Article Text

Abstract
Introduction More than 2 million individuals in the USA have an opioid use disorder (OUD). Methadone maintenance treatment is the gold standard of medication-based treatment for OUD, but high-dose methadone is associated with cardiotoxicity and respiratory complications, among other side effects. These adverse effects make enhancing the effectiveness of lower doses of methadone an attractive therapeutic goal. Long recognised for its capacity to enhance treatment outcomes for a wide range of neuropsychiatric disorders including pain, the placebo effect offers an as-yet untested avenue to such an enhancement. This approach is particularly compelling given that individuals with substance use disorder tend to have higher salience attribution and may thereby be more sensitive to placebo effects. Our study combines two promising clinical methodologies—conditioning/dose-extension and open-label placebo—to investigate whether placebo effects can increase the effective potency of methadone in treatment-seeking OUD patients.
Methods and analysis A total of 120 newly enrolled treatment-seeking OUD patients will be randomly assigned to one of two different groups: either methadone plus daily placebo dose-extension (PDE; treatment group) or methadone/treatment as usual (control). Participants will meet with study team members five times over the course of 3 months of treatment with methadone (baseline, 2 weeks, and 1, 2 and 3 months postbaseline). Throughout this study time period, methadone dosages will be adjusted by an addiction clinician blind to patient assignment, per standard clinical methods. The primary outcome is methadone dose at 3 months. Secondary outcomes include self-report of drug use; 3-month urine toxicology screen results; and treatment retention. Exploratory outcomes include several environmental as well as personality factors associated with OUD and with propensity to demonstrate a placebo effect.
Ethics and dissemination Human subjects oversight for this study is provided by the University of Maryland, Baltimore and University of Maryland, College Park Institutional Review Boards. Additionally, the study protocol is reviewed annually by an independent Data and Safety Monitoring Board. Study results will be disseminated via research conference presentations and peer-reviewed publications.
Trial registration number NCT02941809.
- opioid use disorder
- methadone maintenance
- placebo effects
- opioids
- heroin use disorder
- substance misuse
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first randomised controlled trial designed to assess whether a combined conditioning paradigm and open-label placebo can be harnessed to enhance treatment outcomes in opioid use disorder (OUD).
By employing an open-label transparent design, this study avoids the problematic ethical issues that would arise surrounding concealed or deceptive placebo administration, thus preserving patient autonomy and patient–clinician communication.
Urine specimens will be tested for 240 substances, thus yielding a comprehensive picture of the opioids, new psychoactive drugs and pharmaceutical drugs recently used by patients.
Additionally, a comprehensive patient self-report drug use instrument affords the unique opportunity to assess lifetime and current patterns of licit and illicit substance use (including prescription opioids), which can be used both to determine premorbid drug use patterns and to assess the accuracy of patient self-reports of recent drug use.
As a pilot proof-of-concept study designed to test open-label placebo conditioning on OUD treatment outcomes, this study does not incorporate closed-label (blind) treatment arms; we plan to address this limitation in future follow-up studies.
Introduction
Between 2001 and 2016, the number of opioid-related deaths in the USA increased by 345%, from 9,489 to 42,2451. The incredible surges in overdose deaths and in the prevalence of opioid use disorder (OUD) have caused many federal and state agencies to identify this epidemic as one of the largest looming threats to public health today.2
Methadone maintenance treatment (MMT) is the most highly researched and evidence-based treatment for OUD and has become a mainstay in treatment.3 At appropriate doses, MMT is associated with significant improvements in several outcomes, including decreased drug use and crime, and increased positive health outcomes.3–5 Yet there is great individual variability in MMT response. While some patients can fare well for years on end with low to moderate doses in the range of 30–60 mg/day,6 ,7 many patients need much higher doses of methadone to control craving and drug-seeking behavior.8 9 Unresolved medical debates on whether ‘more is better’ provide no clarity on this issue, and with no generally accepted optimal dose prescription, clinicians titrate MMT dose to a subjective patient behavioural effect—a practice that sometimes translates to the prescription of very high doses of methadone.7
There are several reasons to give serious consideration to adjunctive treatments aimed at prevention of methadone dose escalation. For many patients, the high doses of methadone that seem to be needed for full therapeutic effect come at an unfortunate cost: side effects such as constipation, sedation, nausea and sweating may be so great as to be a major determinant in treatment failures.10 More alarmingly, higher methadone doses have been associated with risk factors for arrhythmias, such as QT interval prolongation and Torsade de pointes,11 and reports of increasing methadone-related deaths have led to greater scrutiny of methadone dosing practices.12 13 And from a patient perspective, an estimated 30% of MMT patients have severe anxiety related to MMT detoxification due to fear of withdrawal and relapse14 15: concerns that theoretically could be eased if those patients could be effectively treated at lower MMT doses. Collectively, these various issues and considerations provide ample rationale to explore options to increase the effectiveness of lower doses of methadone.
