





Abstract
The blue buck or blue antelope (Hippotragus leucophaeus) is an extinct antelope endemic to coastal South Africa. With a much wider distribution in the Pleistocene, it was already in decline before European settlement in the region (c. 400 years ago), and it went extinct in the late 18th century. Due to the short time span between its formal description and the time it went extinct, not much is known about its biology or behaviour, and its phylogenetic placement is still inconclusive. Here we sequence the complete mitochondrial genome of the blue buck from a museum-preserved horn. The data generated in this study allowed the placement of the blue buck as sister species to the sable antelope (Hippotragus niger), with a divergence time of 2.8 Myr. The roan antelope (Hippotragus equinus) is sister taxon to this clade, contradicting some earlier findings.
INTRODUCTION
The blue buck or blue antelope, Hippotragus leucophaeus (Pallas, 1766), was an endemic species of coastal South Africa. Its range, at the time of European settlement, spanned from Caledon to Plettenberg Bay (Fig. 1), but archaeological material indicates a wider past distribution. Fossil bones found in Rose Cottage Cave (1100 km inland in eastern Orange Free State, Plug & Engela, 1992), suggest that the distribution range of the species was larger during the Late Pleistocene, spanning the southern and western coastal plains of the Cape Floristic Region (CFR) into the highlands of Lesotho (Tyler Faith & Thompson, 2013). This is also corroborated by the presence of cave paintings of the blue antelope in a shelter in the Caledon River Valley near Ficksburg (Loubser, Brink & Laurens, 1990). These facts suggest that the species was already in clear decline before European settlement in the Cape Province.
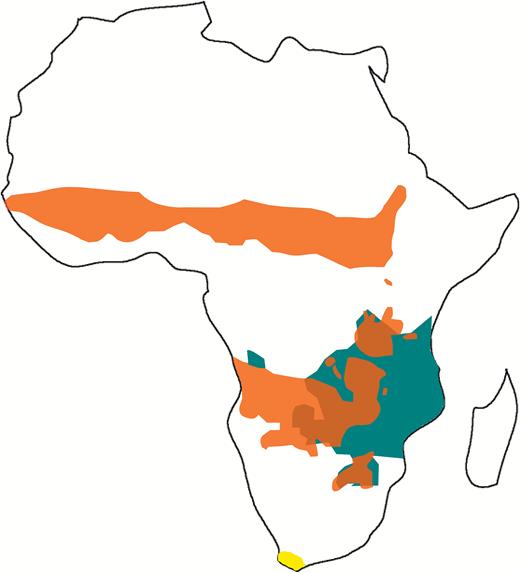
Distribution range of the three species of the genus Hippotragus, Hippotragus equinus in orange, Hippotragus niger in green and Hippotragus leucophaeus in yellow. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The blue antelope was the first African mammal to be hunted to extinction in historical times; it is thought that the last blue buck was killed in the 18th century (Harper, 1945). While it was first mentioned in a publication in 1719, a formal description was only published in 1766 shortly before it became extinct around 1800. The species is very scarce in natural history museum collections, given the short time between its discovery and extinction. Only four complete mounted specimens are known in the museums of Leiden, Paris, Stockholm and Vienna, along with two sets of horns and a few bone remains (Harper, 1945; Rookmaker, 1992). The pair of horns used in this study is the last remaining part of a fifth mounted skin present at the Museum of Evolution, Uppsala University (Rookmaker, 1992).
Not much is known of its biology and behaviour, making it one of the most enigmatic and unstudied big African mammals. Based on fossil and dental records, we can classify this species as a grazer. Dental wear of mid-Pleistocene fossils indicates a diet of grasses (Stynder, 2009). These observations, along with the fact that it is found in the fossil record together with other characteristic grassland species, clearly suggest the blue antelope favoured grasslands. In fact, the presence of the blue antelope in the archaeological record followed a clear pattern, increasing when grass cover increased and declining with the increased dominance of woodland species preferred by its congener, the partially sympatric roan antelope (Klein, 1974).
The blue buck inhabited grassland areas close to water sources, and just like roan and sable antelope, they were probably dependent on fresh water for survival (Matthee & Robinson, 1999). Many calves were attacked by lions, leopards, spotted hyenas and African wild dogs and killed within months of birth.
Reasons behind their demise are still poorly understood. Habitat degradation, as a result of cultivation and overgrazing of grassland, along with disease and hunting, are reasons cited for this species’ demise. Several hypothesis have been formulated for its extinction: overhunting by European colonialists, competition and habitat deterioration due to pastoralism, and long-term environmental change (Klein, 1974). Climate change led to reduction in grasslands around the Cape Province c. 11000 years before present and its replacement with bush, forest and the unique fynbos may have caused the reduction in species range (Robinson et al., 1996). The reduction of grasslands and its replacement by other habitats favoured the expansion of the roan antelope and promoted the decline of blue buck populations due to the reduced availability of suitable food. Being a specialist grazer as well as sensitive to the increasing competition of domestic stock might also have contributed to its extirpation (Kerley et al., 2009). Shedding light into its life history and extinction is very important in the context of big mammal conservation efforts. The quagga (Equus quagga quagga), a subspecies of plains zebra was also a fynbos native and, like the blue buck, went extinct in historical times. After human settlement in the southern cape many herbivores were extensively hunted because they were competing for forage with the recently introduced domestic species.
