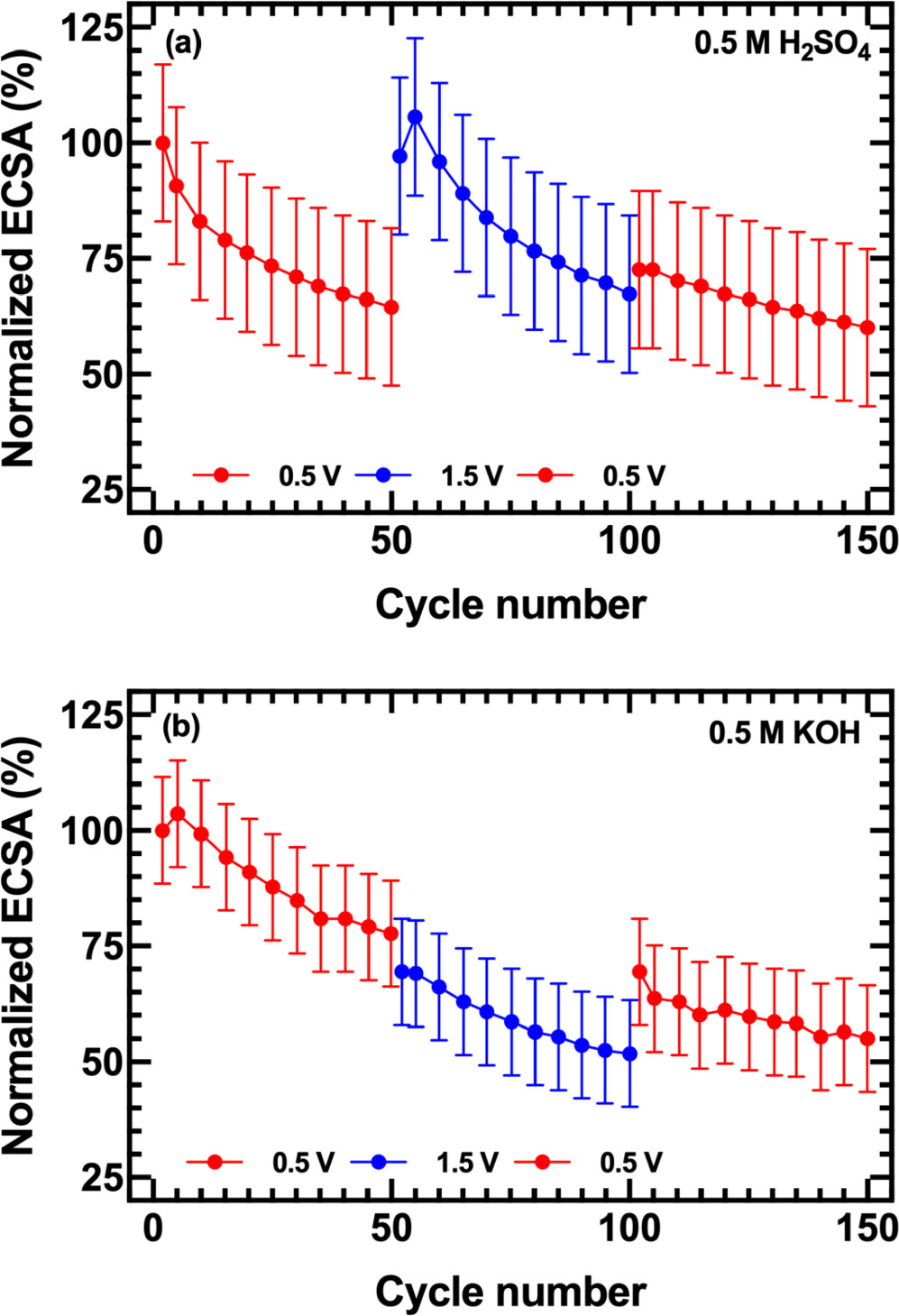
Figure 5. ECSA normalized to the initial value versus cycle number for Pt NP films scanned to a positive potential limit of 0.5 V vs RHE for 50 cycles, followed by 1.5 V for 50 cycles, and again to 0.5 V for 50 cycles in (a) 0.5 M H2SO4 and (b) 0.5 M KOH. Experimental conditions and error bounds were otherwise identical to those in Figs. 2 and 4.