One plausible adjunctive behavioural treatment involves placebo effects. Recent investigations have yielded greater appreciation of the therapeutic potential of harnessing placebo responses that otherwise occur naturally within the frame of medical treatment. Defined as the positive health outcomes derived from an inert substance or device used in the context of medical treatment,16 17 placebo effects are guided by an individual’s conscious or unconscious expectation of salubrious effects and can yield very powerful determinations of health outcomes across many different diseases and encounters.16–19 Studies spanning two decades have shown that it is possible to condition the opioidergic system, the main neurotransmitter receptor system involved in addiction to drugs like heroin and prescription opioids. For example, placebo responses can be elicited by pairing morphine with placebo—an effect that is dependent on the strength of the association paradigm used to create the conditioned response.20–23 Although groundbreaking, clinical practice translation of these studies is limited by methodology incorporating deception: researchers told participants that they would receive drug when in fact they were to receive placebo, and vice versa.
We know of two promising strategies for ethically harnessing placebo effects. The first employs principles of Pavlovian conditioning.24 By pairing placebo pills and clinical contextual cues (conditioned stimuli) with a physiologically active treatment (unconditioned stimuli), researchers have shown that medication dosages can be lowered without decreasing treatment efficacy.25–29 This strategy is often referred to as placebo ‘dose-extension’, due to the fact that the placebo pill can be used to ‘extend’ the efficacy of the medication with which it was paired, and subsequent placebo administration can produce therapeutic effects.
A second strategy is known as open-label placebo administration, in which the placebo is identified as such. Patients are usually told that ‘we know that placebos have powerful effects in double-blind trials and we want to test whether placebos work even when patients know that they are taking placebos’.30 31 This approach has yielded positive results in a variety of somatic and pain-related conditions.32–36 Researchers are proposing that open-label-induced placebo effects may involve aspects of error prediction processing.30 37 Irrespective of mechanism, the available data suggest that open-label methods work, and that they provide a solution to the ethical dilemma of patient-blinded placebo delivery.
In the context of substance use disorders, the placebo effect is interesting due to prima facie overlap in the genes and brain substrates implicated.38 On a practical level, the case for harnessing placebo effects in addiction treatment is supported by a long line of research demonstrating that such effects are strong in drug-dependent individuals. For example, individuals dependent on nicotine,39 40 alcohol41 42 and marijuana43 show differential drug consumption and/or subjective drug effects based on experimentally manipulated expectations. In one of the earliest studies demonstrating this phenomenon, Marlatt showed that drinking behaviour in alcohol-dependent subjects could be manipulated by beliefs concerning the alcohol content of the beverage: when expecting to sample a drink containing alcohol, subjects drank almost twice as much as those expecting to receive only non-alcoholic beverages.44
In a pivotal study, using a ‘balanced placebo’ design, Volkow and colleagues45 administered placebos to both cocaine abusers and non-drug abusing subjects and found a significant effect of modulating expectations; brain metabolic changes were about 50% greater when the subjects were informed about receiving drug, in comparison with the group of subjects who were informed about receiving placebo.38 45 Intriguingly, methadone-treated OUD patients might be particularly sensitive to placebo effects relevant to their treatment.46 To date, however, no group has explicitly tested whether these placebo effects could be used to improve medicalised addiction treatment outcomes. Further, no study has investigated the efficacy of either conditioning or open-label placebo strategies in a methadone maintenance context. This study uniquely combines two validated approaches to harnessing placebo effects in what we call an ‘open-label conditioning dose extension with placebo’ paradigm, or, more succinctly, an ‘open-label placebo dose-extension (PDE)’ paradigm.
Objectives
The broad goal of this study is to improve treatment outcomes for OUD patients who are newly enrolled in a daily outpatient MMT programme. Specifically, we hypothesise that an open-label PDE paradigm will obviate higher-dose methadone treatment for a significant portion of new initiates and will thereby reduce methadone-associated side effects, with no concomitant change in outcomes such as treatment retention, drug use, self-reports and clinical observations of withdrawal, craving or quality of life.
