





Abstract
Inorganic trivalent arsenicals are vicinal thiol-reacting agents, and dithiothreitol (DTT) is a well-known dithiol agent. Interestingly, both decreasing and increasing effects of DTT on arsenic trioxide–induced apoptosis have been reported. We now provide data to show that, at high concentrations, DTT, dimercaptosuccinic acid (DMSA), and dimercaptopropanesulfonic acid (DMPS) decreased arsenic trioxide–induced apoptosis in NB4 cells, a human promyelocytic leukemia cell line. In contrast, at low concentrations DTT, DMSA, and DMPS increased the arsenic trioxide–induced apoptosis. DTT at a high concentration (3 mM) decreased, whereas at a low concentration (0.1 mM), it increased the cell growth inhibition of arsenic trioxide, methylarsonous acid (MMAIII), and dimethylarsinous acid (DMAIII) in NB4 cells. DMSA and DMPS are currently used as antidotes for acute arsenic poisoning. These two dithiol compounds also show an inverse-hormetic effect on arsenic toxicity in terms of DNA damage, micronucleus induction, apoptosis, and colony formation in experiments using human epithelial cell lines derived from arsenic target tissues such as the kidney and bladder. With the oral administration of dithiols, the concentrations of these dithiol compounds in the human body are likely to be low. Therefore, the present results suggest the necessity of reevaluating the therapeutic effect of these dithiol compounds for arsenic poisoning.
Arsenic, an environmental toxicant, is associated with several human diseases, including Blackfoot disease (Tsai et al., 2005), diabetes (Walton et al., 2004), hypertension (Rahman, 2002), and cancers of the skin (Rossman et al., 2004), lung, bladder, kidney, and liver (Chen et al., 1992). Although several hypotheses have been proposed, the mechanism of arsenic toxicity has not been established. Recent studies indicated that reactive oxygen species (Rossman et al., 2004) are involved in arsenite-induced cell signaling (Lynn et al., 2000), DNA damage (Lynn et al., 2000; Wang et al., 2001), gene mutation (Hei et al., 1998), micronuclei (Wang et al., 1997), apoptosis (Gurr et al., 1999; Wang et al., 1996), and cell proliferation (Barchowsky et al., 1999). Similarly, the production of nitric oxide has been demonstrated to be involved in arsenite-induced DNA damage (Liu and Jan, 2000), poly (ADP-ribosylation) (Lynn et al., 1998), micronucleus induction (Gurr et al., 1998), and inhibition of pyrimidine dimer excision (Bau et al., 2001). However, another hypothesis comes from a well-established concept that arsenite has a high affinity for vicinal thiols, and arsenite may bind critical thiols and thus interfere with normal cellular functions.
Arsenite has been shown to complex with the lipoic acid of pyruvate dehydrogenase (Hu et al., 1998), and with three closely spaced cysteines on the rat glucocorticoid receptor (Chakraborti et al., 1992). The binding affinity of arsenite was shown to be inversely related to the distance between the two thiol groups (Delnomdedieu et al., 1993). The active sites of many phosphatases contain adjacent sulfhydral residues (Cavigelli et al., 1996). Many phosphotyrosine phosphatases behave as vicinal thiol proteins and require DTT for activity measurements in vitro. Mammalian cells possess a system that regulates the redox status of cellular thiols and protects SH-containing protein from excessive oxidation. It includes low-molecular-weight donors of SH groups and enzymes, which can catalyze the reduction of SH groups in proteins and detoxify pro-oxidants by conjugation with glutathione. The protective effects of thiols such as glutathione, cysteine (Watson et al., 1996), and dithiol (Aposhian et al., 1984) against the toxic effects of arsenic suggest that arsenic toxicity results from the formation of reversible bonds with the thiol groups of proteins. This view is also consistent with the report that the whole-blood nonprotein sulfhydryl level in arsenic-exposed subjects was 60% of that of controls (Pi et al., 2000), and with reports showing that DTT suppresses As2O3-induced apoptosis (Cai et al., 2000; Zhu et al., 1999). Surprisingly, DTT and several other dithiol compounds were found to enhance As2O3-induced apoptosis in NB4 cells (Gurr et al., 1999). The purpose of this investigation was to scrutinize the apparent contradictory effects of DTT on As2O3-induced apoptosis in NB4 cells.
