Abstract
Short-range ordering in  was investigated with
was investigated with  NMR and first principles structure computations. NMR indicates that the tendency for
NMR and first principles structure computations. NMR indicates that the tendency for  to replace
to replace  in the
in the  layers decreases with decreasing nickel content. Li in the Ni/Mn layers preferentially occupies sites near
layers decreases with decreasing nickel content. Li in the Ni/Mn layers preferentially occupies sites near  and avoids the
and avoids the  ions, leading to nonrandom configurations. Calculations indicate that the ground state of
ions, leading to nonrandom configurations. Calculations indicate that the ground state of  contains zigzag rows of
contains zigzag rows of  and
and  ions. Although a disordering temperature of approximately 1000 K is calculated, ordered fragments persist above the phase transition and these materials contain significant short-range order, even when quenched from high temperature. © 2004 The Electrochemical Society. All rights reserved.
ions. Although a disordering temperature of approximately 1000 K is calculated, ordered fragments persist above the phase transition and these materials contain significant short-range order, even when quenched from high temperature. © 2004 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
Layered O3 lithium nickel manganese oxides  with the
with the  structure, have recently been shown to be promising positive electrode materials for use in lithium-ion rechargeable batteries. Operating between 2 and 4.6 V, these materials give capacities of approximately
structure, have recently been shown to be promising positive electrode materials for use in lithium-ion rechargeable batteries. Operating between 2 and 4.6 V, these materials give capacities of approximately 
 1
2 In theory, the material with
1
2 In theory, the material with  (i.e.,
(i.e.,  is most attractive, because it has the highest theoretical capacity (280 mAh/g). However, in practice, the capacity and polarization of this material depends on its method of preparation. Makimura and Ohzuku obtained a rechargeable capacity of 200 mAh/g between 2.5 and 4.5 V,3 while Lu et al. obtained a capacity of approximately 140 mAh/g between 3.0 and 4.4 V for a material with the same nominal stoichiometry.4 The different electrochemical properties observed by these two groups may arise from differences in the local and long-range ordering of the
is most attractive, because it has the highest theoretical capacity (280 mAh/g). However, in practice, the capacity and polarization of this material depends on its method of preparation. Makimura and Ohzuku obtained a rechargeable capacity of 200 mAh/g between 2.5 and 4.5 V,3 while Lu et al. obtained a capacity of approximately 140 mAh/g between 3.0 and 4.4 V for a material with the same nominal stoichiometry.4 The different electrochemical properties observed by these two groups may arise from differences in the local and long-range ordering of the 
 and
and  cations in the transition metal layers, and from
cations in the transition metal layers, and from  substitution in the Li layers. In this article,
substitution in the Li layers. In this article,  MAS NMR spectroscopy and first principles calculations were applied to investigate the nature of the cation ordering.
MAS NMR spectroscopy and first principles calculations were applied to investigate the nature of the cation ordering.
When multiple ions coexist on a common sublattice, as in the transition metal sites of  the system can either phase separate or form a set of ordered compounds at low temperature. In each case, at high temperature, a disordered solid solution exists, although in some materials, the temperature needed to reach disorder may be above the decomposition temperature of the solid. In the
the system can either phase separate or form a set of ordered compounds at low temperature. In each case, at high temperature, a disordered solid solution exists, although in some materials, the temperature needed to reach disorder may be above the decomposition temperature of the solid. In the  series, only the structure of the end member
series, only the structure of the end member 
 is known in detail and consists of a pure Li layer alternating with an
is known in detail and consists of a pure Li layer alternating with an  layer in which Li and Mn are ordered in a
layer in which Li and Mn are ordered in a  supercell. The remainder of the composition range of
supercell. The remainder of the composition range of  is generally believed to be a solid solution,5 although a recent transmission electron microscopy (TEM) study indicated that some superstructure formation may also occur for
is generally believed to be a solid solution,5 although a recent transmission electron microscopy (TEM) study indicated that some superstructure formation may also occur for 
 6 Even in a solid solution, the ions generally are not distributed randomly. In particular, on frustrated lattices, such as a triangular lattice, significant short-range order often exists for several hundred degrees above the equilibrium order/disorder temperature. Here, we show that when Li is present in the transition metal layer of
6 Even in a solid solution, the ions generally are not distributed randomly. In particular, on frustrated lattices, such as a triangular lattice, significant short-range order often exists for several hundred degrees above the equilibrium order/disorder temperature. Here, we show that when Li is present in the transition metal layer of  it strongly prefers to be surrounded by Mn, leading to a much higher frequency of
it strongly prefers to be surrounded by Mn, leading to a much higher frequency of  environments than is expected from a random distribution of the cations. In addition, we find that Li repels Ni in the transition metal layers.
environments than is expected from a random distribution of the cations. In addition, we find that Li repels Ni in the transition metal layers.
Experimental
Three samples with  1/3, and 1/10 were synthesized from stoichiometric quantities of coprecipitated manganese and nickel double hydroxides with
1/3, and 1/10 were synthesized from stoichiometric quantities of coprecipitated manganese and nickel double hydroxides with  -enriched (Aldrich) lithium hydroxide at 900°C for 24 h in
-enriched (Aldrich) lithium hydroxide at 900°C for 24 h in  3 X-ray diffraction (XRD) confirmed the lack of impurities and the formation of layered materials. The electrochemistry and diffraction patterns of the
3 X-ray diffraction (XRD) confirmed the lack of impurities and the formation of layered materials. The electrochemistry and diffraction patterns of the  and 1/3 samples have been reported elsewhere.7
8
and 1/3 samples have been reported elsewhere.7
8  magic-angle spinning (MAS) NMR experiments were performed at 29.47 MHz on a CMX-200 spectrometer, with rotor synchronized spin-echoes (90°-τ-180°-τ-acq)
magic-angle spinning (MAS) NMR experiments were performed at 29.47 MHz on a CMX-200 spectrometer, with rotor synchronized spin-echoes (90°-τ-180°-τ-acq)  rotor period) and spinning speeds of 38 kHz and were referenced to 1 M LiCl at 0 ppm.
rotor period) and spinning speeds of 38 kHz and were referenced to 1 M LiCl at 0 ppm.
