Article Text

Abstract
It is now well accepted that many tumors undergo a process of clonal selection which means that tumor antigens arising at various stages of tumor progression are likely to be represented in just a subset of tumor cells. This process is thought to be driven by constant immunosurveillance which applies selective pressure by eliminating tumor cells expressing antigens that are recognized by T cells. It is becoming increasingly clear that the same selective pressure may also select for tumor cells that evade immune detection by acquiring deficiencies in their human leucocyte antigen (HLA) presentation pathways, allowing important tumor antigens to persist within cells undetected by the immune system. Deficiencies in antigen presentation pathway can arise by a variety of mechanisms, including genetic and epigenetic changes, and functional antigen presentation is a hard phenomenon to assess using our standard analytical techniques. Nevertheless, it is likely to have profound clinical significance and could well define whether an individual patient will respond to a particular type of therapy or not. In this review we consider the mechanisms by which HLA function may be lost in clinical disease, we assess the implications for current immunotherapy approaches using checkpoint inhibitors and examine the prognostic impact of HLA loss demonstrated in clinical trials so far. Finally, we propose strategies that might be explored for possible patient stratification.
- immunity
- cellular
- immunity
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The major histocompatibility complex class I (MHC-I) proteins in humans are termed as human leucocyte antigen I (HLA-I) and they are divided into classical and non-classical HLA-I subtypes.1 Classical type I HLA molecules (HLA-A, HLA-B, and HLA-C) function to present cellular antigens to T cells and are essential for immunosurveillance and cancer immunotherapy. It is now clear that loss of HLA-I function is an important escape mechanism for tumors from immunotherapy, by a variety of mechanisms2–4 Between 60% and 90% of patients may be affected even before treatment, and this would render them unable to respond to all current immunotherapy approaches.5 On the other hand, a growing number of studies suggest that non-classical HLA-I molecules also play a critical role in cancer immune escape.
Classical HLA class I expression
Classical HLA-I molecules, which function to present cellular antigens to T cells, consist of a highly polymorphic α-heavy chain encoded by HLA-A, HLA-B or HLA-C genes and β−2-microglobulin (β2M) light chain which provides stability to the HLA-I complex.6 Each HLA molecule has a cleft that binds antigen-derived peptides and presents them to T cells. Several cellular components are involved in the antigen processing and presentation mechanism. Following ubiquitinylation intracellular proteins are degraded by the immunoproteasomes to produce short peptides for transportation by the transporter associated with antigen processing (TAP1/TAP2) into the endoplasmic reticulum (ER). The glycoprotein Tapasin, in cooperation with other chaperones (calreticulin, ERp57) mediates an interaction between TAP1/2 and newly synthesized HLA-I molecules, forming peptide/HLA-I complexes which are transported to the cell surface for T cell surveillance and possible antigen-specific recognition (figure 1). Classical HLA-I molecules can also be recognized by killer cell immunoglobulin-like receptors (KIRs) expressed on natural killer (NK) cells and inhibit their HLA-I independent cytotoxic function.7

Possible deficiencies in antigen presenting machinery. (1) Acquired mutations, transcriptional or post-transcriptional regulations in HLA or antigen presentation machinery (APM) genes or epigenetic modifications in their promoter regions. (2, 3) Defects in type-I or type-II interferon pathways, which are direct stimulators of HLA-I expression. (4) Aberrant activation of PI3K-Akt oncogenic pathway interferes with phosphorylation of STAT1 and hinders interferon mediated HLA-I expression. (5) Oncogenic BRAF mutation can drive internalization and endosomal degradation of surface HLA-I antigens. (6) Autophagy cargo receptor NBR1 protein can bind to HLA-I leads to autophagy-mediated degradation. (7) Defects in the proteasome components (LMP2, LMP9 or MECL-1, etc). (8) Defects in the peptide transport or ER peptide loading complex (TAP1/TAP2, ERp57, calnexin, calreticulin). (9) Downregulation of light chain β2M can lead to complete absence of HLA-I. (10) Microenvironmental conditions such as glucose deprivation, hypoxia, acidosis or excessive IL-10, TGF-β levels can also drive loss of HLA. β2M, β−2-microglobulin; ER, endoplasmic reticulum; HLA, human leucocyte antigen; IFNAR1, interferon alpha and beta receptor 1; IL-10, interleukin 10; ISRE, interferon stimulated response element; TAP1, transporter associated with antigen processing 1; TGF-β, transforming growth factor-β.
Non-classical HLA class I expression
In contrast to classical HLA-I molecules, non-classical HLA-E, HLA-F and HLA-G have immunosuppressive functions. HLA-E consists of a heavy chain, β2M and a bound peptide derived from leader peptide sequences of other HLA class I molecules. Its main function appears to be allowing NK cells indirectly to monitor expression of the other class I HLA molecules and prevent NK attack while simultaneously suppressing T cell cytotoxicity.8 It can be expressed in all nucleated cells at low basal levels, although it is often abundant on trophoblast and tumor cells.9 HLA-F and HLA-G were also originally identified as expressed on trophoblastic cells to provide fetal-maternal immune tolerance.10 11 HLA-F has structural similarities to HLA-E and classical HLA-I antigens although its complete function remains to be elucidated, as it is also expressed on activated immune cells.12 Unlike other HLA-I antigens, HLA-G comprises four membrane-bound and three soluble isoforms (sHLA-G) as a result of alternative RNA splicing. Except HLA-G1 and HLA-G5, the other five HLA-G isoforms do not associate with β2M.13 The isoform variations and structural complexity make precise HLA-G measurements particularly challenging.14 HLA-G is known to interact with all the main immune cell subsets and can inhibit their cytotoxicity via various receptors (Expression of HLA-G).15
What happens to HLA-I in cancer?