MATERIALS AND METHODS
Chemicals.
As2O3 and NaAsO2 were from Merck (Darmstadt, Germany). The source of MMAIII was the solid oxide (CH3AsO), and that of DMAIII was the iodide [(CH3)2AsI]. The precursors were prepared following procedures described in the literature (Cullen et al., 1989; Goddard, 1930) and were kept at −20°C. The purities of CH3AsO and (CH3)2AsI exceeded 99% as analyzed by nuclear magnetic resonance spectrometry (AMX400) and a Heraeus CHN-OS rapid element analyzer. CH3AsO and (CH3)2AsI were freshly diluted in deionized water to form CH3As(OH)2 and (CH3)2AsOH, which are the respective precursors for MMAIII and DMAIII. DTT, DMSA, and DMPS were from Sigma (St. Louis, MO).
Cells.
NB4 cells, a human promyelocytic leukemia cell line kindly provided by Dr. C. Y. Liu (Veterans General Hospital, Taipei, Taiwan, ROC), were cultured in RPMI-1640 medium. The 293 cell line, obtained from the American Type Culture Collection (Rockville, MD), is derived from adenovirus-transformed human embryonic kidney epithelial cells. The SV-HUC-1 cell line (ATCC CRL 9520) is a SV40 large T-transformed human urothelial cell line. SV-HUC-1 and 293 cells were cultured in F-12 medium and Dulbecco's modified Eagle medium (DMEM), respectively. All growth media were supplemented with 10% fetal calf serum, penicillin (100 units/ml), streptomycin (100 μg/ml), and 0.03% glutamine. Cultures were maintained at 37°C in a water-saturated atmosphere containing 5% CO2.
Measurement of nuclear fragmentation.
NB4 cells treated with As2O3 and/or dithiols for 48 h were fixed in 70% ethanol and stored at −20°C overnight. They were collected by centrifugation and resuspended in an ethanol–acetate mixture (3:1, v/v). Cells were dropped onto a glass slide, air-dried, and then stained with 20 μl of a 20 μM YOYO®-1 iodide (Molecular Probes, Eugene, OR) solution. Slides were then examined under an epifluorescence microscope with a 365-nm excitation filter, a 400-nm dichroic mirror, and a 435-nm barrier filter. The nuclear integrity of 500 randomly-selected cells was examined for each treatment.
Measurement of cell viability.
The cytotoxic effect of arsenic compounds and/or DTT was measured using a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. One thousand cells in 1 ml of medium were seeded in each well of a 24-well flat-bottom plate and incubated overnight. The medium was then replaced with drug-containing medium for an additional 72 h. One milliliter of the MTT solution (5 mg/ml) was then added to each well, and incubation was continued for an additional 3 h. Then, 100 μl of 100% dimethyl sulfoxide was added and incubated for 5 min in order to solubilize the blue formazan crystals. Absorbance at 570 nm was measured using a scanning multi-well ELISA reader (Sunrise Remote, TECAN, Grödig, Austria).
Measurement of glutathione.
The level of glutathione was measured with the aid of a fluorogenic probe, O-pathaldehyde (Sigma-Aldrich Chemical, Milwaukee, WI), as described by Senft et al. (2000).
Measurement of protein thiol.
Measurement of protein thiol followed the description by Ellman (1959) with some modifications. Briefly, the cell extract (60 μg) was precipitated with 200 μl of saturated ammonium sulfate. The mixture was vortexed and incubated for 15 min at room temperature. The precipitated protein was harvested by centrifugation at 12,000 × g. The supernatant was discarded, and the pellet was redissolved in 150 μl of phosphate-buffered saline and kept for 30 min at room temperature. Fifty μl of protein extracts was mixed with 200 μl of dithionitrobenzoic acid reagent (9.5 mg/ml dithionitrobenzoic acid in 0.1 M K2HPO4, 17.5 mM EDTA adjusted to pH 7.5, and diluted 1:50 with phosphate-buffered saline before use). The mixture was added to 96-well microtiter flat-bottom plates, and the absorbance was read at 412 nm. Results were calculated by subtracting the absorbance of a phosphate buffered saline blank and a dithionitrobenzoic acid blank from the absorbance of the tested sample.