Phylogenetically, this animal is placed in the genus Hippotragus, as it is related to roan (Hippotragus equinus) and sable (Hippotragus niger) antelopes. The blue antelope was nevertheless slightly smaller than its congeneric species. Mohr (1967) placed it as a subspecies of roan antelope. Klein, however, based mainly on fossil horn cores and teeth, described it as a separate species (Klein, 1974). Parsimony and maximum likelihood (ML) phylogenies based on 500 bp of cytochrome b (CYTB) place the blue buck in its own species, H. leucophaeus, and as sister taxon to the clade formed by roan and sable antelopes, however with low bootstrap values, 83.7 and 78%, respectively (Robinson et al., 1996). In contrast, a neighbor-joining (NJ) phylogeny based on the same data, places the blue buck as sister species to the sable antelope, with the roan antelope as sister to this clade (Robinson et al., 1996). The uncertainty and discordance among these results may be due to the use of a limited number of nucleotides from the mitochondrial genome, and resolution can be improved by using either more genes/nucleotides or the entire mitogenome (see for example Duchêne et al., 2011). The use of high throughput shotgun sequencing makes sequencing the entire mitogenome affordable when comparing with targeting specific mitochondrial genes, with the added benefits of also obtaining nuclear DNA sequences, and of working with short DNA fragments such as those that can be recovered from museum-preserved specimens. The availability of mitogenome sequences (and lack of nuclear data) from many of the species of Hippotraginae and Alcelaphinae makes this the most useful marker for phylogenetic analysis.
Natural history collections have been used extensively in the recent past to uncover lost populations and genetic diversity, domestication or taxonomic placement of extinct species (e.g. Campos et al., 2010; Cappellini et al., 2010; Schlumbaum et al., 2010). They are invaluable sources of bone and keratinous material from which DNA can be retrieved (e.g. Campos et al., 2012; Espregueira Themudo, Rufino & Campos, 2015). Here we present the full mitochondrial DNA (mtDNA) of the extinct blue antelope sequenced to a high depth, recovered from a horn core of a historical specimen collected in the 18th century and housed in the Museum of Evolution at Uppsala University. Its mitogenome allowed us to investigate the validity of its taxonomic specific status, to finally resolve congeneric relationships and infer the time of divergence among Hippotragus. Moreover, it provides essential insights into the biogeography of the Southern Cape previous to European colonization.
MATERIAL AND METHODS
Sample collection
The sample was collected in May 2014 in the Museum of Evolution – Zoology, Uppsala University from a pair of horns mounted as a hunting trophy identified as H. leucophaeus (voucher number UPSZMC 78488). This specimen was most probably collected by Carl Peter Thunberg sometime during the 1770s, and it was incorporated into the collection before 1785. Four grams of bone powder were obtained by drilling in loco into the horn core, to reduce the damage to the specimen, using a Dremel Drill, with precautions to avoid contamination (use of gloves, face mask and new drill bit).
DNA extraction
All DNA extractions, library preparations and PCR setups were performed in a dedicated ancient DNA facility, located in a separate building from other DNA labs. All subsequent molecular biology-based laboratory work, such as PCR amplification, Bioanalyzer runs and sequencing, was performed in a separate DNA facility. Total cellular DNA was extracted, in parallel for two replicates, hereafter named Hleuco3 and Hleuco4 according to the following protocol: between 0.01 and 0.09 g of bone powder, obtained by drilling into the horn core with a Dremel Drill, was incubated overnight at 37 °C in 1.0 mL of 0.5 M EDTA and 25 mg/mL proteinase K. To pellet the nondigested powder, the solution was centrifuged at 12000 × g for 5 min. The liquid fraction was then transferred into a Centricon micro-concentrator (30-kDa cutoff) and spun at 4000 × g for 10 min. When the liquid was concentrated down to about 200–250 μL, the DNA was purified using the MinElute PCR purification kit (Qiagen), with the following modifications (Campos & Gilbert, 2012): (1) 10× buffer PB was used for the DNA binding step; (2) spins were done at 8000 rpm with the exception of the final one at 13000 rpm; and (3) in the elution step, spin columns were incubated in 40 μL buffer EB at 37 °C for 10 min, spun down and this step then repeated. The eluates from both rounds of elution were pooled.