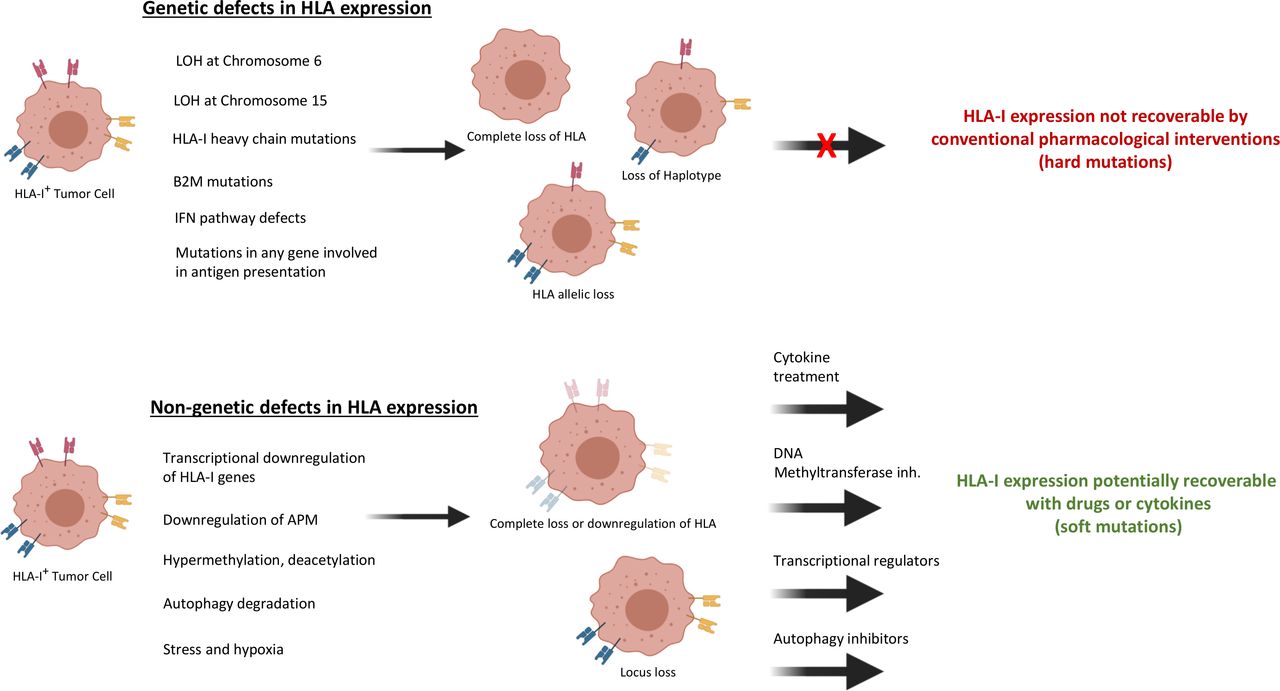
The multistep nature of the antigen-presentation process provides transformed cells with a variety of options to deregulate antigen presentation at genetic, epigenetic, transcriptional or post-transcriptional levels, and thereby to evade immune surveillance (figure 1) 16. In this section, we outline some of the known cancer-associated changes and assess their frequency of occurrence. Tumor heterogeneity means that a range of changes can occur within a single tumor 17, making it hard to state frequencies accurately, nevertheless it is possible to get some insight into the patterns of HLA loss that occur in different cancer types. The types of defects which lead to surface HLA-I aberration can be divided into two main groups: irreversible or reversible (sometimes also referred as hard or soft), reflecting whether restoration of HLA-I expression is possible following cytokine or pharmaceutical treatment.18
Irreversible HLA defects
Molecular changes to the coding regions of HLA or antigen presentation machinery (APM) component genes are often described as ‘irreversible’ defects. They are identified empirically by searching for mutations in HLA class I molecules using DNA sequencing. Recovery of HLA-I expression can be only possible when these mutated genes are compensated with intact ones by gene editing or delivery techniques.
HLA-I gene mutations
The most commonly observed genetic change is loss of heterozygosity (LOH) of the HLA-ABC genes located at chromosome locus 6p21 which is reported to occur at a frequency of around 40%–54% in non-small cell lung carcinoma (NSCLC) and 44% in colorectal cancer19 20 (figure 2A). The HLA gene is the most polymorphic gene in the human genome, essential for its role of presenting a wide range of peptides. LOH at chromosome 6 leads to complete loss of either parental or maternal HLA haplotypes. This means that the repertoire of peptides normally presented by that haplotype is lost, potentially preventing presentation of important neoantigens. Simultaneously maintaining expression of the HLA haplotype from the other parent staves off attack by NK cells. Perhaps the best-defined example of this arose from the TRACERx analysis of early-stage patients with lung cancer, where McGranahan et al showed that 40% of patients had HLA LOH, using a novel computational tool determining HLA-allele specific copy number.20 High levels of neoantigens predicted to bind to the lost HLA allele were identified. Due to its subclonal nature they inferred that HLA LOH was a late event in tumor evolution and occurred under selective pressure as just one mechanism for cancer cells to avoid presenting neoantigens and consequent immune elimination.

Schematic overview of genomic localization of HLA molecules and cisregulatory elements in promoter region of classical and non-classical HLAs. (A) Genes encoding HLAs and APM proteins are localized on the chromosome 6 short arm. (B) Classical HLA genes promoters comprise two major regulatory modules; NF-κB response element and ISRE consisting upstream nodule and SXY enhanceosome nodule. TATA and CCAAT elements controlling basal transcription of these genes. Non classical HLA-E and HLA-G promoter regions have some differences. HLA-E gene can be transactivated by NLRC5 and IFN-γ but not NF-κB. HLA-F regulatory nodules shows high homology to classical HLA-I. Differently, HLA-G promoter region has diverse binding sites such as HSE, HEF, RRE and PRE which overlaps TATA box. HDAC1 can interact with RREB1 increases chromatin condensation. HLA-G promoter NF-κB binding sites can only bind p50 homodimers therefore NF-κB has no transactivator function. NF-κB, kappa-light-chain-enhancer of activated B cells; APM, antigen presentation machinery; HLAs, human leucocyte antigen; HSE, heat shock response element; IFN-γ, interferon-γ; NLRC5, NOD-like receptor caspase protein 5; ISRE, interferon stimulated response element; PRE, progesterone response element.