Calculation Methodology
All calculations were performed in the Generalized Gradient Approximation to Density Functional theory, with core states represented by ultrasoft pseudopotentials, as implemented in the Vienna Ab Initio Simulation Package (VASP).9 The plane wave cutoff was 400 eV and  -points varied from
-points varied from  for the one formula-unit structures to
for the one formula-unit structures to  for the largest unit cells, so as to keep the
for the largest unit cells, so as to keep the  -point density approximately constant. All calculations were performed with ferromagnetic spin polarization. Allowing
-point density approximately constant. All calculations were performed with ferromagnetic spin polarization. Allowing  antiferromagnetic spin ordering lowers the total energy, but does not significantly change the energy difference between the structures.
antiferromagnetic spin ordering lowers the total energy, but does not significantly change the energy difference between the structures.
Results
NMR.—
The  MAS NMR spectra of the three samples
MAS NMR spectra of the three samples  with
with  1/3, and 1/2 are shown in Fig. 1. Two major clusters of resonances are observed, one at approx. 1300-1500 ppm and one at approx. 700 ppm, which have been assigned previously to Li in the transition metal layers,
1/3, and 1/2 are shown in Fig. 1. Two major clusters of resonances are observed, one at approx. 1300-1500 ppm and one at approx. 700 ppm, which have been assigned previously to Li in the transition metal layers,  and Li in the Li layers,
and Li in the Li layers,  respectively.7
10 The large isotropic (Fermi-contact) shifts observed for these resonances are controlled by both the number and type of paramagnetic ions in the nearby cation coordination sphere.11
12 For example, the resonance at 1560 ppm is assigned, based on its shift, to the local environment
respectively.7
10 The large isotropic (Fermi-contact) shifts observed for these resonances are controlled by both the number and type of paramagnetic ions in the nearby cation coordination sphere.11
12 For example, the resonance at 1560 ppm is assigned, based on its shift, to the local environment  which contains six Mn and six Li in the first cation coordination sphere surrounding the central Li atom. Analysis of the shape of the spinning sidebands of this resonance, and comparison with those observed for a similar local environment found in
which contains six Mn and six Li in the first cation coordination sphere surrounding the central Li atom. Analysis of the shape of the spinning sidebands of this resonance, and comparison with those observed for a similar local environment found in  (Fig. 2a), shows that the six nearby
(Fig. 2a), shows that the six nearby  ions must all be located in the same plane.10 Thus, this resonance is ascribed to
ions must all be located in the same plane.10 Thus, this resonance is ascribed to  and is abbreviated as
and is abbreviated as  in the subsequent discussion. The second major, high-frequency resonance at 1365 ppm is assigned to a
in the subsequent discussion. The second major, high-frequency resonance at 1365 ppm is assigned to a  local environment containing five nearby
local environment containing five nearby  ions such as
ions such as  (Fig. 2b). Only a very weak resonance is seen at 1150 ppm due to Li nearby 4
(Fig. 2b). Only a very weak resonance is seen at 1150 ppm due to Li nearby 4  ions, suggesting that the