Library construction and amplification
Double-stranded DNA Illumina libraries were constructed from extracts Hleuco3 and Hleuco4. A blunt-end library was constructed on 21.25 μL of the DNA extract using NEBNext DNA Sample Prep Master Mix Set 2 (New England Biolabs, E6070). The protocols outlined in the kit manual and Raghavan et al. (2014) were followed with the following modification. Reaction volumes were cut down from the manufacturer’s protocol by a quarter in the end-repair step and by half in the ligation and fill-in steps. After the end-repair and ligation incubations, the reaction was purified through MinElute spin columns and eluted in 15 and 21 μL, respectively, after a 5-min incubation at 37 °C with Qiagen EB buffer. Ligation reaction was performed for 25 min at 20 °C using Illumina-specific adapters specified in Meyer and Kircher (2010). Fill-in reaction was performed for 20 min at 65 °C.
The purified libraries were amplified in a two-step manner, where 5-μL PCR product from the first amplification round was transferred into new 50-μL PCR reactions. To increase complexity, the second-round PCRs were set up with four reactions in parallel. PCR products were then pooled and purified through a single Qiagen MinElute spin column, and eluted in 25-μL EB buffer following a 10-min incubation at 37 °C.
The purified libraries were amplified as follows: 25-μL DNA library, 1× high-fidelity PCR buffer, 2 mM MgSO4, 200 μM dNTPs each (Invitrogen, Carlsbad, CA, USA), 200 nM Illumina Multiplexing PCR primer inPE1.0 10 μM, 4 nM Illumina long index primer 10 μM, 1 U of Platinum Taq DNA Polymerase (High Fidelity) (Invitrogen, Carlsbad, CA, USA) and water to 50 μL. Cycling conditions were: initial denaturing at 94 °C for 4 min, 8 cycles of: 94 °C for 30 s, 60 °C for 30 s, 68 °C for 40 s and a final extension at 72 °C for 7 min. PCR products were purified through MinElute spin columns and eluted in 20 μL of Qiagen Buffer EB, following a 10-min incubation at 37 °C. A second round of PCR (four parallel reactions for each library) was set up as follows: 5 μL of purified product from first PCR round, 1× high-fidelity PCR buffer, 2 mM MgSO4, 200 M dNTPs each, 200 nM each of Sol_bridge_P5 and Sol_bridge_P7 10 μM (Maricic, Whitten & Pääbo, 2010), 1 U of Platinum Taq DNA Polymerase (high fidelity) and water to 50 μL. Cycling conditions included an initial denaturing at 94 °C for 4 min, 10 cycles of: 94 °C for 30 s, 58 °C for 30 s, 68 °C for 40 s and a final extension at 72 °C for 7 min. The amplified library was run on Agilent 2100 Bioanalyzer High Sensitivity DNA chip. Hleuco3 and Hleuco4 libraries were pooled with five others and sequenced on one lane on an llumina HiSeq 2000 (100 cycles, single read mode) at the Danish National High-Throughput DNA Sequencing Centre.
Data processing and mapping
Base calling was performed using the Illumina software CASAVA 1.8.2, with the requirement of a 100% match to the 6-nucleotide index used during library preparation. Adapter sequences were trimmed, and filtered for Ns and reads shorter than 30 bp using AdapterRemoval (Lindgreen, 2012). Trimmed reads were initially mapped to the H. niger complete mtDNA genome (GenBank accession number NC_020713) using bwa-0.7.5a-r405, with seed length disabled to improve mapping efficiency in ancient DNA data sets (−l 1000: Schubert et al., 2012). The generated sequence data were also mapped to the nuclear genomes of Bos taurus (version UMD3.1) and Ovis aries (version 3.1.75) using the same procedure. The nuclear and mtDNA alignments were sorted using Samtools (Li et al., 2009) and filtered for PCR duplicates using Picard MarkDuplicates-1.88 (http://picard.sourceforge.net) and for paralogs using the X1 tag (not equal to 0) as defined by BWA. The X1 tag indicates the number of suboptimal hits for a particular read, so removing reads with X1 > 0 eliminates paralogs.
Due to a large number of gaps in the mitochondrial genome sequence compared with the reference, we used MITObim (Hahn, Bachmann & Chevreux, 2013), a software that enables the reconstruction of full mtDNA genomes using a related species as reference, through baiting and iterative mapping, with a maximum number of 30 iterations. Any 5′ and 3′ overhangs in respect to the reference were removed after verification of correct circularity.
Authentication and alignment
To authenticate the sequences obtained as genuine, we ran mapDamage 2.0 (Jónsson et al., 2013) in order to detect DNA damage patterns typical of ancient or degraded DNA. The program uses misincorporation patterns, particularly deamination of cytosine into uracils, within a Bayesian framework. An elevated C to T substitution rate towards sequencing starts (and complementary G to A rate towards the end) is considered indicative of genuine ancient or degraded DNA.
We aligned the complete mitochondrial genome of the blue buck with 18 available mitochondrial genomes of species in the subfamilies Hippotraginae and Alcelaphinae (see Table 1 for GenBank accession numbers) using MUSCLE (Edgar, 2004). The complete mitochondrial genome of Ovis aries was used as outgroup for this analysis. As the position of the blue buck within Hippotraginae is undisputed, Alcelaphinae species serve as an internal outgroup in these analysis. One of the sequences of Oryx dammah (NC_016421) grouped with Addax nasomaculatus (NC_020674) in a preliminary phylogenetic tree and not with other Oryx sp., including the other O. dammah sequence (JN632678). A visual comparison of the sequences revealed that NC_016421 seems chimeric between A. nasomaculatus and O. dammah, with only three regions (ND2 and adjacent tRNAs; the region between CO3 and ND4L; and the control region) being apparently genuine O. dammah (with a top hit in Blast). The remaining sequence is identical to NC_020674. We therefore removed NC_016421 from further analyses.