Loss of a functional HLA alleles by mutation or gene deletion is another but less frequent structural defect. A recent analysis detected non-silent genetic mutations in HLA genes in approximately 3.3% of all tumors, with greater prevalence for head and neck, lung, stomach and colorectal cancer.21 Loss of a functional HLA allele through mutation would have similar consequences as HLA LOH, precluding presentation of immunodominant peptides such as MART-1 following loss of HLA-A2 in melanoma.22 A striking example was observed in a clinical study (NCT01174121) in which patients were screened for tumor antigen profile and then treated with ex vivo expanded antigen specific autologous T cells. One patient who responded to HLA-C*08:02 restricted KRAS G12D targeted T cells therapy subsequently encountered tumor relapse. Whole exome analysis revealed loss of the chromosome 6 haplotype encoding HLA-C*08:02 in progressing tumors.23 This case report shows how cancer cells can acquire significant genetic changes when under strong selective pressure by antigen specific T cells.
Mutations in B2M
Another important irreversible defect is the loss of β2M protein expression. This generally arises by a combination of two events; LOH on chromosome 15 (15q21) and mutation in the remaining β2M allele. This type of alteration results in complete loss of HLA presentation, as β2M is responsible for stability of the HLA-I complex.24 It has been suggested that LOH at chromosome 15 is an earlier event and selective pressure causes mutations in the second β2M copy which leads to complete loss of functional antigen presentation.25 Non-silent mutations in β2M have been reported in a broad range of patients26 and LOH at chromosome 15q21, assessed using two microsatellite markers, was found in 44% of bladder carcinomas (n=69), 35% of colon carcinomas (n=95), 16% of melanomas (n=70) but only 7% of renal cancers (n=45).27–29 Interestingly the rate of mutations was appreciably higher in microsatellite unstable disease (particularly colorectal, gastric and uterine cancers) and was associated with an increased overall burden of mutations for both microsatellite stable and unstable disease, commensurate with the role of the HLA system in presenting acquired mutations for immunosurveillance. Moreover, loss of B2M mediated acquired resistance to immune checkpoint inhibitors (ICIs) has been reported in clinical investigations.30–33
Mutations in interferon pathway genes
Type-I and type-II interferons are strong inducers of HLA-I expression and they have pro-apoptotic effects on tumor cells.34–36 Therefore, not surprisingly, they become important targets for tumors during immune evasion, disease progression and metastasis. Particularly, interferon-γ (IFN-γ) is master regulator of several APM genes and the most potent HLA molecule inducer. It is strongly secreted by activated T cells and upregulates expression of MHC class-I and MHC class-II transactivator (CIITA) proteins, NOD-like receptor caspase recruitment domain containing protein 5 (NLRC5) and MHC CIITA, respectively.37 The HLA heavy chains and APM promoters contain interferon stimulated response element (ISRE) region, the binding site of interferon regulatory factor-1 (IRF1) (figure 2), which is induced via the JAK/STAT pathway following IFN-γ binding to the IFNGR1 extracellular domain.38 In tumor cells, loss of function mutations in JAK/STAT or IRF1 genes can remove this responsiveness to IFN-γ, and JAK/STAT pathway mutations have been reported for several cancer types. They are associated with poor prognosis of the disease and resistance to immunotherapies.39–41 Alternatively, missense mutations in the IFNGR1 gene can cause loss of cell surface IFGNR1, thus inhibiting IFN-γ responsiveness.42 43 In addition, IFN-γ suppressor genes, SOC1 and PIAS4 have also been shown to be upregulated in patients who do not respond to checkpoint blockade.39
Mutations in genes involved in antigen presentation
Mutations in the more obvious HLA and associated genes may be only the tip of the iceberg. Acquired genetic mutations may also be present in other components of antigen presentation, for example in immune proteasomal degradation pathways44 (such as LMP2, LMP9, MECL-1) or the ER peptide loading complex (for example tapasin, ERp57, calnexin, calreticulin).45 46 Hence our knowledge of genetic changes leading to dysfunctional class I antigen presentation is likely still in its infancy, and there may be myriad other genetic changes exploited by tumors that we have not so far identified.
Reversible HLA defects
Loss-of-functional HLA presentation resulting from epigenetic silencing, transcriptional or post-transcriptional/translational modifications of the HLA genes themselves or of any other gene essential for antigen processing, are often termed ‘reversible’ defects. In principle, HLA function might be restored in these cancers using pharmacological interventions (Figure 3).

{kind=link}
{kind=link}
{kind=link}
Type of classical HLA-I alterations in cancer cells. Various HLA-I abnormal cancer phenotypes can form depending on whether these abnormalities are caused by genetic or non-genetic defects. The striking difference between these defects is genetic aberration-originated HLA-I dysregulation cannot be recovered by pharmaceutical or IFN treatment. The most frequently formed genetic abnormality is LOH at chromosome 6 which leads to loss of maternal or paternal HLA haplotypes. Heavy chain mutations or β2M mutations causes complete loss of HLA function. HLA allelic loss may occur in the case of locus-specific mutations. Any mutations in IFN signaling pathway or APM genes may lead to complete loss or down regulation of HLA-I expression. On the other hand, cancer-specific transcriptional down regulation of HLA-I genes or APM genes, epigenetic changes such as methylation or acetylation, autophagy-mediated degradation, stress and hypoxia can lead to HLA-I expression dysregulation. However, HLA-I can be recovered by using different treatment approaches in accordance with the specific type of defect. APM, antigen presentation machinery; β2M, β−2-microglobulin; IFN, interferon; LOH, loss of heterozygosity; HLA, human leucocyte antigen.