ions, suggesting that the  preferentially occupy sites near five or six
preferentially occupy sites near five or six  ions. Possible ordering schemes consistent with the intensities of the different resonances are described in the Discussion.
ions. Possible ordering schemes consistent with the intensities of the different resonances are described in the Discussion.
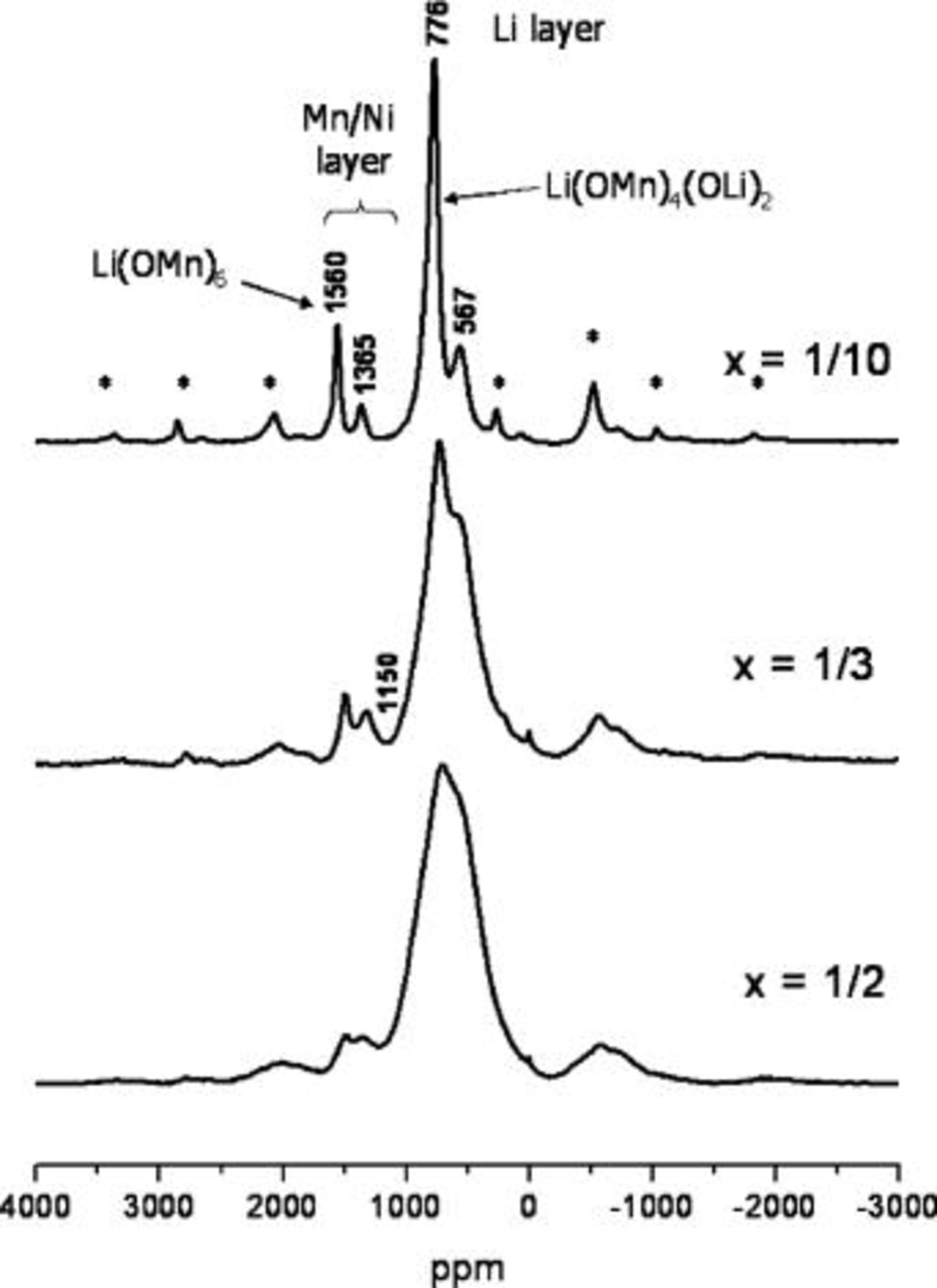
Figure 1.
 MAS NMR spectra of
MAS NMR spectra of  with
with  1/3, and 1/2. The resonances corresponding to the local environments,
1/3, and 1/2. The resonances corresponding to the local environments,  and
and  found in
found in  are marked. The frequencies of the major resonances are indicated; asterisks indicate spinning sidebands.
are marked. The frequencies of the major resonances are indicated; asterisks indicate spinning sidebands.
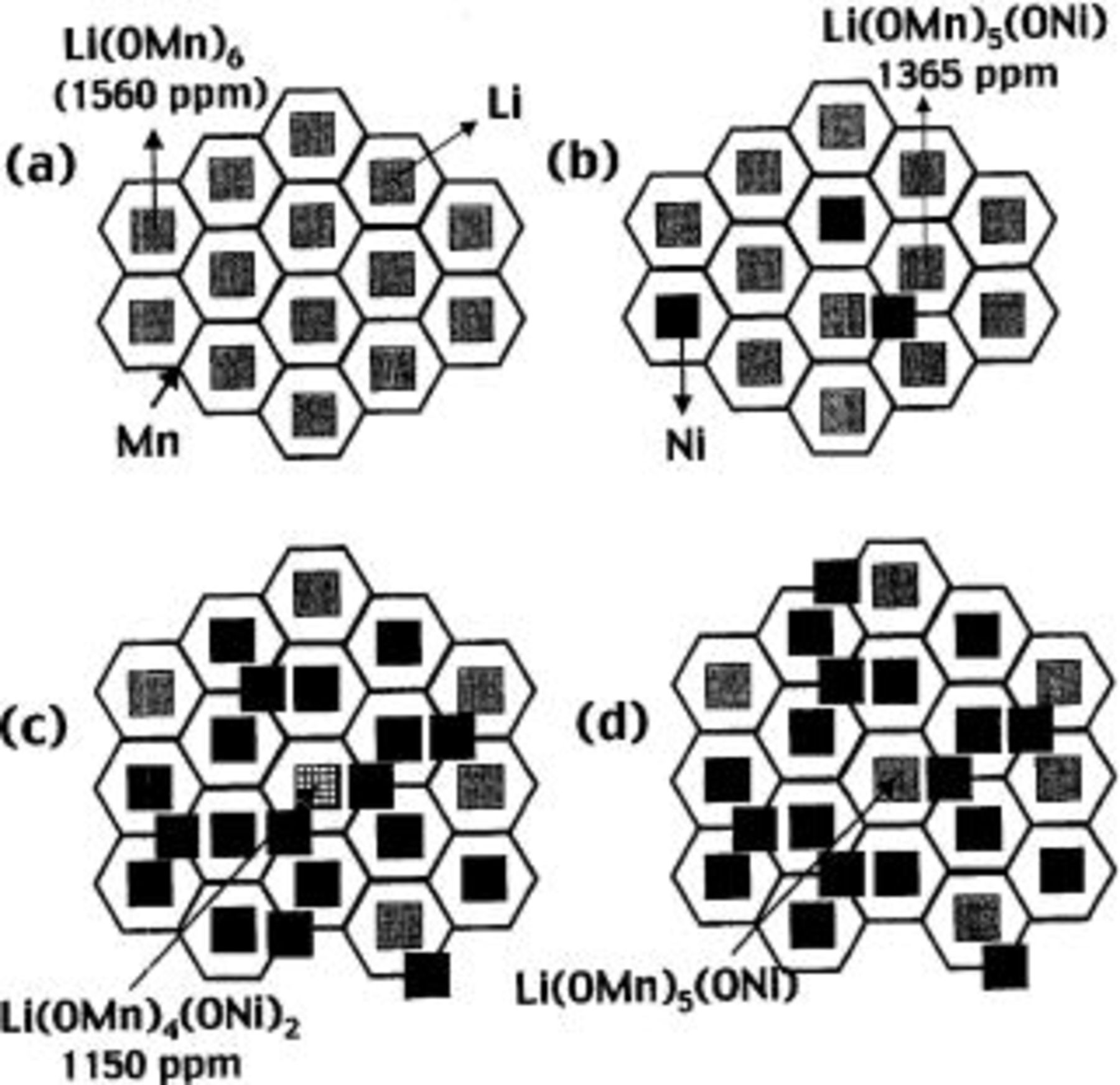
Figure 2. Different 
 and
and  cation ordering schemes for ions in the transition metal layers. A schematic of the ordering scheme found in
cation ordering schemes for ions in the transition metal layers. A schematic of the ordering scheme found in  showing the honeycomb structure formed by Li (gray squares) and Mn (a), the structure derived by replacing two Li and 1 Mn by 3 Ni (black squares), for the
showing the honeycomb structure formed by Li (gray squares) and Mn (a), the structure derived by replacing two Li and 1 Mn by 3 Ni (black squares), for the  (b) and
(b) and  (c) compositions (model II; Table III). (d) An ordering scheme based on
(c) compositions (model II; Table III). (d) An ordering scheme based on  ordering and Li-Ni avoidance (model III; Table III), for the
ordering and Li-Ni avoidance (model III; Table III), for the  composition.
composition.
The frequency with which different Li environments occur can be obtained from a quantitative analysis of the signal intensities. The relative intensities (of the isotropic  sidebands) of the low- (approx. 700 ppm;
sidebands) of the low- (approx. 700 ppm;  sites) and high-frequency resonances (1300-1500 ppm;
sites) and high-frequency resonances (1300-1500 ppm;  (Table I) are close to the values calculated from the formula
(Table I) are close to the values calculated from the formula  for
for  indicating that little exchange occurs between in the Li layer and Ni in the TM layer,
indicating that little exchange occurs between in the Li layer and Ni in the TM layer,  As the value of
As the value of  increases, the experimentally determined concentration of
increases, the experimentally determined concentration of  decreases, but for
decreases, but for  concentration remains similar to the predicted value. At
concentration remains similar to the predicted value. At  however, where negligible concentrations of
however, where negligible concentrations of  and
and  are expected based on the composition, 7% of the Li ions remain in the TM layers. These values are lower than those of Lu et al. ,4
13 who obtained occupancies of 93-94 and 88-89% for
are expected based on the composition, 7% of the Li ions remain in the TM layers. These values are lower than those of Lu et al. ,4
13 who obtained occupancies of 93-94 and 88-89% for  for
for  and 1/2, respectively, from neutron diffraction experiments, implying occupancies for
and 1/2, respectively, from neutron diffraction experiments, implying occupancies for  of 7 and 12%. However, the same group found lower occupancies of 6-5 and 8% for