Subfamily, (sub)species, common name, GenBank accession number and reference of the complete mitogenomes used in this study
| Subfamily | (Sub)Species | Common name | GenBank | Reference |
|---|---|---|---|---|
| Caprinae | Ovis aries | Domestic sheep | NC_001941 | Hiendleder et al. (1998) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | NC_020676 | Hassanin et al. (2012) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | JN632593 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes gnou | Black wildebeest | NC_020698 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | NC_020699 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | JN632627 | Hassanin et al. (2012) |
| Alcelaphinae | Damaliscus pygargus | Bontebok | NC_020627 | Hassanin et al. (2009) |
| Alcelaphinae | Damaliscus lunatus | Tsessebe | NC_023543 | Steiner et al. (2014) |
| Alcelaphinae | Beatragus hunteri | Hirola | NC_023542 | Steiner et al. (2014) |
| Hippotraginae | Addax nasomaculatus | Addax | NC_020674 | Hassanin et al. (2012) |
| Hippotraginae | Oryx beisa | East African oryx | NC_020793 | Hassanin et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | NC_016421 | Zhang et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | JN632677 | Hassanin et al. (2012) |
| Hippotraginae | Oryx gazella | Gemsbok | NC_016422 | Y. Ren, H. Zhang (unpubl. data) |
| Hippotraginae | Oryx gazella | Gemsbok | JN632678 | Hassanin et al. (2012) |
| Hippotraginae | Oryx leucoryx | Arabian oryx | NC_020732 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus equinus | Roan antelope | NC_020712 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger niger | Sable antelope | NC_020713 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger variani | Giant sable antelope | KM_245339 | Espregueira Themudo et al. (2015) |
| Subfamily | (Sub)Species | Common name | GenBank | Reference |
|---|---|---|---|---|
| Caprinae | Ovis aries | Domestic sheep | NC_001941 | Hiendleder et al. (1998) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | NC_020676 | Hassanin et al. (2012) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | JN632593 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes gnou | Black wildebeest | NC_020698 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | NC_020699 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | JN632627 | Hassanin et al. (2012) |
| Alcelaphinae | Damaliscus pygargus | Bontebok | NC_020627 | Hassanin et al. (2009) |
| Alcelaphinae | Damaliscus lunatus | Tsessebe | NC_023543 | Steiner et al. (2014) |
| Alcelaphinae | Beatragus hunteri | Hirola | NC_023542 | Steiner et al. (2014) |
| Hippotraginae | Addax nasomaculatus | Addax | NC_020674 | Hassanin et al. (2012) |
| Hippotraginae | Oryx beisa | East African oryx | NC_020793 | Hassanin et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | NC_016421 | Zhang et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | JN632677 | Hassanin et al. (2012) |
| Hippotraginae | Oryx gazella | Gemsbok | NC_016422 | Y. Ren, H. Zhang (unpubl. data) |
| Hippotraginae | Oryx gazella | Gemsbok | JN632678 | Hassanin et al. (2012) |
| Hippotraginae | Oryx leucoryx | Arabian oryx | NC_020732 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus equinus | Roan antelope | NC_020712 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger niger | Sable antelope | NC_020713 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger variani | Giant sable antelope | KM_245339 | Espregueira Themudo et al. (2015) |
Subfamily, (sub)species, common name, GenBank accession number and reference of the complete mitogenomes used in this study
| Subfamily | (Sub)Species | Common name | GenBank | Reference |
|---|---|---|---|---|
| Caprinae | Ovis aries | Domestic sheep | NC_001941 | Hiendleder et al. (1998) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | NC_020676 | Hassanin et al. (2012) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | JN632593 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes gnou | Black wildebeest | NC_020698 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | NC_020699 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | JN632627 | Hassanin et al. (2012) |
| Alcelaphinae | Damaliscus pygargus | Bontebok | NC_020627 | Hassanin et al. (2009) |
| Alcelaphinae | Damaliscus lunatus | Tsessebe | NC_023543 | Steiner et al. (2014) |
| Alcelaphinae | Beatragus hunteri | Hirola | NC_023542 | Steiner et al. (2014) |
| Hippotraginae | Addax nasomaculatus | Addax | NC_020674 | Hassanin et al. (2012) |
| Hippotraginae | Oryx beisa | East African oryx | NC_020793 | Hassanin et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | NC_016421 | Zhang et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | JN632677 | Hassanin et al. (2012) |
| Hippotraginae | Oryx gazella | Gemsbok | NC_016422 | Y. Ren, H. Zhang (unpubl. data) |
| Hippotraginae | Oryx gazella | Gemsbok | JN632678 | Hassanin et al. (2012) |
| Hippotraginae | Oryx leucoryx | Arabian oryx | NC_020732 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus equinus | Roan antelope | NC_020712 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger niger | Sable antelope | NC_020713 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger variani | Giant sable antelope | KM_245339 | Espregueira Themudo et al. (2015) |