We plan to recruit 120 participants and randomly assign them to one of two conditions: open-label PDE (group PDE) plus methadone or methadone/treatment as usual (TAU). We will follow them for 3 months for a total of five in-person meetings (baseline, and 2 weeks, 1, 2 and 3 months postbaseline). For the first 2 weeks, we will implement principles of pharmacological conditioning24 47 whereby placebo pills are temporally paired with the oral methadone hydrochloride solution that is provided to patients at the clinic (conditioning phase). Having established an association and contextualised the placebo as part of the therapeutic experience, placebos are then used as a dose extension (PDE) pill (dose extension phase, week 3 up to 3 months). Additionally, we are applying an open-label paradigm, giving participants information concerning the placebo pill in an honest and transparent manner. Our primary outcome is methadone dose 3 months after (baseline) entry into treatment; secondary outcomes include several measures of treatment success including comprehensive urine toxicology screens, self-reported drug use and treatment retention. We are also capitalising on this unique patient access opportunity to measure several personality and environmental factors associated with OUD, as well as factors associated with placebo response.
Methods and data analysis
Study setting
Our ongoing study takes place at the University of Maryland Drug Treatment Center, an urban clinic located in West Baltimore, Maryland. The clinic is open 6 days a week (excluding holidays), and in addition to medication-based treatment, the clinic provides counselling and psychiatric services. The majority of our patients reside within one of five zip codes that immediately surround the clinic address, and present to the clinic either by referral or self-admission. Approximately five new patients are enrolled into MMT per week.
Patient and public involvement
Neither patients nor the public were involved in the design, recruitment or conduct of the study.
Patient recruitment
Study participants will be 120 men and women OUD adults newly admitted to the UM Methadone Treatment Programme (MTP). New patients are recruited on the first day of treatment in the clinic (day 0). At the end of initial intake procedures of the first day, the programme intake coordinator asks new patients if they are interested in hearing information about a study that is testing a novel approach to enhancing methadone treatment, for which they would receive compensation. A member of the study team (either the P.I. (AMB) or the primary research coordinator (TOC)) contacts the intake coordinator to receive names of patients who are newly enrolled for MMT treatment and are willing to hear about the study.
Patients who indicate their interest are brought to a private interview room located near the methadone dosing area, where they are screened for eligibility, informed about the study and conceptual basis of the placebo effect and consented for participation. The study is registered with ClinicalTrials.gov. All procedures are performed in accordance with the relevant international and local guidelines and regulations for human research (UMB IRB Protocol # HP-00070829). A written informed consent is obtained from each study participant.
Eligibility criteria
Participants are included in the study if they are newly enrolled (admission within the same day) in the MTP, have not had very recent experience with MMT in a clinic setting (within the last 3 weeks) and do not have any extenuating factors that would have a strong influence on clinical methadone dose determination. Inclusion criteria include (1) adult (aged 18 years or over) and (2) newly admitted to the MTP. Exclusion factors include (1) pregnancy, (2) treatment transfer (patients who have initiated methadone treatment course at another methadone treatment facility), (3) hospital transfers (patients who initiated methadone treatment course in a hospital setting) or (4) criminal justice system referrals.
Study design and procedures
Randomisation and treatment allocation
Prior to study inception, random treatment allocation was generated by an independent investigator, and consists of sequentially numbered opaque envelopes containing treatment assignments drawn from a computer-generated random number sequence. These numbers are used to assign participants to either the open-label PDE arm (PDE group) or a TAU arm, and two stacks of envelopes were created to ensure an even distribution of men and women (n=30/group/sex for a total of 120 random treatment allocations).
Treatment allocation occurs after completion of the assessments. The investigator performs allocation by pulling an envelope from the top of the sex-specific stack. Following day 0 study procedures, and just prior to the first dose of methadone at the treatment window, the investigator conducts a treatment assignment ‘reveal’, opening the envelope and letting the patient know the group to which s/he has been assigned.