of 7 and 12%. However, the same group found lower occupancies of 6-5 and 8% for  in a more recent paper.5 Generally, the cation occupancies determined from diffraction measurements of disordered systems containing multiple types of cations on the same site are not as well determined as implied by the estimated standard deviations quoted in the text (in part because occupancies and thermal parameters are strongly correlated). Nonetheless, the NMR values are in good agreement with diffraction measurements for
in a more recent paper.5 Generally, the cation occupancies determined from diffraction measurements of disordered systems containing multiple types of cations on the same site are not as well determined as implied by the estimated standard deviations quoted in the text (in part because occupancies and thermal parameters are strongly correlated). Nonetheless, the NMR values are in good agreement with diffraction measurements for  and provide an independent measure of Li occupancy in the Ni/Mn layers.
and provide an independent measure of Li occupancy in the Ni/Mn layers.
Table I.
Comparison of the predicted and experimentally determined Li contents in the transition metal layers, as a percentage of the total lithium content, for the compounds  Errors in the experimentally determined values are given in parenthesis. Errors in the experimentally determined values are given in parenthesis. | ||
|---|---|---|
Nickel content
 | Li content in T.M. layers (%) | |
| Predicted | Experiment | |
| 1/2 | 0 | 7(1.5) |
| 1/3 | 10 | 10(2) |
| 1/10 | 21 | 19(2) |
| 0 | 25 | 22(3) |
The  resonances at 1560-1365 ppm are much broader for the
resonances at 1560-1365 ppm are much broader for the  sample than for the other two samples. We tentatively ascribe this to the presence of nearby
sample than for the other two samples. We tentatively ascribe this to the presence of nearby  ions in the Li layers
ions in the Li layers  These
These  ions can be located in either a first or second neighbor position to a Li in the TM layer and are expected to add small (negative) or large (positive) shifts,7
14 respectively, to the total hyperfine shift of the
ions can be located in either a first or second neighbor position to a Li in the TM layer and are expected to add small (negative) or large (positive) shifts,7
14 respectively, to the total hyperfine shift of the  environment. Values for these shifts of approximately
environment. Values for these shifts of approximately  and
and  respectively, per
respectively, per  were determined in recent NMR studies of the layered compound
were determined in recent NMR studies of the layered compound  15 Given that no resonances are observed at frequencies higher than 1560 ppm, the
15 Given that no resonances are observed at frequencies higher than 1560 ppm, the  environments in the Ni/Mn layers do not appear to contain additional
environments in the Ni/Mn layers do not appear to contain additional  ions in their second coordination shells. Furthermore, the resonance at 1560 ppm is sharper than the 1365 ppm resonance, suggesting that most
ions in their second coordination shells. Furthermore, the resonance at 1560 ppm is sharper than the 1365 ppm resonance, suggesting that most  environments do not contain
environments do not contain  ions in their first coordination shell.
ions in their first coordination shell.