| Subfamily | (Sub)Species | Common name | GenBank | Reference |
|---|---|---|---|---|
| Caprinae | Ovis aries | Domestic sheep | NC_001941 | Hiendleder et al. (1998) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | NC_020676 | Hassanin et al. (2012) |
| Alcelaphinae | Alcelaphus buselaphus | Hartebeest | JN632593 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes gnou | Black wildebeest | NC_020698 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | NC_020699 | Hassanin et al. (2012) |
| Alcelaphinae | Connochaetes taurinus | Brindled gnu | JN632627 | Hassanin et al. (2012) |
| Alcelaphinae | Damaliscus pygargus | Bontebok | NC_020627 | Hassanin et al. (2009) |
| Alcelaphinae | Damaliscus lunatus | Tsessebe | NC_023543 | Steiner et al. (2014) |
| Alcelaphinae | Beatragus hunteri | Hirola | NC_023542 | Steiner et al. (2014) |
| Hippotraginae | Addax nasomaculatus | Addax | NC_020674 | Hassanin et al. (2012) |
| Hippotraginae | Oryx beisa | East African oryx | NC_020793 | Hassanin et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | NC_016421 | Zhang et al. (2012) |
| Hippotraginae | Oryx dammah | Scimitar-horned oryx | JN632677 | Hassanin et al. (2012) |
| Hippotraginae | Oryx gazella | Gemsbok | NC_016422 | Y. Ren, H. Zhang (unpubl. data) |
| Hippotraginae | Oryx gazella | Gemsbok | JN632678 | Hassanin et al. (2012) |
| Hippotraginae | Oryx leucoryx | Arabian oryx | NC_020732 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus equinus | Roan antelope | NC_020712 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger niger | Sable antelope | NC_020713 | Hassanin et al. (2012) |
| Hippotraginae | Hippotragus niger variani | Giant sable antelope | KM_245339 | Espregueira Themudo et al. (2015) |
Phylogeny and divergence time
We used MrBayes v3.2 (Ronquist & Huelsenbeck, 2003) to place our specimen of blue buck within the phylogeny of Hippotraginae and therefore confirm which of the two congeneric species is most closely related to this extinct taxon. The data set was partitioned by gene, according to the annotation of H. niger from GenBank (NC020713). Intergenic base pairs (bp) and the control region were excluded from the analysis, as these may present gaps and misalignments that could potentially introduce bias or uncertainty in the phylogenetic reconstruction.
For the Bayesian inference, we used the general time reversible model for nucleotide evolution with a gamma distribution on the rate of variation and a proportion of invariable sites (GTR + G + I). We have chosen this more general model, as model parameters may converge on simpler nested models. Preliminary analysis with other models such as HKY or using the Bayesian model jumping approach produced identical tree topologies and branch posterior probabilities. The final model in this latest approach was similar to the TN93 model with an extra parameter for G ↔ C substitutions. The Markov chain Monte Carlo (MCMC) ran for 10 million generations along four heated chains (heated chain temperature = 0.2), with a subsample frequency of 1000 and a burn-in length of 25%. Branch lengths were left unconstrained. Consensus trees resulting from the analyses were then drawn using FigTree v1.4 (Rambaut, 2012).
In order to date the divergence between H. leucophaeus from the other congeneric species, we also reconstructed the phylogeny of the Hippotraginae and Alcelaphinae subfamilies using the strict molecular clock in Beast v1.8.0 (Drummond, Rambaut & Suchard, 2013). We chose a strict clock as we do not have many calibration points and the species/subspecies are sufficiently closely related that we can assume that rates do not differ significantly between lineages. We also tested both exponential and uncorrelated lognormal clock models, and both failed to converge on a solution based on low Effective Sample Size (ESS). We calibrated the molecular clock by placing informative priors on the nodes leading to Hippotraginae (median = 6.4 Mya; 95% range = 6.4–13 Mya) and Alcelaphinae (median = 4.5 Mya; 95% range = 4.5–7.0 Mya). These fossil calibrations were set after Bibi (2013) and references therein (Gentry, 1980, 2011; Hendey, 1981; Vrba & Gatesy, 1994; Vrba, 1997; Geraads et al., 2008; Lebatard et al., 2008; Deino, 2011). For each taxon, we placed a constraint on the date of origin of the internal node following a lognormal distribution with the corresponding median and 95% range. As in MrBayes, we assumed the GTR + G + I model of nucleotide evolution, and a constant rate birth–death speciation model for the splitting of lineages within the tree (Yule, 1925; Gernhard, 2008). We ran the MCMC chain for 10 million iterations. The first quarter was discarded as burn-in. The ESS was monitored using Tracer 1.5 (Rambaut & Drummond, 2007), and after verifying that ESSs for all parameters were above 200, a maximum clade credibility tree was generated with TreeAnnotator v1.8 (part of the BEAST software package) and visualized in FigTree v1.4.