Download figure:
Standard image High-resolution imageBy contrast to the results in acid, when cycled in alkaline electrolyte, the ECSA did not recover when the voltage range was modulated from 0.5 to 1.5 V and back to 0.5 V. Instead we observed a nearly continuous decrease in surface area over 150 cycles, with a systematic negative offset in apparent ECSA for data collected to 1.5 V vs RHE attributable to systematic errors resulting from the Pt surface oxide reduction feature in the voltammogram (see Supplementary Data).
Our observations agree with prior reports that electrochemical oxidation is highly effective at removing catalyst poisons from Pt in acid. 70 Many organic (e.g. hydrocarbons) and inorganic (e.g. sulfate, chloride) species from the synthesis mixture, the electrolyte, or the atmosphere may adsorb to the Pt surface and can be removed by cycling to positive potentials. 70–72 By contrast, oxidative electrochemical cleaning did not renew the catalyst surface in base. There are at least three possible explanations for this latter observation. First, the catalyst surface may become contaminated with an intractable poison over all potential windows. A second explanation is that alkaline conditions facilitate detachment of nanoparticles from the electrode surface, and this may proceed at some relatively constant rate regardless of cycling conditions. A third explanation is that the Pt NPs undergo continuous particle growth in alkaline conditions under potential cycling that again depends weakly, if at all, on the positive scan limit.
To further differentiate between loss of surface area via particle growth, dissolution, and detachment, we performed identical location transmission electron microscopy (IL-TEM) experiments on catalyst thin films before and after they were subjected to CV cycling over a range of potential windows for 50 cycles at 100 mV s−1. Figure 6 compiles the results of these experiments in acid electrolyte. At a positive limit of 0.5 V vs RHE, the TEM images before and after cycling show minimal changes in particle morphology. The overall particle size distribution also did not change before and after cycling. Similar results were observed when Pt NPs were scanned to 1 V vs RHE in acid. However, a stark difference was observed when the particles were cycled to 1.5 V vs RHE, where the TEM images clearly show particle growth. The particle size distribution data confirmed that the mean particle diameter increased by slightly more than two-fold, in rough agreement with the nearly 50% loss in ECSA for NP films on glassy carbon substrates cycled under the same conditions. A similar analysis was also performed in alkaline electrolyte, where scanning at 0.5 V and 1 V vs RHE did not result in a significant increase in particle size but cycling at 1.5 V vs RHE resulted in a ∼30% increase in mean particle diameter (see Supplementary Data), also in rough agreement with the ECSA loss observed for Pt NP films on Au.
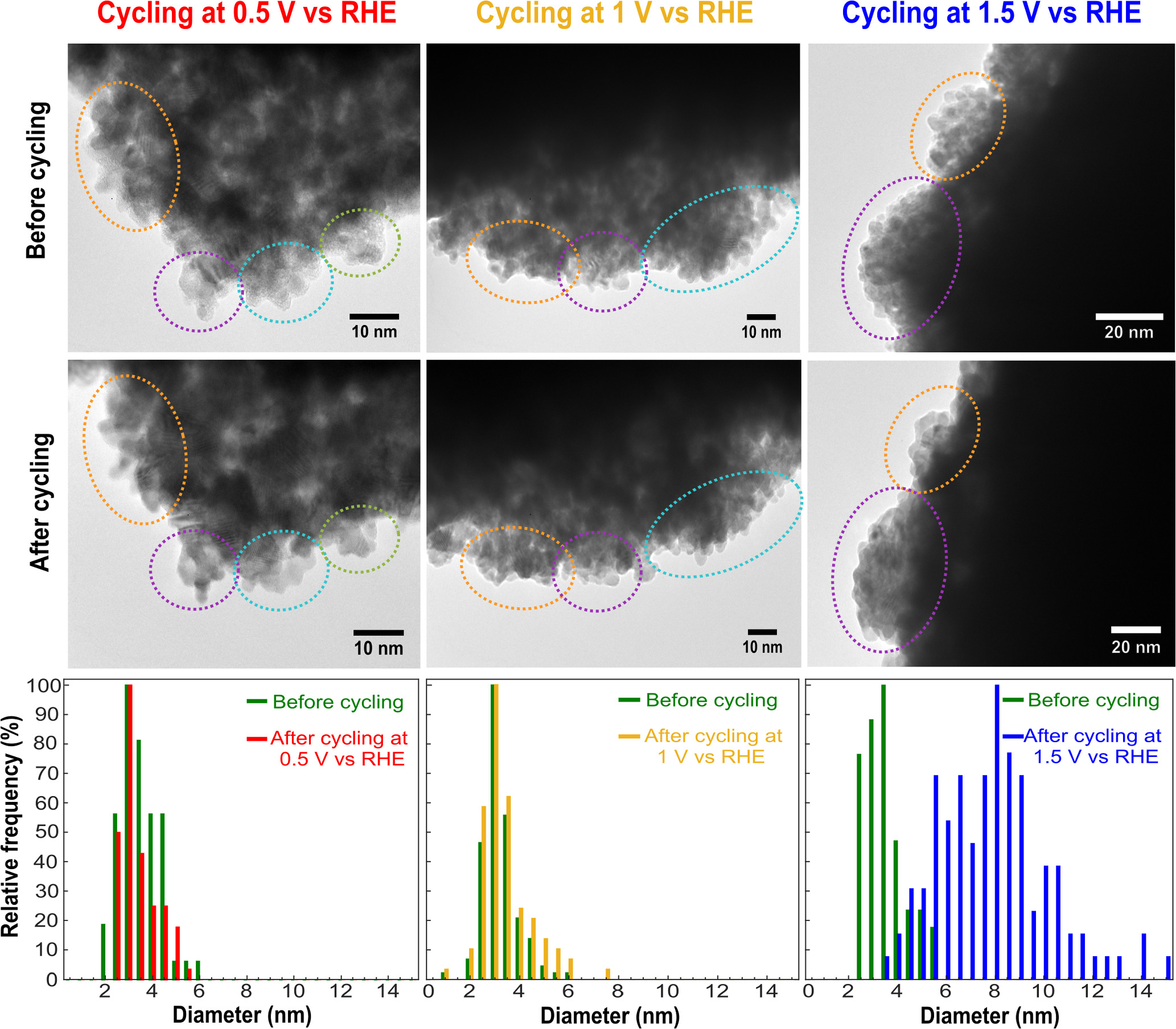
Figure 6. Identical location TEM images and particle size distributions of Pt nanoparticles before and after cycling at three different positive limit potentials in 0.5 M H2SO4.
Download figure:
Standard image High-resolution imageCombining the ECSA and IL-TEM analysis shows that when the potential is cycled up to 1 V vs RHE, catalyst poisoning is the dominant mechanism by which ECSA diminishes. This is evidenced by the fact that we saw no changes in particle size before and after cycling. But when samples were cycled to 1.5 V vs RHE, the particle size increased significantly in acid and base. We therefore attribute the growth of Pt nanoparticles to Ostwald ripening, which may be facilitated by electrochemical Pt dissolution and redeposition. 33
It is striking that, despite evidence that at least two different potential and pH-dependent mechanisms are involved, the observed rate of Pt NP deactivation was remarkably consistent—tens of percent loss in ECSA over tens of minutes—across all conditions studied. Absent evidence to the contrary, we conclude that this consistent rate of ECSA loss is coincidental, and in fact the comparative rates of catalyst poisoning and particle growth could be different in more highly purified or more severely contaminated electrolytes. Indeed, prior work on Pt NPs under scrupulously clean conditions showed that stable cycling can be achieved when the potential is maintained negative of 1 V vs RHE. 43,54 However, it is challenging and costly to obtain this level of cleanliness; hence, it may be preferable in many cases to simply refresh the Pt NP film by removing "spent" particles from the electrode substrate and replacing with a fresh NP film. This approach is directly analogous to the ubiquitous use of abrasive polishing to clean polycrystalline Pt electrodes between experiments, with the added benefit of the ability to control the Pt loading, and hence the areal density of active sites.
The effect of Pt deactivation on benchmark measurements
Alongside efforts to study degradation processes in unsupported Pt NPs, we assessed their suitability for routine measurements associated with HER/HOR catalyst benchmarking. Specifically, we sought to identify experimental timescales over which catalyst degradation translates to diminished activity toward hydrogen evolution/oxidation in reagent-grade electrolytes.
Figure 7 shows representative HER/HOR polarization curves of Pt nanoparticles compared to a commercial Pt/C catalyst at Pt mass loading of 0.2 mg/cm2. Note that these electrodes initially exhibit roughness factors (electroactive surface area normalized to superficial area of the electrode) of ∼50, assuming accessibility of surface sites in the porous film matches that implied by HUPD measurements. We also included a simulated concentration overpotential curve (simulation details can be found in the Supplementary Data), which represents the current-overpotential response of an ideal nonpolarizable electrode whose activity is limited by mass transfer of H2(aq) to and from the electrode surface. In acid, the HER activity of Pt/C and our unsupported Pt NPs are nominally identical and overlay with the concentration overpotential curve. This means that the hydrogen evolution/oxidation reaction on Pt is purely transport limited, and the kinetic behavior of the catalyst is not directly measurable. We then cycled the unsupported Pt NPs in the HER/HOR region for ∼30 min from −0.15 to +0.1 V vs RHE at 100 mV s−1 and measured the HER/HOR performance again. No change was observed in the current-overpotential data after cycling, which suggests that any loss in catalytic activity was not sufficient to induce a kinetic limitation.