First principles computations of ordering in  .—
.—
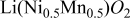
To understand the Ni-Mn interactions better, which are difficult to investigate by using NMR experiments a series of first principles calculations were performed on stoichiometric  with twelve different Ni-Mn arrangements. Of these, the zigzag ordering of Fig. 3a was most stable. The two arrangements with closest energy to this ground state are also shown in Fig. 3. Ordering in rows along the hexagonal axis (Fig. 3b), as for example, is, observed16 and predicted17
18 for Li and vacancies in
with twelve different Ni-Mn arrangements. Of these, the zigzag ordering of Fig. 3a was most stable. The two arrangements with closest energy to this ground state are also shown in Fig. 3. Ordering in rows along the hexagonal axis (Fig. 3b), as for example, is, observed16 and predicted17
18 for Li and vacancies in  is considerably less stable. Whether Ni/Mn ordering is present in actual samples depends on the synthesis temperature, and the equilibrium order/disorder temperature. We studied finite temperature disorder with the cluster expansion technique,19
20 whereby the energies of the calculated Ni/Mn arrangements are fit to interactions in an Ising-like model that describes the occupation of each lattice site by Ni or Mn. Previously, this approach has been applied to study Li-vacancy ordering in
is considerably less stable. Whether Ni/Mn ordering is present in actual samples depends on the synthesis temperature, and the equilibrium order/disorder temperature. We studied finite temperature disorder with the cluster expansion technique,19
20 whereby the energies of the calculated Ni/Mn arrangements are fit to interactions in an Ising-like model that describes the occupation of each lattice site by Ni or Mn. Previously, this approach has been applied to study Li-vacancy ordering in  17 and
17 and  21 and more details can be found in these papers. The interactions in the Ising-like model are given in Table II. Most in-plane pair interactions are positive reflecting the ordering tendency of Ni-Mn. Finite temperature Monte Carlo simulation with this set of interactions indicates that the system undergoes a weak first-order order/disorder transition at approximately 1000 K, although the exact temperature depends somewhat on the details of the interaction fit. Figure 4 shows a snapshot from the Monte Carlo simulation at 1200 K. Although the system is in a disordered phase at this temperature, significant short-range order exists, with Ni and Mn forming small fragments of the structures with low energy: When this material is cooled through the transition, some small ordered fragments grow and form larger well-ordered regions, depending on the diffusivities of Ni and Mn and the cooling rates. Assuming a 1 eV activation barrier for diffusion, and a 0.1% vacancy fraction, the hopping rate for the cations at the transition temperature of 1000 K is still about
21 and more details can be found in these papers. The interactions in the Ising-like model are given in Table II. Most in-plane pair interactions are positive reflecting the ordering tendency of Ni-Mn. Finite temperature Monte Carlo simulation with this set of interactions indicates that the system undergoes a weak first-order order/disorder transition at approximately 1000 K, although the exact temperature depends somewhat on the details of the interaction fit. Figure 4 shows a snapshot from the Monte Carlo simulation at 1200 K. Although the system is in a disordered phase at this temperature, significant short-range order exists, with Ni and Mn forming small fragments of the structures with low energy: When this material is cooled through the transition, some small ordered fragments grow and form larger well-ordered regions, depending on the diffusivities of Ni and Mn and the cooling rates. Assuming a 1 eV activation barrier for diffusion, and a 0.1% vacancy fraction, the hopping rate for the cations at the transition temperature of 1000 K is still about  When such a hopping rate is applied in our Monte Carlo simulation, the systems become well ordered over long distances at less than 1 s. Even for an activation barrier of 1.5 eV, the hopping rate is several hundreds per second at 1000 K, more than enough for small domains of well-ordered material to form.
When such a hopping rate is applied in our Monte Carlo simulation, the systems become well ordered over long distances at less than 1 s. Even for an activation barrier of 1.5 eV, the hopping rate is several hundreds per second at 1000 K, more than enough for small domains of well-ordered material to form.
Figure 3. Different in-plane orderings of Ni and Mn in  and their calculated energy (with respect to the most stable structure). The structures can be stacked in multiple ways along the
and their calculated energy (with respect to the most stable structure). The structures can be stacked in multiple ways along the  axis, and the energy is given for the stacking with lowest energy. Dark (white) octahedral indicate Ni (Mn).
axis, and the energy is given for the stacking with lowest energy. Dark (white) octahedral indicate Ni (Mn).
Table II.