Epigenetic silencing
Histone deacetylation and DNA methylation are epigenetic gene regulatory mechanisms which are crucial for tissue specific gene expression and homeostasis. Histone deacetylase (HDAC) and DNA methyltransferase (DNMT) act as gene silencers by condensing the chromatin structure. Cancer cells can use these mechanisms to inhibit tumor suppressor genes or apoptosis47 alongside other mechanisms that can lead to down regulation of β2M and other APM proteins.48 49 Type-I and type-II IFNs levels were shown to be upregulated following inhibition of these epigenetic mechanisms.50 For example, decreased STAT1 expression in squamous cell carcinoma of head and neck (SCCHN) was associated with promoter methylation, and levels could be restored in SCCHN cell lines using the DNMT inhibitor azacytidine.51 Several studies have now shown that HLA expression can sometimes be restored using HDAC or DNMT inhibitors.52 Recently, synergistic effects of the US Food and Drug Administration (FDA) approved HDAC inhibitors and checkpoint inhibitors have been reported in preclinical studies, suggesting HDAC inhibitors can restore functional HLA in some patients53–56 and additional clinical studies are underway.57 58 At the data cut-off, objective response rates were reported as 31%, 19%, and 18% in patients with uveal melanoma (n=29), NSCLC (n=57), and melanoma (n=53), respectively, in phase 2 studies.59–61 It is perhaps worth noting that Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) has become an effective technology in cancer epigenetics research62 and it gives the opportunity to investigate small sample sizes, which is a common issue in clinical studies. Adapting such techniques to ongoing studies should provide a better understanding of the impact of epigenetic therapy on HLA-I expression.
Transcriptional silencing
Transcription of classical HLA and other antigen presentation pathway genes is controlled by two regulatory modules (figure 2). The upstream module consists of the enhancer A containing κB1 and κB2 binding sites for NF-κB family members p50, p65, and c-Rel and ISRE with binding sites for IRF1 and IRF2, together with binding sites for USF1 and USF2 and Sp1. The downstream ‘SXY’ module has binding sites for a multiprotein complex of RFX, CREB/ATF and NFY. It was recently reported that NLRC5 translocates to the SXY-module, forms an enhanceosome together with RFX-complex and activates HLA-I transcription.37 63 Together these two modules provide constitutive and regulatory control of classical HLA-I gene expression64 (figure 2B). Due to this transactivator role of NLRC5 it is suggested to be a key target for immune evasion in many cancer types.65 Analysis in large cohort human cancer samples revealed that high NLRC5 expression is correlated with higher HLA expression and better survival.66 Another regulator of HLA class I expression is suggested to be tumor suppressor Fhit protein.67 Although its mechanism has not been clearly identified, Fhit-transfected cancer cells recovered MHC-I expression independent of NLRC5. Additionally, loss of Fhit expression in human breast tumors is associated with loss of HLA-I molecules.68
The non-classical HLA-I molecules (notably HLA-E and HLA-G) have slightly different regulatory sequences, providing the opportunity for differential expression of classical and non-classical HLA genes through transcription factor variation (figure 2B). Both regulatory modules mentioned above can be identified in the HLA-E and HLA-F promoter regions, however in HLA-E the κB1 and κB2 and ISRE sites are different from the classical genes meaning that HLA-E is thought to be not induced by NF-κB, although an upstream GAS site provides strong inducibility by IFN-γ. HLA-E shows some induction by binding of NLRC5 to the SXY domain. The HLA-F promoter structure exhibits strong homology to classical HLA-I antigens, consisting of one NF-κB binding site followed by ISRE and SXY nodule. On the other hand, in HLA-G the κB sites bind only the p50 subunit of NF-κB, meaning the promoter is not activated by NF-κB, and part of the ISRE is deleted removing the sensitivity to IRF1 and meaning HLA-G is not induced by IFN-γ either.64 HLA-G appears to be completely non-responsive to NLRC5, although it is induced by the DNA demethylating agent 5-aza-2′-deoxycytidine.69 Distinctly, the HLA-G promoter region comprises heat shock response element (HSE), hypoxia stimulation response element (HSE), Ras response element and progesterone response element, showing that expression of HLA-G is regulated by several factors. This may give an advantage to HLA-G over other HLA-I antigens allowing better induction under stressful conditions.
Taken together this provides tremendous potential for transcriptional downregulation of classical HLA gene expression in cancer, perhaps with upregulation of non classical HLA. In line with these observations, low classical HLA expression in neuroblastoma was found to be associated with low transcriptional availability of NF-κB.70
Post-transcriptional silencing
Expression of HLA-I antigens is also controlled at the RNA level. Some HLA alleles have variable sites in the three prime untranslated region (3ʹUTR) that are recognized by RNA binding proteins and miRNAs. Particularly, non-classical HLA-I molecules have been shown to be strongly regulated by miRNAs. The high tissue tropism of HLA-G is not only related to its unique promoter region but also its distinctive 3ʹUTR.71 For example, inverse correlation between miR-628–5 p and HLA-G expression has been identified in renal cell carcinoma.72 Upregulation of some miRNAs has also been reported to affect classical HLA-I expression by silencing HLA or components of APM. For example, Mari et al reported that overexpression of miR-148a in esophageal carcinoma cell lines reduced expression of HLA-ABC. They also showed miR-125 decreased the level TAP2 which affected HLA expression.73 Similarly, Lazaridou et al found that miR200a-5p can target TAP1 and its overexpression in melanoma patients resulting in impaired HLA-I expression.74 In colorectal cancer cell lines overexpression of miR-27a repressed HLA-I expression by targeting calreticulin.75 The effects of miRNAs on cancer immune escape have been documented in more detail in a recent review by Yi et al.76
Another post-transcriptional mechanism that regulates HLA-I expression is mediated by RNA binding proteins. According to a study by Huang et al, aberrant expression of the RNA binding protein MEX3B in colorectal cancer patients downregulated HLA-A expression and led to resistance to immunotherapy. In a similar vein, The Cancer Genome Atlas (TCGA) melanoma database analysis indicates that anti-PD1 resistant melanoma patient tumor samples have significantly higher MEX3B expression levels compared with the checkpoint inhibitor responsive tumors.77 Interestingly, MEX3C, which is another member of the MEX3 family, has been reported to downregulate only HLA-A by binding its 3ʹUTR, without affecting other class I HLAs.78 Another RNA binding protein, the heterogeneous nuclear ribonucleoprotein R (HNRNPR) has been also shown to positively regulate classical and non-classical HLA-I expression by binding to their 3ʹUTR and stabilizing them.79