Figure 7. Representative HER/HOR polarization data for fresh films of Pt NPs before and after cycling in the HER/HOR region for 30 min. Comparisons are between our unsupported Pt NPs, commercial 10 wt% Pt/C (bound to the electrode with Nafion ionomer), and a simulated diffusion overpotential curve under hydrogen gas purge at 1600 rpm in (a) 0.5 M H2SO4 and (b) in 0.5 M KOH. The Pt mass loading was held constant at 0.2 mg cm−2.
Download figure:
Standard image High-resolution imageThis result illustrates several important properties of Pt catalysts when they are used for hydrogen evolution/oxidation measurements in acid. First, a clean, high surface area Pt catalyst film should generally yield transport-limited activity for the HER/HOR in aqueous acid unless steps are taken to greatly increase the mass transfer coefficient beyond what is achievable using conventional techniques like RDE voltammetry. 73,74 Such a nanostructured Pt catalyst will continue to exhibit transport-limited activity even after it becomes poisoned or otherwise deactivated, provided the remaining active site density remains sufficiently high. A recent report showed that areal loadings of unsupported 3.8 nm Pt NPs needed to be reduced to <1 μg cm−2 (i.e., sub-nm equivalent thickness) to reduce the observed rate of HER per superficial electrode area. 21 This would be roughly equivalent to a >99.5% loss in ECSA at the 0.2 mg cm−2 loading we used here. Accordingly, unsupported Pt NPs and commercial Pt/C catalysts can certainly be used to benchmark the activity of a new HER/HOR catalyst, even in unpurified reagent-grade electrolyte, provided steps are taken to avoid catalyst contamination by dissolved Pt. 75 However, if a novel catalyst is found to exhibit higher activity toward the HER or HOR than a Pt control in acid solution, it is in fact very unlikely that the catalyst of interest is "better than Pt," but rather the Pt control has likely been severely contaminated by electrolyte impurities or another source. 21
We also performed HER/HOR measurements in alkaline electrolyte, which gave considerably different results. The activity of Pt/C and Pt nanoparticles was similar, and both were lower than the simulated concentration overpotential. This is consistent with numerous prior reports showing that Pt is less active toward hydrogen evolution/oxidation under alkaline conditions than in acid. 15,18,74 Cycling Pt nanoparticles in the HER region from −0.2 V to +0.2 V vs RHE for ∼30 min also resulted in ∼30% reduction in current