Values of n th neighbor pair  interactions in an Ising-like model for the Ni-Mn distributions in the transition-metal layers. interactions in an Ising-like model for the Ni-Mn distributions in the transition-metal layers. | ||||||
|---|---|---|---|---|---|---|
| meV | In-plane interactions | Interplane interactions | ||||
| Constant |
 |
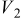 |
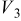 |
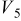 |
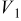 |
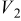 |
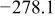 | 78.0 | 8.1 | 14.8 |
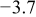 |
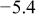 | 4.9 |
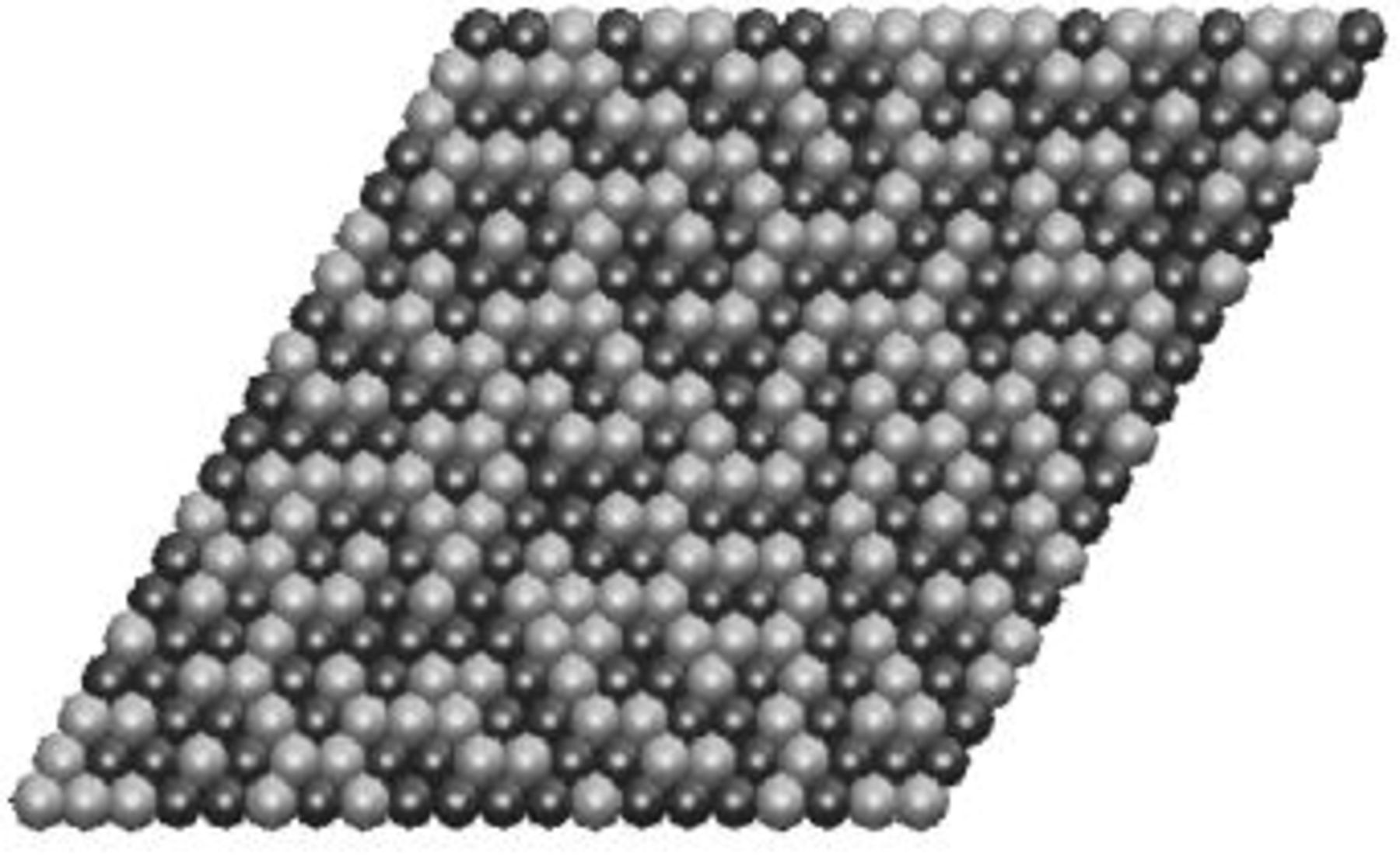
Figure 4. Snapshot of the Monte Carlo simulation of Ni, Mn disorder in  at 1200 K.
at 1200 K.
Discussion and Models
Both the NMR and first principles results indicate that substantial short-range order exists in these materials. The NMR spectra of these materials are remarkably simple, indicating that a small number of local environments dominate, in contrast to what is expected from a random solid solution of Li, Ni, and Mn in the TM layer. Because NMR probes only a local coordination sphere, it cannot be excluded that the strong preferences for particular  configurations are the result of long-range order. A more detailed study with TEM, XRD, or neutron diffraction is needed to fully characterize any long-range order that may exist.
configurations are the result of long-range order. A more detailed study with TEM, XRD, or neutron diffraction is needed to fully characterize any long-range order that may exist.
For stoichiometric  with no Li in the TM layer, calculations predict that the zigzag structure has the lowest energy. Whether this structure can persist in a typical real
with no Li in the TM layer, calculations predict that the zigzag structure has the lowest energy. Whether this structure can persist in a typical real  sample depends on the amount of Li in the TM layer and how strongly this Li perturbs that ordering. A high order/disorder temperature (1000 K) is predicted, which has significant implications for understanding the effect of processing on the properties of these materials. Using low-temperature processing routes, one may actually be in the ordered phase region and be able to synthesize materials with substantial long-range order. However, even for materials synthesized above the order/disorder temperature, significant short-range order can exist.
sample depends on the amount of Li in the TM layer and how strongly this Li perturbs that ordering. A high order/disorder temperature (1000 K) is predicted, which has significant implications for understanding the effect of processing on the properties of these materials. Using low-temperature processing routes, one may actually be in the ordered phase region and be able to synthesize materials with substantial long-range order. However, even for materials synthesized above the order/disorder temperature, significant short-range order can exist.