Determination of trolox equivalent antioxidant capacity.
The trolox equivalent antioxidant capacity was measured by assessing the ability of hydrogen-donating antioxidants to scavenge the radical cations generated by 2,2′-azinobis(3-ethylbenzothiazoline)-6-sulfonic acid (ABTS) (Miller et al., 1993). Briefly, cells (1 × 106) after treatment were washed with phosphate-buffered saline, suspended in 200 μl of ice-cold lysis buffer (10 mM Tris–HCl (pH 7.4), 1 mM EDTA, 0.5 mM PMSF, 0.5 μg/ml leupeptin, and 0.5 μg/ml pepstatin), and sonicated for 3 min with 9-s pulses alternating with 1 s off. The lysate was centrifuged at 10,000 rpm for 10 min, and the protein concentration of the supernatant fraction was determined with a Bio-Rad Protein Assay Kit (Hercules, CA). Bovine serum albumin was used as a standard. ABTS dissolved in water (at a final concentration of 7 mM) was oxidized with 2.45 mM potassium persulfate for at least 1 h in darkness. The ABTS·+ (positively charged ABTS) solution was diluted to an absorbance of 0.8 ± 0.05 at 734 nm with 10 mM phosphate-buffered saline (pH 7.4). The absorbance was measured 15 min after the initial mixing of the cell extracts of different concentrations or the trolox standard with 1 ml of the ABTS·+ solution. The total antioxidant capacity of cell extracts was expressed as the trolox equivalent antioxidant capacity, calculated from at least three different concentrations of extract tested in the assay which gave a linear response.
Measurement of H2O2 production.
Cellular levels of H2O2 were measured with the aid of the fluorogenic probe, Amplex red, as previously described (Liu and Jan, 2000).
Measurement of DNA damage.
The comet assay, involving incubation with NB4 cell nuclear extract, was carried out as described by Wang et al. (2005). Briefly, the slides containing As2O3, DMSA, and DMPS-treated 293 or SV-HUC-1 cells were lysed and then incubated with nuclear extract prepared from NB4 cells to digest the DNA adducts and to transform them into DNA strand breaks. Migration of DNA from the nucleus in each cell was measured with Comet Assay III software (www.perceptive.co.uk). Fifty individual cells were measured for each treatment.
Measurement of micronucleus induction.
Cells seeded overnight were treated with 4 μg/ml cytochalasin B plus arsenite and/or dithiols for 24 h. Cells were removed by trypsinization, incubated in 0.05% KCl for 5 min, and fixed in a methanol–acetic acid mixture (3:1 v/v) for 3 min. The cells were dropped onto slides, air-dried, and stained with 500× SYBR green (Molecular Probes, Eugene, OR) for 10 min. The numbers of mononucleated cells, binucleated cells, and micronuclei in 1000 binucleated cells were counted under an epifluorescence microscope.
Measurement of sub-G1 populations.
Cells (1 × 106) seeded overnight were treated with As2O3 and/or dithiols for 48 h, then harvested by trypsinization, washed with phosphate-buffered saline, and fixed in 70% ethanol at −20°C overnight. Prior to analysis, cells were washed again with phosphate-buffered saline and resuspended in a solution containing 0.05 mg/ml propidium iodide (Sigma-Aldrich Chemical), 1 mM EDTA, 0.1% Triton X-100, and 1 mg/ml RNase A in phosphate-buffered saline. The suspension was mixed, and the DNA contents of the cells were measured with an EPICS® XL-MCL flow cytometer (Beckman-Coulter, Fullerton, CA) with excitation at 488 nm and emission at 620 nm.
Colony formation assay.