density. This agrees well with ECSA data showing that accessible Pt surface sites decreased by tens of % over tens of minutes of continuous cycling. It is also consistent with the expectation that Pt HER/HOR electrocatalysis is kinetically limited in base, even for electrodes with relatively high Pt surface areas; hence, catalyst deactivation manifests as a proportional decrease in activity. These results collectively suggest that Pt-based control measurements in alkaline electrolytes are considerably more sensitive to sample history than those in acid. Comparisons between Pt and novel HER/HOR catalysts in alkaline conditions are also more relevant, since Pt remains limited by reaction kinetics over a wide range of catalyst loadings and it is indeed possible to obtain catalytic activities exceeding that of pure Pt. 76–78
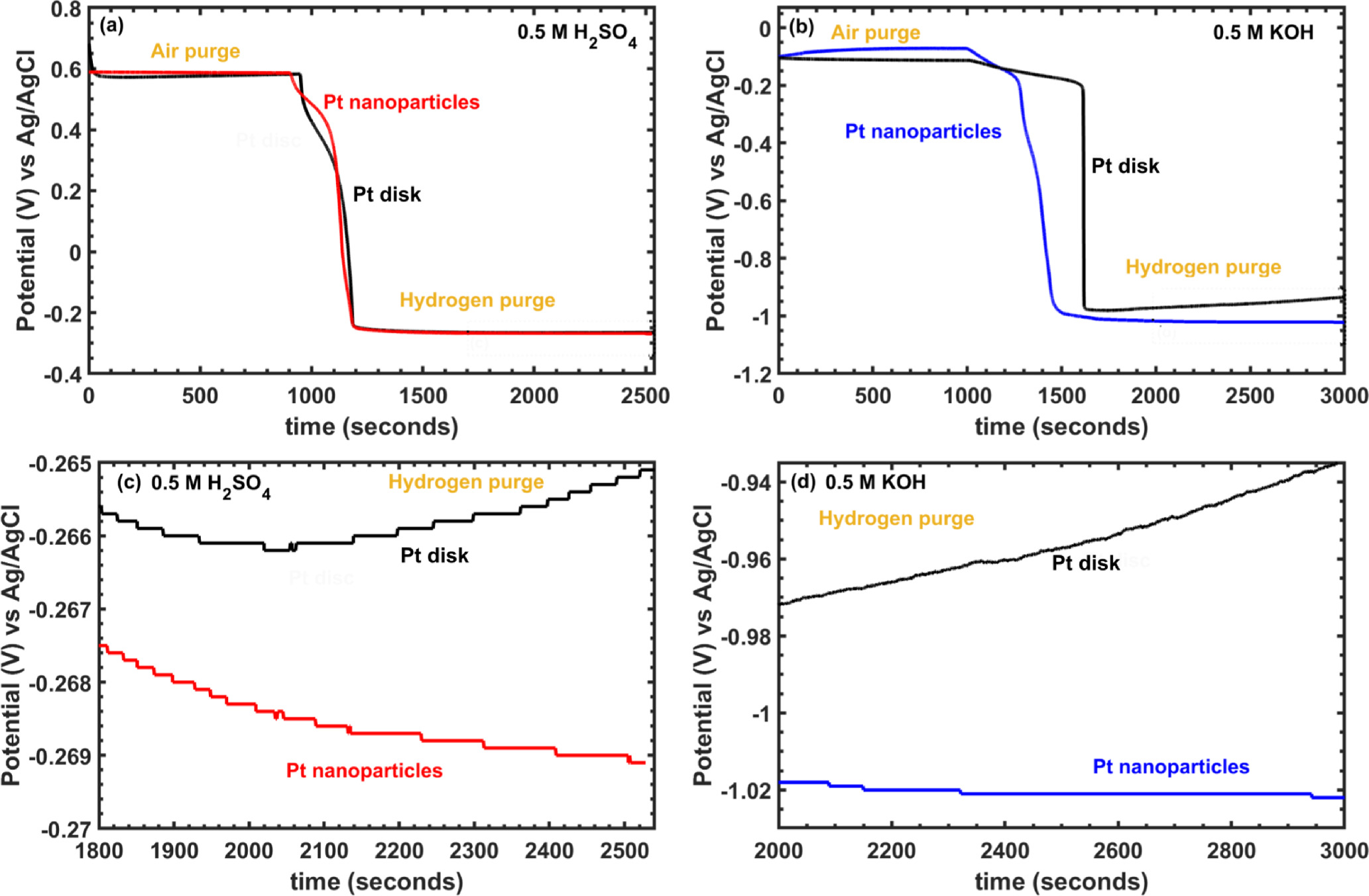
As a final assessment of the utility of our Pt NPs for catalyst benchmarking, we used open-circuit potential (OCP) measurements to calibrate Ag/AgCl reference electrodes to the reversible hydrogen electrode (RHE) potential. This procedure is crucial in characterizing catalysts for reactions involving proton-electron transfer because it enables accurate determination of catalyst overpotentials relative to an empirically measured, pH-dependent formal reduction potential. Figure 8 depicts representative RHE calibrations using Pt NPs alongside polycrystalline Pt disk electrodes in acid and base. For consistency, these measurements were initiated in electrolytes that were first purged with atmospheric air for several minutes (∼1000 seconds) until a stable potential was attained. The purge gas was then switched to hydrogen at the same flow rate. The open-circuit potential was found to decrease over several minutes as dissolved oxygen was replaced with hydrogen until it asymptotically approached a stable potential in the expected range for a reversible hydrogen electrode (near −0.25 V vs Ag/AgCl in 0.5 M H2SO4 and −1.0 V in 0.5 M KOH). Figures 8c, 8d show detailed views of a narrow range of potentials in the asymptotic region.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Open circuit potential measurements of Pt nanoparticles and Pt disk electrodes under air purge followed by H2 purge (beginning in each case at approx. t = 1000 s) in (a) 0.5 M H2SO4 and (b) 0.5 M KOH. Panels (c) and (d) are details of the potential vs time data in (a) and (b), respectively, over a narrower range of potentials after the solution became saturated with H2.