The NMR results suggest that a degree of ordering persists even in the Li-excess materials. The integrated intensities of the NMR resonances gives quantitative information on the relative frequency of various environments around  and thereby provide constraints on which ordering models are possible. The concentrations of different Li local environments in the Ni/Mn layers were determined from the intensities of the 1560, 1365 (and the weaker 1150) ppm resonances in the
and thereby provide constraints on which ordering models are possible. The concentrations of different Li local environments in the Ni/Mn layers were determined from the intensities of the 1560, 1365 (and the weaker 1150) ppm resonances in the  and 1/10 samples (Table III) and compared to the different local environments that can arise from various Li/Mn/Ni ordering schemes in the TM layers. The first model, in which the
and 1/10 samples (Table III) and compared to the different local environments that can arise from various Li/Mn/Ni ordering schemes in the TM layers. The first model, in which the 
 and
and  ions are distributed randomly on the sites of the TM layer, fails to account for the observed NMR intensities. In particular, the local environment
ions are distributed randomly on the sites of the TM layer, fails to account for the observed NMR intensities. In particular, the local environment  which is observed in the end-member compound
which is observed in the end-member compound  is present experimentally in much higher concentrations than predicted based on the random model. In model II (Table III; Fig. 2b), we considered an ordering scheme derived from that found in the manganese layers of
is present experimentally in much higher concentrations than predicted based on the random model. In model II (Table III; Fig. 2b), we considered an ordering scheme derived from that found in the manganese layers of  (i.e.,
(i.e.,  . In this compound, the Li and Mn ions are ordered so that each Li ion is surrounded by six manganese ions, while the manganese ions are surrounded by only three other Mn ions in the
. In this compound, the Li and Mn ions are ordered so that each Li ion is surrounded by six manganese ions, while the manganese ions are surrounded by only three other Mn ions in the  layers, to form a honey-comb arrangement with two different cation sites (Fig. 2a). To accommodate
layers, to form a honey-comb arrangement with two different cation sites (Fig. 2a). To accommodate  in the TM layer, one
in the TM layer, one  and two
and two  ions are randomly replaced by three
ions are randomly replaced by three  ions on the
ions on the  array, to maintain charge neutrality (i.e., the
array, to maintain charge neutrality (i.e., the  occupancy of the Li and Mn sites is 2:1), (Fig. 2b). The fit between this model and the intensities obtained by NMR is good for the sample with lower nickel content and provides supporting evidence for the assignment of the 1360 ppm NMR resonance to the
occupancy of the Li and Mn sites is 2:1), (Fig. 2b). The fit between this model and the intensities obtained by NMR is good for the sample with lower nickel content and provides supporting evidence for the assignment of the 1360 ppm NMR resonance to the  local environment. The model is consistent with the intensities and shifts of the
local environment. The model is consistent with the intensities and shifts of the  resonances, which are also dominated by the "
resonances, which are also dominated by the " "-like local environments (776 ppm), and are not consistent with random Ni/Mn/Li cation ordering (model I).
"-like local environments (776 ppm), and are not consistent with random Ni/Mn/Li cation ordering (model I).
Table III.
| Concentrations of the different Li environments in the transition-metal layers obtained from NMR and predicted from three different models for Li/Mn/Ni ordering. Concentrations are expressed as fractions of the total Li content in the transition metal layers. | |||||
|---|---|---|---|---|---|
NiContent
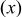 | LocalEnvironmenta | ExperimentalIntensityb | Probabilityc | ||
| Model I(Random) | Model II | Model III(a)-(b) | |||
| 1/10 |
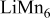 | 0.72 (1560) | 0.06 | 0.74 | 0.74-0.77 |
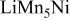 | 0.22 (1365) | 0.06 | 0.23 | 0.26-0.23 | |
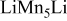 | 0.16 | 0 | 0 | ||
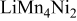 | 0.06 (1150) | 0.02 | 0.03 | 0 | |
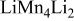 | 0.17 | 0 | 0 | ||
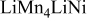 | 0.13 | 0 | 0 | ||
| 1/3 |
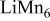 | 0.5 (1560) | 0.03 | 0.34 | 0.34-0.60 |
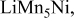 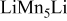 | 0.41 (1365) | 0.15 | 0.40 | 0.60-0.40 | |
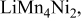 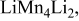 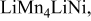 |
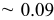 , (1150) , (1150) | 0.33 | 0.20 | 0.05-0.0 | |
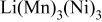 | - | 0.13 | 0.05 | 0 | |
a The two local environments that, in principle, may contribute to the intensity of the resonance at 1365 ppm are listed together. Similarly environments that resonate at lower frequencies, such as 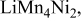 containing 4Mn and 2Ni in the Li cation first coordination shell (where the 6 additional Li in the first cation coordination shell, in the Li layers are omitted for clarity) are grouped together. Since the second cation coordination shell contains only Li, these are not considered. containing 4Mn and 2Ni in the Li cation first coordination shell (where the 6 additional Li in the first cation coordination shell, in the Li layers are omitted for clarity) are grouped together. Since the second cation coordination shell contains only Li, these are not considered. | |||||
| b Shifts of the resonances (in ppm) are given in parentheses. | |||||
| c See Ref. 22 for details on the probability calculations. The values were calculated by assuming that all the Ni is in the transition metal layers. | |||||
The agreement is less good for the sample with a higher  content (Fig. 2c). In particular, model II predicts that the environment
content (Fig. 2c). In particular, model II predicts that the environment  should be present in a higher concentration than was observed experimentally. In model III, we constrain the system to have no Li environments
should be present in a higher concentration than was observed experimentally. In model III, we constrain the system to have no Li environments  with
with  This can be done by either creating more
This can be done by either creating more  environments (model IIIa) or by putting the Ni in environments surrounded by only Mn (i.e., no Li), as for stoichiometric
environments (model IIIa) or by putting the Ni in environments surrounded by only Mn (i.e., no Li), as for stoichiometric  (model IIIb). The experimental data lie between the two extremes of model III, indicating that the tendency for Ni and Li avoidance, in the first cation coordination shell is strong. At higher values of
(model IIIb). The experimental data lie between the two extremes of model III, indicating that the tendency for Ni and Li avoidance, in the first cation coordination shell is strong. At higher values of  most
most  ion in the Mn sites are located nearby Li sites in the
ion in the Mn sites are located nearby Li sites in the  layer substituted by Ni. This results in "zigzag" ordered regions that are similar to those found in the disordered
layer substituted by Ni. This results in "zigzag" ordered regions that are similar to those found in the disordered  (Fig. 4).