Cells were seeded onto 60-mm Petri dishes and incubated for 6 h to allow cell attachment. The number of cells seeded per dish was varied so that about 100 colonies were counted after 12 days of incubation. For each treatment, three dishes of cells were plated. At the end of the incubation, colonies were fixed for 10 min in 100% methanol and stained with a 10% Giemsa solution for 10 min. Colonies containing over 50 cells were counted. Percent colony formation was calculated by assigning untreated cultures as 100%. The percent colony formation of treated cells was calculated using the following formula: percent colony formation of treated cells = (colony number of treated cells / colony number of untreated cells) × 100.
Statistical analysis.
All experiments were performed independently at least three times. The mean of each experiment was calculated and is expressed as
RESULTS
DTT decreased the percentage of nuclear fragmentation in arsenite-treated NB4 cells at high doses (1, 2, 3, and 4 mM), while the opposite effect was true at lower doses (12.5, 25, and 50 μM) (Figs. 1A and 1D). Similar results were also obtained with NB4 cells exposed to arsenic in combination with either DMSA or DMPS (Figs. 1B, 1C, 1E, and 1F). To confirm this inverse-hormetic effect of dithiols on arsenite toxicity, the effects of high and low concentrations of DTT on the viability of cells treated with different trivalent arsenical compounds were examined. The results indicated that DTT at a high concentration (3 mM) increased but at a low concentration (100 μM) decreased the cell viability in As2O3-, NaAsO2-, MMAIII-, and DMAIII-treated cells (Figs. 2A–2D).
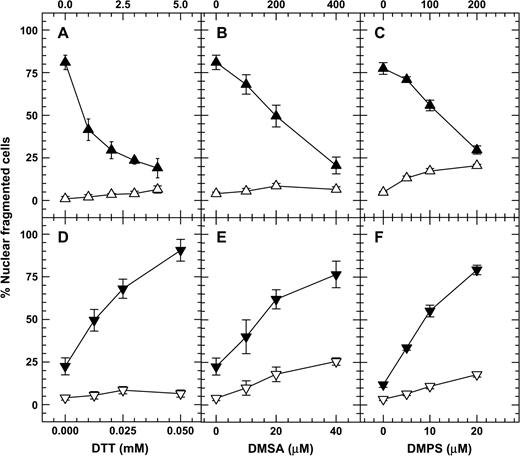
Dithiol compounds at high concentrations decreased but at low concentrations increased the nuclear fragmentation of As2O3-treated NB4 cells. Cells were treated for 48 h with high concentrations of dithiols alone (open triangles) or dithiols plus 3 μM As2O3 (filled triangles, A, B, C), and with low concentrations of dithiols alone (open inverted triangles) or dithiols plus 1 μM As2O3 (filled inverted triangles, D, E, F). To demonstrate the decreasing effect of DTT, a high concentration (3 μM) of As2O3 was used. To demonstrate the increasing effect of DTT, a low concentration (1 μM) of As2O3 was used.
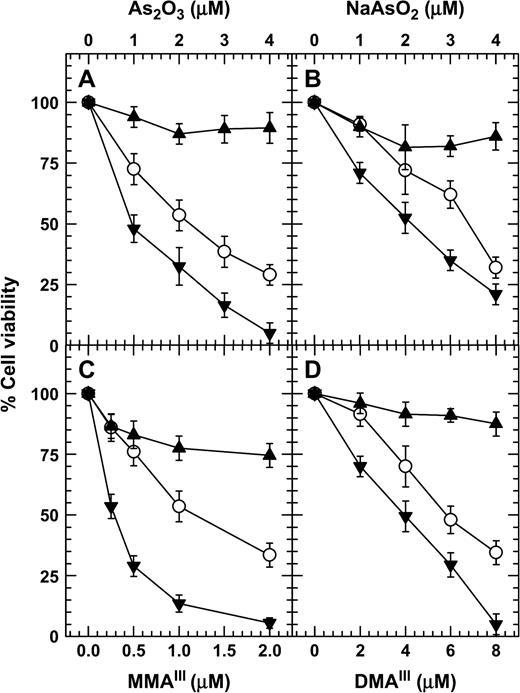
DTT at high concentrations increased but at low concentrations decreased the viability of cells treated with different trivalent arsenicals in NB4 cells. Cells were treated for 72 h with either different concentrations of arsenic alone (open circle), arsenic plus 3 mM DTT (filled triangles), or arsenic plus 100 μM DTT (filled inverted triangles). The cell viability was determined by MTT method.