Download figure:
Standard image High-resolution image{kind=link}
In acid, OCP values for the Pt disk electrode and unsupported Pt NPs remained stable within ±2 mV over the last 10 min of the measurement. The minimum OCP values, which can be interpreted as an estimate of 0 V vs RHE, were −0.266 and −0.269 V vs Ag/AgCl; the modest difference between these values constitutes uncertainty in the RHE calibration attributable to, e.g. reference electrode drift or incomplete exclusion of atmospheric air during the hydrogen purge. These data show that clean Pt electrodes of varying surface roughness remain sufficiently catalytic over the measurement interval to obtain a stable equilibrium between hydrogen and protons.
In alkaline solution, Pt nanoparticles gave a similarly stable final OCP value, but the Pt disk was found to drift positive by ∼30 mV over several minutes. This indicates that the surface composition this electrode is sufficiently perturbed on the timescale of the RHE calibration to exhibit a mixed potential likely involving equilibration to the HER/HOR equilibrium and one or more additional redox reactions associated with impurities in solution or adsorbed on the electrode. 79 These data again exemplify the greater sensitivity of Pt to deactivation in base, which results from its lower catalytic activity toward hydrogen evolution/oxidation. Moreover, the ability to ameliorate this instability by increasing the areal density of Pt surface sites suggests the deactivation mechanism involves surface chemistry where the rate of the deactivation is not directly proportional to the areal density of surface sites. For example, transport-limited surface poisoning reactions involving low concentrations of impurities with high sticking coefficients would be expected to give this behavior.
Conclusions
In summary, we synthesized uniform, ∼3 nm unsupported Pt nanoparticles via a straightforward aqueous route involving ascorbic acid and polyacrylate as reductant and capping agent, respectively. The resulting Pt NP colloidal suspension can be purified by basifying the reaction mixture, which substantially removes the polyacrylate capping agent. The purified particles remain stably suspended in pure water, which lends well to their use as a laboratory standard for electrochemical hydrogen evolution/oxidation and potentially in numerous other applications. We further showed these particles lose tens of percent of their accessible surface sites for H-adsorption over tens of minutes during continuous electrochemical cycling in unpurified, reagent grade aqueous acid and base. We contextualized these results in terms of the use of these Pt NPs to benchmark novel HER/HOR catalysts and calibrate reference electrodes against the reversible hydrogen electrode. We further demonstrated that Pt deactivation has a much greater negative impact on these measurements in base than in acid.
Considering together all the results reported here, we conclude that it is indeed possible to collect accurate Pt HER/HOR activity benchmark data and to calibrate reference electrodes without the need to rigorously purify electrolytes or otherwise maintain scrupulously clean conditions. To do so consistently, we recommend adopting the following practices:
- (i)When possible, maximize the areal density of Pt active sites by using Pt NPs with mass loading ≥10μgcm−2 or platinized Pt electrodes with roughness factors >10.
- (ii)When using Pt NPs, adequate surface cleanliness can be inferred from HUPD measurements in acid that yield ECSA values in agreement with the surface area expected from independently measured particle size/shape as the mass loading decreases.
- (iii)When operating in an acidic environment, Pt catalysts may be further cleaned by scanning to oxidizing potentials on the order of 1.5 V vs RHE. However, these treatments should be limited to a short duration (no more than a few minutes), after which particle films should simply be removed from the electrode substrate and a fresh film deposited.
- (iv)Avoid the use of potential cycling to clean Pt catalysts in unpurified alkaline electrolytes, as it has limited beneficial effect. Instead, measurements should be limited to a few tens of minutes or less, between which catalyst films should be removed and refreshed. Experiments requiring clean Pt surfaces that require longer durations will likely require electrolyte purification.
- (v)Regularly execute internal checks for catalyst deactivation. These include hydrogen evolution/oxidation measurements in acid, which should give consistent activity (commensurate with a mass-transfer limit in acid), and RHE calibrations, which should yield little to no potential drift over at least several minutes.
Acknowledgments
We acknowledge the Arnold and Mabel Beckman Foundation and the University of Pittsburgh Mascaro Center for Sustainable Innovation for financial support of this work. The TEM work was performed at the Nanoscale Fabrication and Characterization Facility, a laboratory of the Gertrude E. and John M. Petersen Institute of NanoScience and Engineering, housed at the University of Pittsburgh.
Supplementary data (3.6 MB PDF)