Microenvironmental regulation of HLA expression
There is a complex interplay between cancer cells and the tumor microenvironment (TME) which generally favors tumor progression by suppressing immune function.80 The best characterized microenvironmental conditions are hypoxia, acidosis, glucose deprivation and the presence of excess immunosuppressive cytokines. These conditions not only limit the activity of effector T cells but also dysregulate antigen presentation and HLA-I expression.81 For example, interleukin 10 (IL-10) and transforming growth factor-β (TGF-β) are two major immunosuppressive cytokines that are frequently overexpressed in the TME.82 IL-10 inhibits activity of Th1 cells, NK cells, macrophages and proinflammatory cytokines, such as IFN-γ. It has also been shown that IL-10 downregulates HLA class-I molecules, suppresses antigen presentation and induces HLA-G expression in cancer cells.83–85 Similar findings have been reported for TGF-β.86–88 A recent study showed that TGF-β inhibits MHC-I expression by downregulating β2M via a Smad-dependent pathway.89 Lee et al examined PD1 inhibitor-resistant melanoma biopsies and found that HLA-I downregulation was associated with TGF-β activity in 31% of progressive tumors.90
Uncontrolled tumor growth leads to insufficient blood perfusion which causes formation of hypoxic, glucose deprived and acidic conditions in the TME. Each of these conditions can induce ER stress and lead to accumulation of unfolded or misfolded proteins in the ER, which consequently, causes a significant disruption of antigen processing and presentation machinery.91 Once cells experience hypoxia, hypoxia-inducible factor 1-alpha accumulates in the cytoplasm, subsequently translocates to the nucleus and induces transcription of several genes.92 Recently, Marijt et al showed that hypoxic and glucose-deprived conditions disrupt IFNγ/STAT1 signaling by metabolic stress-induced activation of PI3K-Akt pathway.93 An earlier study reported that transcription of HLA-I heavy chains, TAPs and LMPs were downregulated in vitro and in vivo under hypoxia, while upregulation was observed when cells were later incubated with high oxygen levels.94 Intriguingly, non-classical HLA-E and HLA-G have been shown to be overexpressed on cancer cells due to hypoxia and glucose deprivation, providing another immune escape mechanism.94–96 Cancer cells tend to produce energy by glycolysis rather than oxidative phosphorylation even in the presence of oxygen (Warburg effect). This phenomenon results in accumulation of excessive concentrations of lactate as byproduct which decreases the pH in the TME. Low pH has been shown to abrogate IFN-γ secretion from NK cells and T cells which hinders IFN-mediated HLA-I expression.97 98 A similar scenario has been recently described for the physiological regulation of T cells in the lymph nodes, providing further insight into the critical role of pH on immune function.99 Neutralization of tumor extracellular pH remains as an intriguing approach to improve the outcome of cancer immunotherapy.100 101
Type-I IFNs (IFN- and IFN-β) activate an antiviral state in cells by upregulating HLA-I expression to promote antigen presentation,102 103 while simultaneously stimulating immune cells and mediating antiproliferative effects.104 One of the major cancer-specific type-I IFN signaling defects is TME stress-mediated down-regulation of interferon alpha and beta receptor 1 (IFNAR1),105 leading to disruption of antigen presentation both in cancer and antigen presenting cells and inhibiting IFN-induced apoptosis in cancer cells. Moreover, down regulation of IFNAR1 was reported to reduce intratumoral T cell viability and activity due to disruption of the STAT3/granzyme B pathway.106 107 A recent study by Cho et al showed that IFNAR1 deficient cancer-associated fibroblasts play a significant role in stromagenesis and contribute to rapid tumor growth.108 Conversely, when IFNAR1 is intact, conventional therapies can induce IFN-β and lead to induction of HLA-I expression.109 110 For example, Wan et al observed HLA-I upregulation in breast cancer cell lines following topotecan treatment due to elevated levels of IFN-β signaling.111 Similarly radiation induced IFN-β led to increased HLA-I levels and subsequently sensitized cancer cells to anti-PD1 therapy, an effect that was reversed by blockade of IFNAR1.112 IFN-β monotherapy was also reported to increase HLA-I expression in melanoma cell lines and upregulate presentation of tumor associated antigens, such as Melan-A/Mart-1, gp100, MAGE-A1, although this effect was not observed when cells were treated with IFN-γ or IFN-.113
Oncogene-mediated regulation of HLA expression
Oncogene activation can also modulate surface HLA expression (figure 1). For example, abnormal activity of the MAPK pathway in tumor cells has been reported to down-regulate antigen processing and presentation.114 The MAPK pathway consists of the Ras/Raf/MEK/ERK signaling cascade and can be activated by growth factor receptors, such as HER2 and EGFR, which are known to be selectively overexpressed in many tumor types. Activation of MAPK signaling, either by overexpression of growth factor receptors, or by gain-of-function mutations in signaling proteins can lead to loss of HLA.115–118 Similarly HLA downregulation and APM deficiencies were found to be more frequent in K-Ras mutated NSCLC and colorectal cancer,119 120 although application of MAPK or EGFR inhibitors could recover cell surface HLA expression and restore TAP1, TAP2, and β2M.120 121 Ras/MAPK activity has been correlated with reduced level of tumor-infiltrating lymphocytes (TILs) together with low HLA expression in triple negative breast cancer. Intriguingly, simultaneous inhibition of programmed death-ligand 1 (PD-L1) and MEK enhanced antitumor immune response in breast cancer-bearing mice.122 Similarly the oncogenic BRAF V600 mutation is very frequent in advanced melanoma and recent studies have shown that BRAF inhibitors can relieve immune suppression in TME by upregulating HLA expression.123 124 Bradley et al reported that in melanoma cells BRAF V600E mutation drives internalization of surface HLA molecules and subsequent degradation by endocytic compartments. This process related to phosphorylation of highly conserved Serine-335 site within the HLA-I cytoplasmic tail.125 Additionally, immunosuppressive HLA-G upregulation was detected in BRAF V600E mutated papillary thyroid carcinoma.126