Because arsenite is a thiol-binding agent, and DTT is a thiol donor, we examined the effects of treating NB4 cells with arsenite, DTT, and arsenite plus DTT on the cellular glutathione content and the levels of protein thiol. Treatment with arsenite decreased cellular glutathione content and protein thiol and reached a low level around 16 h (data not shown). For this reason, the effects of DTT on the level of glutathione and protein thiol were measured at 16 h after drug treatment. Treatment with As2O3 decreased the cellular glutathione content and the protein thiol levels (Figs. 3A and 3B). DTT at both high and low concentrations increased the cellular glutathione content and the protein thiol level in As2O3-treated cells (Figs. 3A and 3B). Because arsenite is known to express its toxicity through the generation of reactive oxygen species, we also examined the effect of treatment with arsenite plus DTT on the cellular total antioxidant capacity. Treatment with arsenite decreased the cellular total antioxidant capacity, which reached a low level around 16 h (data not shown). For this reason, the effects of DTT on the level of cellular total antioxidant capacity was measured at 16 h after drug treatment. The results indicated that treatment with As2O3 decreased the cellular total antioxidant capacity (Fig. 3C). DTT at high concentrations (1 and 2 mM) increased but low concentrations (25 and 50 μM) decreased the cellular total antioxidant capacity of As2O3-treated cells (Fig. 3C). High concentrations (1 and 2 mM) of DTT alone increased but low concentrations (50 μM) of DTT alone decreased the cellular total antioxidant capacity of NB4 cells (Fig. 3C). Treatment with As2O3 increased H2O2 production (Fig. 3D) and reached a peak around 2 h (data not shown). Treatment with low concentrations of DTT, DMSA, and DMPS further increased H2O2 production in As2O3-treated NB4 cells (Fig. 3D).
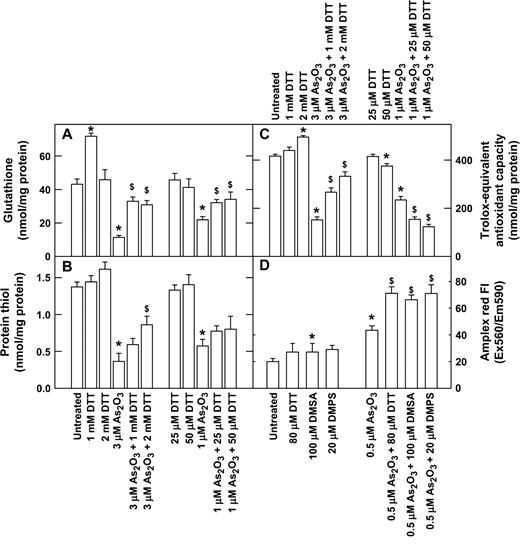
Effect of low and high concentrations of dithiol on the cellular reduced glutathione content (A), the level of protein thiol content (B), the cellular total antioxidant capacity (C), and the intracellular H2O2 level of As2O3-treated NB4 cells (D). In A, B, and C, cells were treated for 16 h with drugs. In D, cells were treated for 2 h with DTT, DMSA, DMPS, As2O3, or As2O3 plus dithiol, and intracellular H2O2 levels were measured using Amplex red. *p < 0.01 compared to untreated samples; $p < 0.01 compared to samples without dithiol.