We also reconstructed the phylogeny based only on the CYTB fragment used in Robinson et al. (1996) including the sequences released in the aforementioned study (GenBank accessions AH006574–80). For comparison with the original publication, we reconstructed the phylogeny using NJ and ML methods, in addition to the Bayesian inference. These analyses were done in Geneious v.7.1.9 (http://www.geneious.com; Kearse et al., 2012) and PhyML (Guindon & Gascuel, 2003) with 10000, and 1000 bootstrap replicates, respectively, and a transition/transversion ratio of 1:1.6 in the ML analysis.
Selection
In order to detect positive and balancing selection, Ka/Ks ratio was calculated between the blue buck sequence and 12 other antelopes using DNAsp v5 (Librado & Rozas, 2009). Ratios were calculated over the entire mitogenome and using a sliding window of 100 bp moving 50 bp at a time. DNAsp considers both substitutions in non-coding regions, such as those coding for tRNAs and rRNAs, and synonymous substitutions in coding regions as silent, and calculates Ks accordingly. We also tried to detect sites subject to episodic diversifying selection, which happened in only some of the branches of the tree, using the mixed effects model of evolution (MEME; Murrell et al., 2012) through the Datamonkey webserver (Pond & Frost, 2005) implementing the HyPhy package (Pond, Frost & Muse, 2005). The model also calculates empirical Bayes factor for the existence of selection at a particular site at a given branch, although only for exploratory purposes.
RESULTS
Mitochondrial genome
The initial mapping resulted in an incomplete mitochondrial sequence, with 71 gaps where coverage was less than 20×. These gaps had an average length of 90 bp, adding up to a total of 6390 bp without sufficient coverage. The iterative baiting and mapping procedure implemented in MITObim resulted in a complete assembly of the mitogenome. From a total of 271705238 quality control-passed reads, 102870 mapped to the mitochondrial genome of H. niger, corresponding to 12% of all the mapped reads (mitochondrial and nuclear). After removing duplicates, these were further collapsed to 36225 reads. The average depth of coverage of the mitochondrial genome was 146×, with 99.86% of the genome covered. According to mapDamage 2.0, damage patterns in the mapped reads show an increase in the frequency of thymine and adenine in the 5′ and the 3′ end, respectively (Supporting Information, Fig. S1).
The mitochondrial genome of H. leucophaeus is 16507 bp long, similar to the mitochondrial genome of the reference (H. niger niger). Pairwise identity between the reference and the blue buck mtDNA sequence is 98.5%. There are 116 amino acid differences in coding regions of the mtDNA between the blue buck and sable antelope: 5 in ND6, 14 in ATP6, 5 in ATP8, 1 in CO1, 2 in CO2, 5 in CO3, 13 in CYTB, 7 in ND1, 13 in ND2, 6 in ND3, 14 in ND4, 2 in ND4L and 29 in ND5. All other nucleotide differences are synonymous or in non-coding regions.
In the mitogenomic phylogeny, H. leucophaeus is sister taxon of the sable antelope clade, formed by the nominal subspecies and the giant sable antelope. The branch leading to H. leucophaeus is supported with a posterior probability (pp) of 1. A list of synapomorphies supporting this clade is provided in Supporting Information, Table S2. The sable clade and the blue buck diverged, assuming the priors specified above, at ~2.8 Mya, with a 95% HPD between 2.31 and 3.32 Mya (Fig. 2). The divergence time between the two sable subspecies, niger and variani, is 109 Kyr with a 95% HPD 77.5–146.7, pushing forward previous estimates of 170 Kyr (Espregueira Themudo et al., 2015). The inclusion of the blue buck in the phylogeny also narrowed the previous HPD interval of 100–260 Kyr. According to these results, H. equinus diverged from the ancestor of these two species 4.17 Mya (3.47–4.92, 95% HPD).
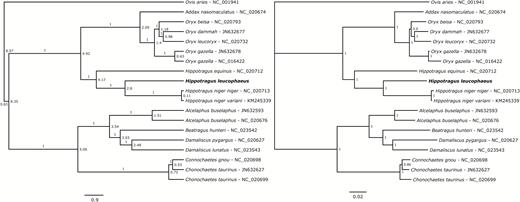
A, time-calibrated Bayesian phylogenetic tree of complete mitochondrial genomes of 18 bovids, of the subfamilies Alcelaphinae and Hippotraginae, including the blue antelope (Hippotragus leucophaeus in bold). Tips are assumed to be sampled in the present and so are vertically aligned. The mitogenome of the domestic sheep (Ovis aries; NC 001941) was used as outgroup. Node labels indicate divergence times (in Myr). Fossil calibrations used are described in the text. Branch labels indicate posterior probabilities. Scale bar indicates time in millions of years. B, phylogram of the same data set, inferred in MrBayes. Scale bar indicates substitutions per site.