Blinding
Clinic staff are independent of the research study implementation. Correspondingly, members of the study team responsible for administration of assessments, delivery of placebo pills or data analysis play no role in dose increase/decrease determinations. Methadone dose adjustments are made based on two criteria: (1) scores on a validated subjective withdrawal symptom checklist and (2) treatment team consensus. (1) The subjective opioid withdrawal scale (SOWS) is an assessment of the severity of symptoms of withdrawal and is delivered outside of the study frame (the SOWS measurements that are obtained as part of the baseline, 2 week and 1-month, 2-month and 3-month study team meetings are distinct, kept separate from the clinical SOWS assessment for dose change determination). All patients in the clinic are asked to submit their responses on this checklist at a time point corresponding with their achievement of an initial stabilisation dose, generally 2–4 weeks following entry into treatment. This assessment is considered as one factor in dose change determinations. (2) Treatment teams meet weekly to discuss individual patients’ progress, and consensus must be obtained between the treating physician, the counsellor and the nurse practitioner (NP) to recommend a dose increase. Primary goals in increasing the methadone dose include suppression of withdrawal symptoms, tempering of intrusive drug cravings and agonist blockade. Physicians and the NP evaluate and document the relative risks and benefits of any proposed dosage change, with attention paid to several factors including over-sedation, drug–drug interactions, cardiac side effects and adherence to daily treatment. Treating physicians and the NP, including the facility’s medical director (AG) are all blind to study enrolment and randomisation. Patient study participation is not discussed during treatment team meetings, and the counsellors, NP and physicians are asked not to probe patients about their involvement and experience with the study. Thus, it is unlikely that a physician or NP would become unblinded to treatment allocation. Regardless, given the myriad variables that determine methadone dose changes, it is unlikely that this knowledge would factor in the calculus of whether to make a dose adjustment, as the clinic’s standard of care dictates that the participant’s well-being is the primary consideration in any clinical course of action. If, however, a physician or the NP becomes unblinded to a patient’s study treatment allocation, they are asked to communicate that to a member of the study team.
As an open-label clinical trial, it is not possible to blind participants to their treatment allocation. However, the scripted information that is delivered to the patients as part of the informed consent procedures (detailed in the Script and study information provided section) do not guide participant expectations regarding the effects that the PDE intervention may have on methadone dose per se. Instead, participants are informed of the non-specific therapeutic benefits that a pharmacologically conditioned PDE protocol may afford, with the primary outcome of interest (methadone dose) never explicitly mentioned to the participant. Additionally, because treatment allocation occurs only after day 0 (baseline) assessments are complete, study team members are blind to treatment assignment for all of day 0 procedures. Finally, data analysts are blind to treatment allocation.
Script and study information provided
Patients are fully debriefed of all study procedures during an informed consent process. Participants are informed that their participation in the trial will have no effect on ongoing treatment afforded by the clinic, and further, that they have the right to withdraw from the study at any time with no impact on their clinical treatment. During and following consent, the notion is reinforced to the patients that the research study is ‘designed to investigate the efficacy of methadone treatment that is enhanced via inner healing processes using placebo effects’. As implemented in previous studies,31 an Institutional Review Board (IRB)-approved script is used as a conversational guide to inform patients of the study rationale and procedures. This script has a positive framing and describes in lay terms the science that underlies placebo effects and pharmacological conditioning, with an aim to facilitate the placebo response in a non-deceitful manner. Following the conversational reading of the script, the investigator asks the participant to view a video of a CBS New York News piece48 that describes scientific studies of the successful use of the placebo phenomenon as a therapeutic intervention for irritable bowel syndrome. The participant then completes the day 0 (baseline) assessments (described in the Placebo pills section).
Placebo pills
The PDE pill is produced by the University of Maryland School of Pharmacy Good Manufacturing Practice facility. The pills are composed of microcrystalline cellulose PH-102, magnesium stearate and D&C Red 7 Ca Lake (inert chemicals and a food colourant, ingredients commonly contained in placebo pills manufactured by the pharmaceutical industry). PDE pills are stored in a locked medications cabinet maintained within the nurses’ station.
Intervention: daily PDE pill dispensing
Following treatment assignment on day 0, the investigator fills a placebo pill dispensing form indicating treatment assignment, and the patient is walked to the methadone dosing station. Placebo pills are stored and dispensed by the nursing staff of the MTP. If the patient is in group PDE, the investigator observes the participant taking the PDE pill. In phase I of the study (first 2 weeks), participants assigned to group PDE are given one pill, to be taken concurrently with the methadone. In phase II (3 weeks up to 3 months), PDE participants continue to take the single (morning, or AM) pill, and are given a second pill in a bottle as a take-home. They are instructed to take this second pill 12 hours following the first pill, ‘at home, or wherever they may be’. Participants are asked to return the take-home pill bottle every day for refill. Circumstances may occur under which a participant may need to be withdrawn from the protocol and include not following instructions given by team members, or repeatedly missing appointments without contacting study staff. Adherence to the instructions to take the AM pill will be monitored by study team nursing staff.