Along with the MAPK pathway, the PI3K-Akt pathway is also activated by receptor tyrosine kinases and frequently dysregulated in cancer cells.127 Recent studies have shown that increased signaling of the PI3K-Akt pathway down regulates class I HLA expression due to a complex interplay between PI3K and STAT1.128 129 As discussed above, hypoxic and glucose-deprived conditions have been shown to induce PI3K-Akt activation, which may contribute to the loss of HLA in cancers.93 Pharmacological inhibition of PI3K restored IFNγ/STAT1 signaling transduction and surface class I HLA expression in vitro.93 129 A study by Sivaram et al investigating the impact of mutational activation of PI3K in an orthotopic pancreas mouse model showed that inhibition of the PI3K pathway increases MHC-I expression, leading to tumor regression due to T cell infiltration.130 Importantly TCGA database analysis also reveals a negative correlation between activated PI3K-Akt and HLA-I-mediated regression in head and neck cancer, lung squamous carcinoma, and pancreatic adenocarcinoma.129 130
Post-translational silencing
Recently, Yamamoto et al described another mechanism that cancer cells exploit to downregulate HLA-I expression. Even in tumors with intact HLA and APM genes, surface MHC levels could be significantly reduced by selectively targeting HLA-I molecules for lysosomal degradation via an autophagy dependent mechanism. Inhibition of autophagy using the anti-malaria drug chloroquine restored the cell surface MHC expression, improved antitumor T cell response in animals and synergized with immune checkpoint therapy.131
Cancer stem cells and HLA
Cancer stem cells (CSC) are thought to represent a minor component of many tumors, described to have crucial roles in formation, sustenance, metastasis and recurrence of the disease.132 A growing number of studies suggests that complete recovery can only be possible when CSC are eradicated.133 While the interaction between CSC and immune function remains to be explored, it has been suggested that they can evade immune surveillance by down regulating HLA-I expression.134 Morrison et al developed a CSC-enriched murine lung tumor sphere model and compared it with non-CSCs counterparts in terms of class I MHC expression level and susceptibility to antitumor immune response. They found that CSC-enriched tumors had lower MHC expression and were more aggressive in mice.135 In clinical settings, loss of class I HLA in CSC has been reported in surgical samples of glioblastoma, melanoma and colorectal cancers.136–138 Yang et al showed that HLA-I down regulation in glioma stem cells was associated with aberrant Wnt/β-catenin activity, which is a physiological regulator of pluripotency and self-renewal of stem cells. Treatment with HDAC inhibitors restored HLA-I expression partly by inactivating Wnt/β-catenin.139 Importantly, while ex vivo experiments have shown that CSC and also cancer cells can be susceptible to NK cell-mediated killing due to low HLA-I expression,137 140 141 these cells still survive and promote tumorigenesis in vivo, indicating the presence of another immune escape mechanism for CSC. In the next section, we discuss this pathological phenomenon of resistance of tumor cells to NK cell-mediated killing.
Upregulation of non-classical HLA-I molecules E, F and G
Tumorigenesis and fetal development show several similarities in their life cycles.142 Even though tumors are a complex process of unplanned cellular and molecular dysregulations, while pregnancy is a programmed physiological process, they both develop strategies to avoid immune detection while surviving in an oxygen-deficient microenvironment. Non-classical HLA-E, HLA-F and HLA-G can suppress immune cells by interacting with inhibitory receptors. Physiologically they are upregulated in placenta or trophoblast and help to protect the embryo against attack by the maternal immune system. Growing evidence indicates that cancer cells exploit this same phenomenon—to evade immune detection they not only down regulate classical HLA-I antigens, they also upregulate non-classical ones similar to fetal tissues.
Expression of HLA-E
HLA-E is a non-classical HLA class I heavy chain paralogue that exists on the cell surface as a trimer with β2M and a bound peptide for presentation. Under normal conditions HLA-E selectively binds and presents the peptides produced following post-translational cleavage of the conserved leader sequences associated with the other class I HLA molecules (including HLA-G) and its main function appears to be in binding the inhibitory receptor NKG2A/CD94 on NK and T cells, thereby allowing NK cells indirectly to monitor expression of the other class I HLA molecules and prevent NK attack.8 There are increasing data showing that tumors can exploit expression of HLA-E as a means to avoid recognition by NK cells, even when classical class I HLA is not present. Aberrant HLA-E expression has been detected in colorectal cancer (65%), gastric cancer (45%), ovarian (89%), breast (50%) and associated with poor prognosis.143–146 In a study evaluating an anti-NKG2A antibody (monalizumab) as a novel checkpoint inhibitor, André et al observed HLA-E expression in many different tumor types, sometimes at higher levels than PD-L1. Disruption of its binding to NKG2A with monalizumab boosted T cell and NK cell activity in vitro and in vivo.92 Intriguingly, it has been also suggested that HLA-E can present pathogen-derived or tumor-derived peptides to unconventional T cells147 148 and at least one report has shown high HLA-E expression in colorectal cancer patients associated with favorable prognosis.149
Expression of HLA-F
HLA-F is the least investigated and characterized non-classical HLA-I antigen. Physiologically it is known to be expressed on activated B cells, T cells and placental tissues.150–152 However, recent studies have shown that it can be upregulated in cases of infection, autoimmune disorders and cancer.153 HLA-F is expressed in two forms, as an empty open conformer which lacks β2M association or as a complex with β2M and peptide, similar to classical HLA-I molecules. However, it is still uncertain what mediate the transformation between these two forms. While open conformers can be recognized by activating or inhibitory KIRs, the peptide-β2M complex form has been suggested to bind inhibitory receptors ILT2 and ILT4.154 Indeed, while overexpression of HLA-F has been reported for various cancer types such as breast cancer (40%), NSCLC (24%), nasopharyngeal cancer (18%), esophageal squamous carcinoma (21%), and gastric cancer (43%) by immunohistochemistry (IHC) analysis, the impact on disease prognosis remains controversial.155–160 It is important to note that analysis of HLA-F mRNA levels may give misleading information as HLA-F is partially retained intracellularly.