Because DTT is known to decrease arsenite toxicity, and DTT was also reported to enhance As2O3-induced apoptosis in NB4 cells, we have started the experiments with NB4 cells, and the above results indicated that low concentrations of DTT increased whereas high concentrations of DTT decreased the toxicity of arsenite. Because dithiol compounds such as DMSA and DMPS are currently used as antidotes for acute arsenic poisoning, we were interested in determining if the inverse-hormetic effect of these dithiol compounds also occurs in cells of arsenic target tissues. Since a large portion of arsenic is excreted through urine, 293 cells, an adenovirus-transformed human embryonic kidney epithelial cell line, and SV-HUC-1 cells, an SV40 large T-transformed human urothelial cell line, were used as experimental materials. Appreciable amounts of DNA damage were detected by the comet assay with NB4 nuclear extract incubation in 293 and SV-HUC-1 cells treated with 1 μM As2O3 for 6 h (Figs. 4A and 4C). Cotreatment with 200 μM DMSA plus 1 μM As2O3 resulted in a lower level of DNA damage; however, cotreatment with 20 μM DMSA plus 1 μM As2O3 resulted in a higher level of DNA damage compared to the treatment with 1 μM As2O3 alone (Figs. 4A and 4C). Similarly, cotreatment with 100 μM DMPS plus 1 μM As2O3 resulted in a lower level of DNA damage; however, cotreatment with 10 μM DMPS plus 1 μM As2O3 resulted in a higher level of DNA damage compared to treatment with 1 μM As2O3 alone (Figs. 4B and 4D).
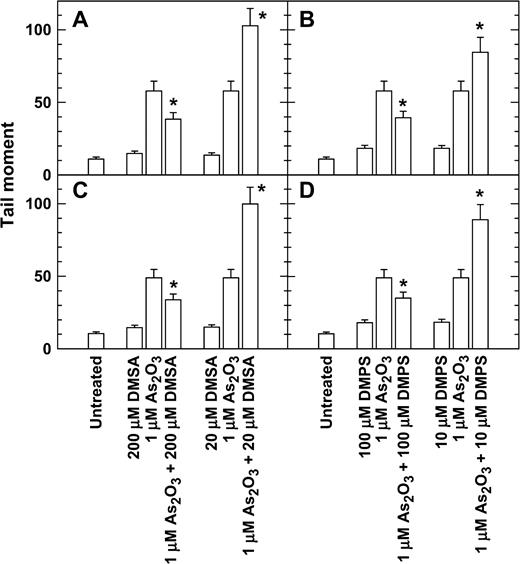
DMSA and DMPS at high concentrations decreased but at low concentrations increased the DNA damage of As2O3-treated 293 (A, B) and SV-HUC-1 cells (C, D). Exponentially growing cells were treated with As2O3, DMSA, and DMPS alone or in combination for 6 h, and the DNA damage was analyzed by a comet assay with nuclear extraction incubation. *p < 0.01 with versus without dithiol.
In cells treated with As2O3, MMAIII, and DMAIII, 200 μM DMSA or 100 μM DMPS decreased micronucleus induction, but 20 μM DMSA or 10 μM DMPS increased micronucleus induction (Fig. 5). Cotreatment with 200 μM DMSA or 100 μM DMPS plus As2O3 also reduced the proportion of cells with sub-G1 DNA content compared to cells treated with 2 μM As2O3 alone (Fig. 6). Cotreatment with 20 μM DMSA or 10 μM DMPS plus As2O3 also resulted in higher levels of cells with sub-G1 DNA content than did treatment with 2 μM As2O3 alone (Fig. 6). The inverse-hormetic effects of DMSA and DMPS were also observed in clonogenic survival studies (Fig. 7). Cotreatment with 200 μM DMSA or 100 μM DMPS plus As2O3 also resulted in higher levels of clonogenic survival, whereas cotreatment with 20 μM DMSA or 10 μM DMPS plus As2O3 resulted in lower levels of clonogenic survival compared to treatment with As2O3 alone.
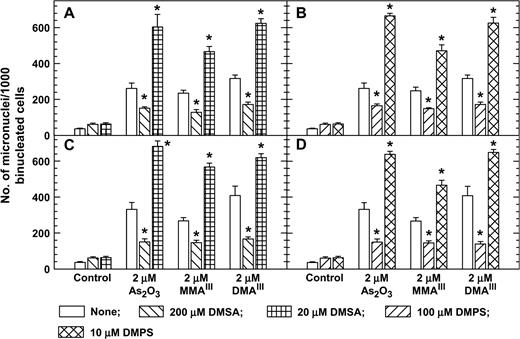
DMSA and DMPS at high concentrations decreased but at low concentrations increased micronucleus formation of As2O3-, MMAIII-, and DMAIII-treated 293 (A, B) and SV-HUC-1 cells (C, D). Exponentially growing cells were treated with arsenic compounds, DMSA, and DMPS plus 4 μg/ml cytochalasin B for 24 h. * p < 0.01 with vs. without dithiol. The percent of binucleated cells was >70%.