Urine toxicology screens
Urine drug screening occurs via two methods: (1) a point-of-care Quik-tox screen (11 panel; LabCorp) conducted by clinic staff, results of which are used for clinical decision making, conducted at baseline and then at monthly (random) intervals postbaseline; (2) and liquid chromatography–tandem mass spectrometry (LC-MS/MS) testing for a panel of >240 drugs, including new psychoactive substances, as well as other illicit and prescription drugs. All testing is conducted by the Division of Forensic Toxicology, Armed Forces Medical Examiner System and coordinated by the University of Maryland, College Park Center for Substance Abuse Research (CESAR) staff. Urine is collected from participants during each of the five meeting times, and the Quik-tox screen is conducted only on samples from meeting one (baseline); the LC-MS/MS testing is conducted on all samples (baseline and 2 weeks, 1, 2 and 3 months postbaseline screening).
Primary outcomes
The primary outcome is final methadone dose at the 3-month time point. MTP clinicians adhere strictly to the Substance Abuse and Mental Health Services Administration’s standards for medication-based treatment of OUD. Patients entering treatment are given an initial evaluation that results in the prescription of an induction (initial dose ≤30 mg/day, followed by a gradual dose up-titration) and initial stabilisation dose (typically 50–70 mg), which is usually reached by the third week of treatment. At 2–3 weeks, treatment progress is evaluated, and an order is written to either continue or increase the initially prescribed maintenance dose. As noted above, clinicians are blinded to the patient’s experimental-group assignment. In the MTP, ~75% of new patients who enter for methadone treatment are given a recommendation to titrate up to a higher dose (personal communications with staff). We will be documenting the induction (starting), maintenance as well as the ultimate stabilisation dose. These data are obtained from patient charts documenting daily records of methadone dose dispensed.
Secondary outcomes
Secondary outcomes include self-report of drug use, baseline and 3-month urine screen results, and treatment retention. Self-report of drug use is assessed with an instrument developed by CESAR that asks lifetime and recent (last 24–48 hours) use of over 50 different licit and illicit drugs. Drug toxicology results of the urinalyses conducted by the Methadone Clinic (monthly random drug testing) and the independent research laboratory, as well as treatment retention (number of days in treatment prior to discharge) will be measured as secondary outcomes at baseline and at the 3-month time point. Other self-report outcomes include scores on withdrawal and craving scales, quality of life and sleep patterns.
Exploratory outcomes
The unique longitudinal nature of this study allows us to assess several factors of interest, including personality factors associated with SUD and those associated with propensity to demonstrate a placebo effect, placebo intervention expectation and compliance, as well as measures of impulsivity and catastrophising. All assessments are administered by a member of our study team.
Clinical assessments
Baseline and postbaseline drug use history and assessment (CESAR): a comprehensive assessment of substance use history and treatment, environmental and psychosocial risk factors, and recent use of >30 commonly used licit and illicit drugs; (see table 1).
Adapted credibility/expectancy questionnaires 49: a three-item assessment of participant beliefs that the PDE would improve their symptoms (day 0), and a 3-item to 4-item group-dependent assessment of participant beliefs that the PDE is helping or would help improve their symptoms (days 14, 28, 56 and 84).
Subjective Opioid Withdrawal Scale (SOWS)50: a 16-item patient self-report instrument to assess common subjective symptoms of craving and withdrawal.
Objective Opioid Withdrawal Scale (OOWS)50: a 13-item clinical assessment of physiological signs of withdrawal.
Craving assessment 51 52: an adapted one-item visual-analogue scale.
WHO Quality of Life Scale—Brief (WHOQOL-BREF): a 26-item assessment of life satisfaction.
Compliance: a visual-analogue scale assessing compliance with instructions to take the PDE pill.
Last 2-week substance use: self-reported frequency of use of four broad classes of drugs.
Methadone symptom severity checklist 53: symptom severity of 38 commonly reported methadone treatment-associated side effects.
Cleveland clinic constipation scoring system 54: an 8-item scale of constipation severity.
Assessment time line
Psychological measurements
Monetary Choice Questionnaire 55: a 27-item assessment of delay discounting that asks participants to make hypothetical choices between a smaller-sooner amount of money available today or a larger-later amount of money available after a delay.