Expression of HLA-G
HLA-G expression in tumors has been described as another immune escape mechanism due to its negative regulation of both adaptive and innate immunity.161 HLA-G binds to several receptors, such as KIR2DL4 (expressed by NK cells), ILT4 (expressed by APC) and ILT2 (expressed by all immune cell subsets)162 and has been implicated in multiple stages of immunoediting.163 Notably, HLA-G expression is induced under hypoxia, which is often present in advanced cancer,95 164 and overexpression has been reported for several cancer types, including esophageal squamous cell carcinoma (90%), colorectal cancer (70%), hepatocellular carcinoma (65%), glioma (70%), breast cancer (66%), gastric cancer (75%), melanoma (36%), ovarian cancer (55%), and lung cancer (75%).165–172 Besides, HLA-G mRNA expression data provided from a TCGA database cohort confirmed that HLA-G gene is expressed in several tumors. A correlation between high proinflammatory transcripts and high HLA-G mRNA expression was used to infer that HLA-G may be important in controlling immune recognition of proinflammatory events during tumor progression.173 However, HLA-G mRNA data should be interpreted cautiously as post-transcriptional regulation, particularly by miRNAs,174 175 is thought to be central to regulating protein expression. On the other hand, a sHLA-G protein form has been detected in plasma, serum and malignant ascites of cancer patients and correlated with poor prognosis.176–179 Taken together, HLA-G has been described as a new checkpoint inhibitor and molecular target for immunotherapy.180 181 It has already been proposed that renal carcinoma patients may benefit from anti-HLA-G/ILT2 and anti-HLA-G/ILT4 therapy.182 183
MHC-I status of preclinical animal models
Syngeneic animal cancer models are a staple component of pre-clinical development for cancer immunotherapies, however, the MHC-I status of the animal tumor cells and the degree to which this may reflect human patients is rarely discussed. Among the most popular preclinical models, B16 murine melanoma cells normally have low levels of basal MHC-I expression both in vitro and in vivo, but this is substantially upregulated by exposure to interferons, reversing their intrinsic insensitivity to cytotoxic T cells.184 185 Accordingly, it may not be surprizing to observe favorable efficacy in B16 models while exploring the use of immune adjuvants that trigger interferon pathways such as STING agonists.186–192 Although both MHC-I upregulation and CD8+ priming may be contributing to the therapeutic outcomes observed in these studies, the former is seldom discussed.
For melanoma patients who have a reversible IFNγ-reversible MHC (HLA) phenotype, B16 tumors might be an appropriate pre-clinical model. It follows that, ideally, patient populations of the appropriate HLA status would be recruited for clinical studies supported by relevant preclinical models. Other syngeneic models frequently used in immunotherapy studies, including CT26, RENCA, EMT6, 4T1, MC38 and EL-4, have much higher levels of basal MHC-I function relative to B16193 194 although whether this makes them more or less relevant to the diverse population of patients is currently unclear.
When it comes to animal models of human cancer that have more durable and defined levels of dysfunction in antigen presentation, the choices are rather slim. B78H1, a subclone of B16 is particularly interesting because its low basal MHC-I expression is not restored by gamma interferon. Furthermore, it also expresses Qa-2 the murine homologue of HLA-G.195 This could make it a very suitable model for some patients.
Overall interest in murine homologues of non-classical HLA-E and -G (known as Qa-1 and Qa-2, respectively)196–198 is extremely limited, despite their likely importance in the human setting. They are seldom considered when interpreting immunotherapy data in the most frequently used translational models.
In summary, it is not obvious that the antigen presentation status of commonly used animal models reflects the phenotypes found in clinical disease. We believe it is likely that increased focus on aligning antigen presentation competencies in laboratory models and patients could improve the effective translation of new immunotherapies.
Effects on clinical practice
Insights into the critical role of the immune system in prevention of tumorigenesis paved the way for development of several immunotherapeutics, such as checkpoint inhibitors, cancer vaccines and adoptive T cell therapy. During the past decade use of checkpoint inhibitors was approved by FDA for the treatment of metastatic melanoma and undoubtedly brought new hope to many. However, the number of patients who benefit from the treatment has remained low, according to a recent study the number is only about 12%.199 Various factors can influence the responsiveness to immunotherapy, and it is important to identify these factors to guide efficient development of new drugs capable of activating the immune system against the cancer.
HLA-I diversity as a predictive biomarker for immunotherapy
Tumor mutation burden (TMB) and inflammation status of the TME have been proposed as potential predictive biomarkers of response to ICIs.200 201 However, high TMB has been detected both in ICI responsive and non-responsive patients, indicating an important impact of other parameters.202 203 Notably, recognition of the HLA peptide complex by the TCR is essential for T cell mediated cell killing. Illustrating clinical importance, HLA-I heterozygosity has been correlated with improved overall survival of patients who received checkpoint inhibitors, while HLA-I homozygosity in at least one locus reduced survival significantly.204 Moreover, an association between high mutational burden and better survival was found to be further increased in patients with all loci HLA-I heterozygosity, indicating the importance of diverse HLA-I expression on outcome of checkpoint blockade therapy.204 The TRACERx study showed that cancer cells with LOH at HLA loci had higher mutational burdens, however, loss of HLA-I meant that these neoantigens could not be presented to T cells which could contribute toward resistance to ICIs. Other groups also observed similar correlations between high TMB and HLA-LOH, and combinational analysis of TMB and HLA-I heterozygosity has been proposed as a possible predictive biomarker for responsiveness to ICI therapy.205 206
Prognostic effect of HLA-I status
Although the vast majority of studies so far have shown that loss of HLA-I is correlated with poor overall survival, some opposite results have also been published even for the same types of cancer. For example, Hiraoka et al stated in their retrospective study that high HLA levels shorten the overall survival of patients with pancreatic ductal adenocarcinoma (PDAC), but in an earlier study, Imai et al demonstrated that HLA loss was associated with poor survival in PDAC patients.207 208 A similar discrepancy has been reported for gastric patients,209 210 but in these two studies the HLA-I levels were investigated in different stages of the disease. While in advanced stages of gastric cancer (MSI-high) HLA-I expression was not a prognostic factor, in earlier stages of gastric cancer (MSS and MSI-low) loss of HLA-I caused a worse survival rate. Reports for colorectal cancer and esophageal cancer are other examples of controversial observations in terms of HLA status and disease prognosis.211 212 The reason for high HLA expression to be associated with poor prognosis was mostly considered to relate to NK-cell inhibition. However, taken together, the most predominant observations are that loss of HLA-I combined with high PD-L1 expression levels gives poor prognosis and that there is a positive correlation between classical HLA-I expression and the density of TILs in the TME. Clinical implications of HLA-I status are summarized in table 1.