When using only a fragment of CYTB, support for individual nodes decreases substantially, as expected when using a limited number of characters. Unlike in Robinson et al., in most analyses sequences of the blue buck group with sable with low support (63 and 53% bootstrap in NJ; % in ML), with the Robinson sequence closer to H. niger and with a longer branch than ours. The only exception is in the Bayesian tree where the Robinson sequence is placed outside the ingroup, while our sequence groups with the sable antelope (Supporting Information, Fig. S2).
The Ka/Ks ratio between the blue buck and related species ranges from 0.038 to 0.058, indicating purifying selection (Supporting Information, Table S1). Scans across 100 bp regions show Ka/Ks ratios below 1 in most of the mitogenome, with the exception of regions in ND2 and ATP8 in a few comparisons (not shown). MEME identified several codons showing evidence of episodic diversifying selection, in CO2 (codon 218), ND2 (5 and 125), ATP8 (32), ND4 (23), ND5 (14, 20, 121, 217 and 451), CYTB (233 and 238) and ND6 (88). However, only the underlined sites seem to have evidence to suggest these events happened in the branches immediately leading to the blue buck.
Nuclear genome assembly
Mapping of the sequence reads to reference nuclear genomes indicates that 516604 reads map uniquely to the genome of B. taurus and 883793 to the genome of O. aries. The distribution of mapped reads among the different chromosomes is roughly proportional to the size of the chromosome (Supporting Information, Table S3). The average depth of the mapped regions in the nuclear genome is 1.03473× and 1.03002×, and the breadth of coverage is 3.22 and 1.85%, respectively. These, of course, represent only a fraction of the genome of the blue buck, and it would require much more sequencing to recover the complete nuclear genome of the species. As there is no genome from any closer related species than the cow or sheep, it is also possible that some of the unmapped reads also originate from the blue buck, but did not map well to these genomes, and therefore the breadth of coverage may be underestimated.
DISCUSSION
We are confident that we sequenced the genuine blue buck mtDNA, and not modern contaminants, due not only to the damage patterns observed at both the 3′ and 5′ end of the reads but also because we obtained a high depth of coverage (146×) when compared to the depth of coverage of the nuclear genome (only 1–3% is covered with c. 1× of depth).
The mapping to the sable antelope mitogenome was not very successful, with many gaps in the assembled sequence. This was most likely due to the phylogenetic distance between the blue buck and the sable antelope used for reference, which made it difficult to align variable regions. The iterative baiting and mapping procedure implemented in MITObim resulted in a complete assembly of the mitogenome, as the consensus of the assembled reads in each step replaced the reference genome in the following iteration. This allowed the progressive extension of the mapped region to variable areas of the genome, finally filling up the gaps in the assembly. Using a different initial reference genome (H. equinus; see Table 1) or a fragment of CYTB from H. leucophaeus (see below) produced an identical result. This same strategy has been used to recover the mitochondrial and chloroplast genome sequence of several flatworms (Hahn et al., 2013), crustaceans (Zhang et al., 2015), plants (Lin et al., 2015) and other groups, with similarly accurate results.
The phylogeny based on the full mitogenomes places the blue antelope as a sister taxon to the sable antelopes, with the roan antelope as outgroup of this clade. The overall topology of the tree matches that of Hassanin et al. (2012), including the detection of mtDNA introgression between the two Connochaetes species, and with the exception of the branching order within Oryx. The grouping of the hirola (Beatragus hunteri) with Damaliscus corroborates the finding of Steiner et al. (2014) that originally released this sequence of hirola. Our result regarding the phylogenetic position of the blue buck contradicts previous results. Robinson et al. (1996) sequenced 502 bp of the mtDNA cytochrome b gene (CYTB) recovered from a skin sample of a mounted specimen.
Maximum parsimony and ML phylogeny reconstruction indicated that the blue buck is a sister taxon to the clade formed by the roan and the sable antelopes. However, those branches have very low bootstrap support (83.7 and 78%, respectively). An NJ phylogeny of the same data set recovered the same topology as this study: H. equinus (H. niger, H. leucophaeus) but with extremely low bootstrap support, which could potentially be explained by the small amount of data used. A direct comparison of the sequence from Robinson et al. (1996) with the corresponding fragment retrieved in our study reveals a relatively low similarity of c. 88%. This could have several possible explanations. As our mitogenome reconstruction used H. niger as a reference it is possible we have biased our results towards reads that are more similar to the reference. We have therefore repeated our analysis using H. equinus (NC020712) and using the first part of Robinson et al.’s CYTB sequence (AH006574) as the starting reference for MITObim. The sequences produced are identical to the first result. Alternatively, it is possible that either our sequence or Robinson et al.’s is not a true mitochondrial sequence but rather a nuclear pseudogene (Numt). However, we have above discarded that hypothesis for our data. The Robinson sequence does not show evidence of stop codons, typical of nuclear pseudogenes. It does have 12 autapomorphies, with 4 of these reflecting changes in amino acid constitution of the respective protein, compared with three autapomorphies in our sequence and no change in amino acids.