Barratt Impulsivity Scale, version 11 56: a 30-item assessment that yields information regarding several three facets of trait impulsivity (ie, attentional impulsivity, motor impulsivity and non-planning impulsivity), wherein participants indicate on a four-point Likert-like scale the extent to which each of 30 items describes their overall behaviour.
Behavioural Inhibition/Activation System Scales (BIS/BAS) 57: a 24-item assessment of behavioural inhibition (BIS) and approach (BAS), wherein participants indicate on a 4-point Likert-like scale the extent to which each of 24 items describes their behavioural style.
Pain Catastrophising Scale 58: a 13-item assessment of how pain is subjectively experienced, wherein participants indicate on a 4-point Likert-like scale the extent to which each of the 13 items describe the thoughts and feelings they have when they are experiencing pain.
Pittsburgh Sleep Quality Index 59: a 9-item assessment of sleep satisfaction.
PEG pain screening tool 60: a 3-item assessment of pain intensity and interference.
Exit survey: a 7-item quantitative and qualitative assessment of how the PDE pill was experienced by participants and their thoughts about their experience participating in the study.
A detailed time line of all outcome assessments is provided in table 1.
Sample size calculation
We anticipate that dose escalations will be recommended at dose evaluation (~3 weeks following entry into treatment) for ~70% of participants in the TAU control group. With 60 participants per group, we will have power of 0.80 to detect a difference between groups if the corresponding rate in the intervention group is 44% or lower (ie, a maximum of 26/60 participants), using a Fisher exact test with a two-tailed alpha of 0.05. This is a medium-to-large effect, equivalent to an OR of 3.03 or a Cohen’s d of 0.61.
For our non-inferiority tests on outcome indicators, power depends on the rate of occurrence (for dichotomous outcomes) and on the sample SD (for continuous outcomes). Thus, if a dichotomous outcome (such as reports of withdrawal) occurs in 15% of each group, we will have power of 0.90 to conclude that the real difference in proportions is no >19%, using a one-sided 95% CI, as is appropriate for a non-inferiority test. If reports of withdrawal occur in 5% of each group, we will have power of 0.90 to conclude that the real difference in proportions is no >12%, using a one-sided 95% CI. Similarly, if scores on a measure of withdrawal severity have a SD of 0.5 with no observed difference in means between groups, we will have power of 0.90 to conclude that the real difference in means is no > 0.27 SD, using a one-sided 95% CI. If the scores have a SD of 0.8 with no observed difference in means between groups, we will have power of 0.90 to conclude that the real difference in means is no > 0.43 SD, using a one-sided 95% CI. To help ensure that we obtain usable data from 120 completed participants (60 per group), we plan to screen and enrol 240 participants. Attrition within the first 3 months of treatment at the MTP is not heavy, but we want to take a conservative approach to the possibility of noncompliance with study measures and procedures.
Data analysis
We will use a between-group (PDE vs TAU) comparison of the proportion of patients who move up to a higher methadone dose, and a comparison of scores or counts on all other measures. We will make these comparisons with an exact test unless demographic comparisons of the two groups suggest that we need to control for potential confounding covariables such as sex, race, age or baseline indicators of addiction severity, in which case we will use multiple logistic regression. We will also test for non-inferiority on the following outcome measures: (1) frequency of positive drug testing (urine and self-report), (2) SOWS score, (3) OOWS score, (4) Craving score, (5) WHOQOL-BREF, (6) days in treatment, (7) methadone side effects checklist and (8) Constipation Severity Score. The analytic method will be determined by the distribution of the data on the outcome measure (eg, generalised linear mixed models for frequency of positive urine drug tests, and general linear mixed models for questionnaire scores), but the hypothesis of non-inferiority will always be tested by comparison of one-sided 95% CIs for the parameter estimates.
Anticipated outcomes
We anticipate that a substantial proportion of patients in the PDE group will not need to be escalated to the higher doses of methadone (see figure 1). We also expect that patients in this group will have clinical improvements at least as good as those in the control group, in terms of decreased craving, withdrawal, drug use and urine-positive drug tests, increased scores on a quality of life assessment, and treatment retention at the 3-month (84 days) time point—with fewer side effects from methadone. Finally, we anticipate that primary and secondary outcomes will scale with both expectancy and compliance.