(Fig. 4).
So far, we have not considered the effect of  doping in the Li layers on the
doping in the Li layers on the  hyperfine shifts. The effect is negligible for the
hyperfine shifts. The effect is negligible for the  sample, and small for the
sample, and small for the  sample, where, at most 2% of the
sample, where, at most 2% of the  sites are occupied by
sites are occupied by  (based on errors in the measurements of the Li signals). Li environments such as
(based on errors in the measurements of the Li signals). Li environments such as  which in model II represent only 2% of the total sites in the Ni/Mn layers, should resonate at higher frequencies than predicted in Table II, if
which in model II represent only 2% of the total sites in the Ni/Mn layers, should resonate at higher frequencies than predicted in Table II, if  ions are present in the second cation coordination shell. The effect on the
ions are present in the second cation coordination shell. The effect on the  spectrum is negligible, if Ni occupancy of the Li layers is random. However, if the location of the
spectrum is negligible, if Ni occupancy of the Li layers is random. However, if the location of the  ions is strongly correlated with higher Ni contents in the Ni/Mn layers, then a small decrease in the intensity of the resonance assigned to
ions is strongly correlated with higher Ni contents in the Ni/Mn layers, then a small decrease in the intensity of the resonance assigned to  (and an increase in the intensity of the higher frequency resonances) results. This effect is more important for the
(and an increase in the intensity of the higher frequency resonances) results. This effect is more important for the  sample, and may provide an additional explanation for the weak intensity of the 1150 ppm resonance and the breadth of the resonance at 1350 ppm, in comparison to the 1560 ppm resonance.
sample, and may provide an additional explanation for the weak intensity of the 1150 ppm resonance and the breadth of the resonance at 1350 ppm, in comparison to the 1560 ppm resonance.
The NMR spectra are extremely sensitive to cation substitution in the first and second cation coordination shell and thus only probe a length scale of approx. 8-9 Å, and hence one cannot make inference about long-range order or separation into two phases with different compositions.5 However, local environments such as  are clearly present, although these have been suggested to be "statistically very unlikely." 5 The frequency of different
are clearly present, although these have been suggested to be "statistically very unlikely." 5 The frequency of different  environments obtained from NMR are consistent with ordering based on a
environments obtained from NMR are consistent with ordering based on a 
 lattice, which has been proposed based on diffraction and TEM experiments, for the
lattice, which has been proposed based on diffraction and TEM experiments, for the  and
and  samples, respectively.6
samples, respectively.6
Conclusions
Both first principles calculations and NMR on  and NMR on
and NMR on  indicate a strong tendency for cation ordering in the TM layers. There appear to be two competing stable ordering schemes for the
indicate a strong tendency for cation ordering in the TM layers. There appear to be two competing stable ordering schemes for the  structure, one involving
structure, one involving  -type ordering (allowed only if accompanied by Ni substitution of the Li layers) and the other, Ni/Mn zigzag ordering. The structure of this compound likely reflects a balance between these two competing driving forces for ordering. Even when the material is synthesized above the long-range ordering temperature, short-range order exists, which can grow as the material is cooled. This kinetic development of long-range order may lead to a larger than usual dependence of the structure (and possibly electrochemical properties) on processing differences. The NMR spectra indicate that nonrandom cation distributions persist in the Li-excess material. A model for the material whereby
-type ordering (allowed only if accompanied by Ni substitution of the Li layers) and the other, Ni/Mn zigzag ordering. The structure of this compound likely reflects a balance between these two competing driving forces for ordering. Even when the material is synthesized above the long-range ordering temperature, short-range order exists, which can grow as the material is cooled. This kinetic development of long-range order may lead to a larger than usual dependence of the structure (and possibly electrochemical properties) on processing differences. The NMR spectra indicate that nonrandom cation distributions persist in the Li-excess material. A model for the material whereby  -like domains are substituted by Ni seems to be consistent with the integrated NMR intensities. Further NMR analysis suggests some clustering of the
-like domains are substituted by Ni seems to be consistent with the integrated NMR intensities. Further NMR analysis suggests some clustering of the  ions, particularly in the samples containing higher
ions, particularly in the samples containing higher  contents. The NMR intensities may also be used to estimate
contents. The NMR intensities may also be used to estimate  doping in the Ni/Mn layers.
doping in the Ni/Mn layers.
Acknowledgments
This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Freedom CAR and Vehicle Technologies of the U.S. Department of Energy under contract no. DE-AC03-76SF00098, subcontract no. 6517748 and 6517749 with the Lawrence Berkeley National Laboratory. S.I. thanks the NSF for support via the Solid State Chemistry REU program.
The State University of New York at Stony Brook assisted in meeting the publication costs of this article.