Clinical implications of tumor HLA-I expression
HLA analysis methods in clinical studies
Standardized protocols for detection and analysis of functional HLA are urgently needed. The most commonly used HLA analysis technique is IHC, and this gives no real indication of functionality.213 IHC tests also face several practical issues, such as preparation of tissue sections, selection of appropriate antibodies and determination of cut-off value for assessing positivity. Cut-off values are usually set according to HLA-I expression levels in tumor stroma or adjacent tissues, however, there is no established guideline for scoring and interpreting the results. Moreover, when non-classical HLA-I molecules are taken into account it becomes even more complicated. The majority of commercially available antibodies against non-classical HLA-I antigens are reported to show low specificity, meaning that HLA-E, HLA-F or HLA-G expression may be inaccurately assessed.214 On the other hand, whole genome sequencing and whole exome sequencing are next generation sequencing approaches which have been used to detect HLA-I mutations. Targeted sequencing, such as MSK-IMPACT panel is also widely used to capture HLA-A, -B and -C.215 One of the greatest advantages of these techniques over IHC is the ability to detect allele-specific losses. As mentioned above, particularly during selective pressure, HLA alleles capable of presenting neoantigens may be lost, and a change that is not detectable by IHC. The great majority of the studies, as shown in table 1, were conducted with IHC analysis, which may in fact be one of the main reasons restricting us from seeing the true picture of the consequences of HLA loss. Importantly, IHC does not give any insight into whether loss of HLA is based on irreversible or reversible defects. This simple information would provide a platform for more rational individualized treatment design.
Conclusion
Major advances in immunotherapy in recent years have brought new perspective to cancer treatment. However, it is now clear that the number of patients responding to immunotherapy is relatively limited. Moreover, acquired resistance in patients who initially responded to treatment reveals an important obstacle that needs to be solved. Although the resistance mechanisms developed by cancer cells against immunotherapy have not been fully characterized, cell surface HLA-I expression and antigen processing, which are indispensable for normal immune function, undoubtedly play an important role. Antigen processing and presentation is a mechanism which needs harmonious function of several different cellular compartments, providing an array of possibilities for cancer cells to disrupt the system.
Genetic mutations in HLA-I expression or in APM compartments are classified as irreversible defects. In these cases, either HLA function must be restored using gene therapy or gene editing techniques, or treatment approaches should focus on classical chemoradiotherapy or the use of HLA-independent immunotherapy, such as CAR-T cells or bispecific T-cell engagers. Each of these approaches is showing rapid progress currently, and careful stratification of patients for HLA function should enable more precise scrutiny of their mechanisms of action and further accelerate clinical progress.
The more common mechanisms of HLA-I losses are reversible defects, involving transcriptional, post-transcriptional or epigenetic changes, where the HLA-I level may sometimes be restored by pharmacological intervention. Particularly, the application of type-I or type-II interferons often results in a significant increase in HLA-I levels in many cells with such reversible defects. Likewise, in recent years, preclinical epigenetic therapy approaches, such as HDAC inhibition, have also been shown to increase cell surface HLA-I levels and to exhibit synergistic effects with immunotherapy.
The other side of the medallion reflects the non-classical HLA-I antigens, whose impact on tumorigenesis remains controversial. The number of reports indicating a role of non-classical HLA-I molecules on cancer development and immunoediting is increasing rapidly. Some studies even suggest that HLA-E or HLA-G are expressed at higher levels than PD-L1 and therefore may play a substantial regulatory role as immune checkpoints.181 216 In future clinical studies, HLA-E and HLA-G levels should be routinely analyzed to generate more extensive data to underpin rational treatment strategies. Undoubtedly, the critical point here is that a standardized analysis protocol has not yet been established for detection of classical and non-classical HLA-I antigens. There is also an urgent need for development of HLA-E and HLA-G specific monoclonal antibodies to be used routinely in clinical settings.
Immunotherapy, like most cancer drugs, can cause serious undesired side effects. Checkpoint inhibitor antibodies have become the most applied type of immunotherapy in the clinic and also the most widely investigated immunotherapy approach in preclinical studies. Most studies focus on how to increase the efficiency of checkpoint inhibitors or to determine which other treatments can create synergistic effects to enhance their benefit. However, still there is a need for predictive biomarkers for selection of patients who will receive the therapy. Although HLA-I expression profile alone does not seem to be a sufficient parameter for successful ICI response in most of the clinical studies, it has a predictive value when tumor PD-L1 levels and TMB are considered together. However, most of the clinical observations are made on the basis of small biopsy materials from single tumor sites, despite the heterogeneous nature of cancer. In addition, the fact that a functional HLA-I analysis method has not yet been established probably underpins the poor understanding of the real effect of classical HLA-I loss or non-classical HLA-I upregulation on patients' response to immunotherapy.
Ethics statements
Footnotes
Contributors All of the authors contributed to the production of ideas supporting this manuscript and each of them was also involved in helping to write the manuscript.
Funding This study was funded by Cancer Research UK (C552/A29106).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.