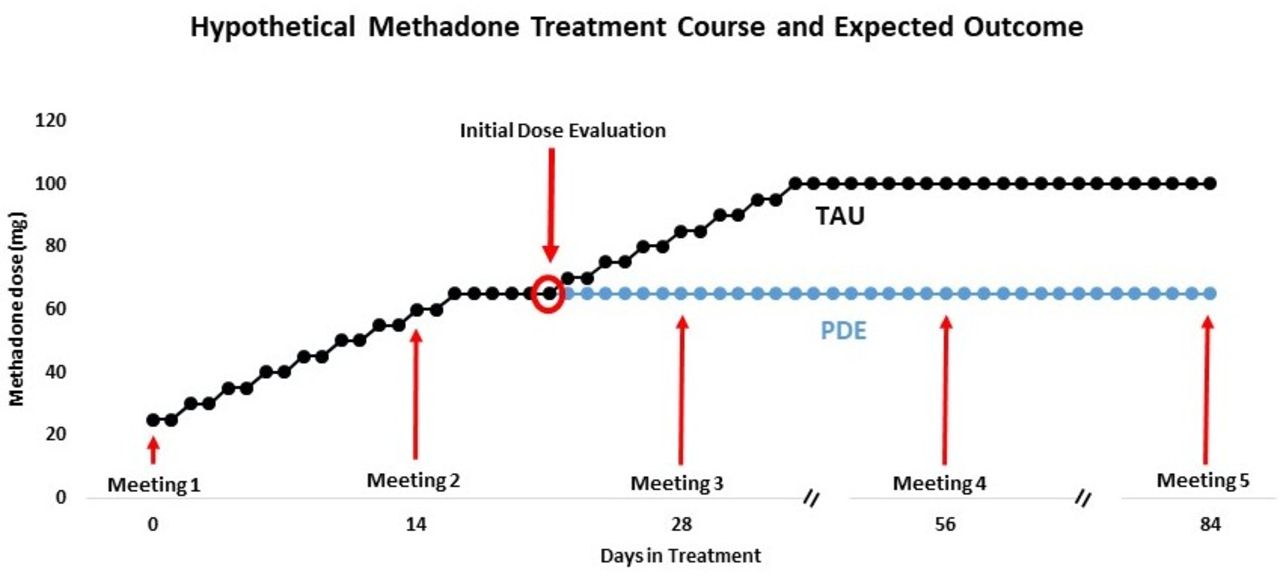{kind=link}
Hypothetical methadone treatment course and expected outcome. TAU, treatment as usual; PDE, placebo dose extension.
Data collection: retention, quality management and storage
Participants are given an appointment card that serves as a reminder of the next date that they are to meet with study staff. Additionally, a member of the study team calls participants 1 day prior to the designated meeting day to remind them of their appointment. Data are collected in an in-person meeting on paper for each instrument. Following the meeting, data are recorded electronically in an ad hoc project created in RedCAP,61 a secure data collection and management application hosted at the University of Maryland, Baltimore. Once recorded, data are verified by a secondary independent observer and subsequently locked to prevent changes from being made. Missing data due to missed meetings are coded as incomplete. The resulting database is imported into SPSS V25 and logical consistency checks are conducted and addressed, and missing values designated. Data collected on paper are deidentified with a study identification number and stored in a locked cabinet in an off-site location. Any electronic identifying information will be password-protected on an encrypted, HIPAA-compliant drive and all study authors will have access to verified, cleaned and deidentified datasets.
Data monitoring
The study is reviewed annually by an independent Data Safety and Monitoring Board. Progress reports include reporting of adverse events, updates on enrolment, raw data reporting and any outcomes and preliminary analyses.
Unanticipated/adverse/reportable new information event reporting
Although the likelihood of an adverse event is exceedingly unlikely, participants in the PDE group will receive a pill instruction handout (attached under ‘relevant materials’ above). This handout will contain explicit information on who to contact in case of an adverse event situation. The Methadone Clinic is staffed daily by a work force of doctors, nurses and clinical counsellors who are specifically trained to work with this population of individuals suffering from substance use disorder. In the unlikely event that a participant shows signs of crisis (eg, heightened anxiety) due to discomfort with any aspect of the assessments, a counsellor or doctor on the floor will be engaged to intervene promptly.