These new results confirm the specific status of H. leucophaeus as has been independently suggested by the fossil record (mainly horn cores and teeth) (Klein, 1974) and molecular data (Robinson et al., 1996) and so contradicts Mohr (1967) who, based on the features of mounted specimens, first described this taxon as a subspecies of roan antelope. Here for the first time we were able to estimate the time of divergence between the three species in the genus Hippotragus. Based on the full mtDNA genome (except control region and tRNAs), H. niger and H. leucophaeus diverged 2.8 Mya, while H. equinus diverged from the ancestor of these two species 4.17 Mya. In a previous study, Espregueira Themudo et al. (2015) found a divergence time between H. niger and H. equinus of 3.08 Mya. The inclusion of the blue buck in the phylogeny pushed this divergence time to 4.17 Mya. This can be explained by incomplete sampling in the phylogeny previously used to date the divergence. In a test of several molecular dating methods, Linder, Hardy & Rutschmann (2005) found that undersampling invariably underestimated divergence time. One other explanation could be that the calibration with fossil data is inaccurate or the calibration points are too distant from the study group; however, the same calibration was used in both phylogenies, as well as in previous more comprehensive studies (Hassanin et al., 2012) and so it seems unlikely that this should cause the discrepancy.
Between 3.5 and 2 Mya Africa was going through a series of climatic oscillations. A study of faunal remains in South Tanzania suggest a general climatic change with the progressive opening of the environment (Gibernau & Montuire, 1996), which might have facilitated the expansion of large ungulates like Hippotragus. Alternating climates of this period are likely to have been relevant for the speciation of many mammals, including hominins, as it led to the fragmentation of both forested and non-forested areas into temporary islands that expanded and contracted. Some studies indicate a substantial shift in the diet of several species of Australopithecines from southern Africa around 3.5 Mya, from a C3 to a C4 diet, possibly due to changes in the surrounding habitat and the availability of food (Sponheimer et al., 2013). Our results indicate that the blue buck has been under purifying selection, which may indicate that the blue buck was highly adapted to its habitat (Ballard & Pichaud, 2014). So it is possible that the diet shifts occurred with Hippotragus, prompting diet specialization from available food sources in different locations. This type of specialization and geographic isolation for periods of time might have contributed to speciation of blue buck, roan and sable antelopes. Furthermore, our results show some evidence of positive selection in ND2 and ATP8. It has been suggested that mutations in subunits of the NADH complex may interfere with the efficiency of the proton-pumping process across the inner membrane of the mitochondria and that ATP8 has some regulatory role in the assembly of the proton channel (da Fonseca et al., 2008). This may have occurred in response to climate change, with optimization of cellular functions to new environmental conditions.
The African continent has been under major geographic and climatic oscillations in the recent past, including mountain formation, volcanic eruptions, temperature oscillations that led to major variation in animal and plant communities (e.g. Trauth, Larrasoaña & Mudelsee, 2009). During cold periods arid savannah habitats were expanding while in warmer periods they were being replaced by tropical forests (e.g. Flagstad et al., 2001). These oscillations between warm and humid and cold and dry periods caused habitat fragmentation, isolating several animal and plant species/populations, increasing biodiversity (Hewitt, 2004). Even though the different cycles are well documented specially the Pleistocene ones, their impact on southern African fauna is still poorly understood. However, previous studies have shown that species tend to contract during warm periods and expand during glacial periods (Flagstad et al., 2001; Lorenzen, Heller & Siegismund, 2012). This trend is the opposite of that seen in the Northern hemisphere where species contract during glacial periods and expand during interglacials (Hewitt, 2004). Periods of retraction usually give rise to spatial and temporal differentiation while expansion periods promote the establishment of secondary contact zones.
In one of the colder events, the ancestral population that gave rise to both H. niger and H. leucophaeus was probably divided, creating an island in the south of Africa. That population then grew in isolation and eventually became a new species. More specimens or nuclear data would be necessary to trace the fate of this extinct antelope; however, the paucity of fossils and museum material makes this a demanding job.
ACKNOWLEDGEMENTS
GET was partially funded by FEDER and COMPETE and by FCT (Portuguese Foundation for Science and Technology), under the project PTDC/BIA-BIC/4177/2012. We thank Dr. Hans Mejlon PhD, Curator of Entomology, Museum of Evolution – Zoology, Uppsala University for giving us access to the sample in question. We also thank Emeritus Professor Alfredo Rasteiro (University of Coimbra) for all his help in locating information on the H. niger variani specimen. Finally, we thank Vicente Campos Themudo for inspiration and allowing us the time to continue this research. Consensus mitochondrial genome sequence of H. leucophaeus has been deposited in GenBank with accession number MF043256. Bam files of the sequences generated in this project are available from NCBI’s Short Read Archive within BioProject PRJNA362292.

{kind=link}
{kind=link}