Ethics and dissemination
All activities associated with this protocol are conducted in full compliance with current University of Maryland, Baltimore and University of Maryland, College Park Human Research Protection Programmes and Institutional Review Board policies and procedures while maintaining compliance with federal regulations. This protocol is approved and is active with IRBs of both universities. Written informed consent is obtained from every participant. Important protocol modifications will be communicated to relevant members of the research team via Collaborative Institutional Comprehensive Evaluation of Research Online, the University of Maryland School of Medicine’s Research Evaluation Portal. Any results from this trial (publications, conference presentations) will be published in peer-reviewed journals and conference proceedings.
Discussion
To our knowledge, this is the first randomised, controlled clinical trial that implements methods of inducing ethically appropriate placebo responses in an addiction treatment context. The findings obtained will provide crucial pilot data concerning the effectiveness of placebo interventions implemented in the context of OUD treatment. Additionally, understanding the capacity for pharmacological conditioning to impact outcomes in this patient population could have very significant clinical and translational implications, and could provide strong justification for its use in the clinic as a method of increasing the ratio of benefits to side effects in MMT.
In addition to exploring the therapeutic potential of a harnessed placebo response, through comprehensive urine screen analysis, this ongoing clinical trial will yield important information regarding the precise types of drugs that are being consumed in a West Baltimore neighbourhood, currently ‘Ground Zero’ in the nation’s current opioid epidemic.62 In a time of rapid changes in the synthetic drugs available on the street, patients often report having no awareness of the substances they have ingested (AMB, EWe, AG, personal communications, 2018). Our comprehensive urinalysis will help inform current trends in use and will help realise the scope of compounds contained in street drugs. Additionally, patient self-report of lifetime history of drug use will afford a unique opportunity to understand drug use patterns (particularly, prescription opioid use) that may have predated use of other opioids such as heroin.
This clinical trial study is designed to explore whether open-label placebo can be integrated into treatment with salubrious effects. Because the goal is to move the needle very quickly to enhance medical addiction treatment, we wanted to take the simplest approach to obtaining an answer on whether placebo pills might be a useful tool in this arsenal. As such, we chose to restrict ourselves to a two-arm design (PDE vs TAU). A limitation of this approach, however, is that we will not be able to produce data on how patients would respond to placebo pills delivered blindly: in other words, traditional placebo effects invoked using non-transparent methods. We plan to address this gap in future follow-up studies.
As numbers of new individuals afflicted with OUD continue to rise, the National Institute of Health has called on the scientific community to deliver effective and sustainable solutions to stave this formidable public health challenge.63 Medication-based treatment is the only treatment strategy with a scientific basis, and the treatment of choice for OUD, endorsed fully by the medical community. Harnessing a potentially effective placebo response to enhance methadone treatment of OUD is responsive to this NIH call, with the additional boon of very low financial burden and risk to patients. If our randomised controlled trial outcome is successful, it would represent an important first step towards a safe, inexpensive and quick-to-launch adjunct to methadone that could feasibly change front-line addiction medicine treatment of OUD.
Acknowledgments
Authors would like to acknowledge the following individuals for their contributions to study implementation: Riti Kotamarti and Olivia Pettingill (assistance with study conduct); Ray Velencia (patient recruitment assistance); Denisha Pendleton (coordination of nurses’ study roles); and the entire nursing and counseling staff of the University of Maryland Methadone Treatment Program. We would also like to acknowledge the contributions made by staff at the Center for Substance Abuse Research (CESAR), University of Maryland for the following contributions to the study: Jonathan Lewis and Julie Factor (assistance with instrument development, database development, data analysis); Lynn Wagner and Theresa Hippolyte in the Division of Forensic Toxicology at the Armed Forces Medical Examiner System (urinalyses). We used the SPIRIT checklist when writing our report.
References
Footnotes
Contributors AMB and TOC conduct the study. AMB, TOC, AG, EWe, DE, ZKK, CJW, EWi, TK and LC participated in the original design of the study protocol. SWH is responsible for placebo pill manufacture. DE was responsible for the statistical design of the study. MW, ASB, EM, EWi, EMW and KH helped to design or choose the study instruments, develop the database and will conduct data analysis for the study. AMB wrote the first draft of the manuscript and all authors contributed revisions and approved the final manuscript.
Funding This work was supported by the Foundation for the Science of the Therapeutic Encounter (AMB), funds from the University of Maryland MPowering the State Opioid Use Disorders Initiative (AMB, EWi, LC), and the Intramural Research Program of the National Institute on Drug Abuse of the National Institutes of Health (DE). These funding sources had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data or decision to submit results.
Competing interests None declared.
Ethics approval The study has been approved by the University of Maryland Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.