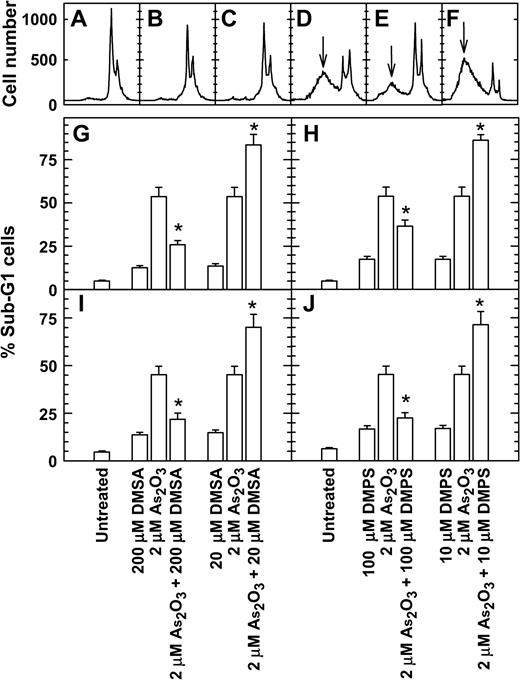
DMSA and DMPS at high concentrations decreased but at low concentration increased the proportion of cells with sub-G1 DNA content in As2O3-treated 293 and SV-HUC-1 cells. Exponentially growing cells were treated with drugs for 48 h, and the DNA content was analyzed by flow cytometry. (A–F) Representative DNA histograms. (A) Untreated 293 cells; (B) treated with 200 μM DMSA; (C) with 20 μM DMSA; (D) with 2 μM As2O3; (E) with 200 μM DMSA plus 2 μM As2O3; (F) with 200 μM DMSA plus 2 μM As2O3. ↓ indicates the sub-G1 peak. G and H, 293 cells; I and J, SV-HUC-1 cells. *p < 0.01 with versus without dithiol.
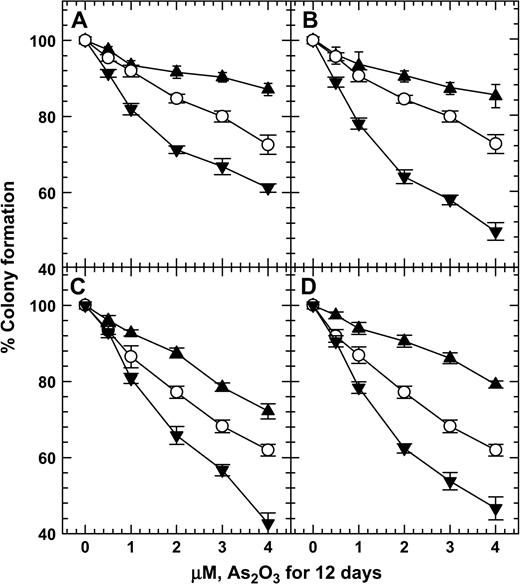
DMSA and DMPS at high concentrations increased but at low concentrations decreased the clonogenic survival of As2O3-treated 293 (A, B) and SV-HUC-1 cells (C, D). Open circles, treatment with As2O3; filled triangles, treatment with As2O3 plus 200 μM DMSA (A, C) or As2O3 plus 100 μM DMPS (B, D); filled inverted triangles, treatment with As2O3 plus 20 μM DMSA (A, C) or As2O3 plus 10 μM DMPS (B, D). The plating efficiencies of untreated 293 cells and 293 cells treated with 20 μM DMSA, 200 μM DMSA, 10 μM DMPS, or 100 μM DMPS were 81.7% ± 5.1%, 75.6% ± 2.0%, 66.7% ± 1.6%, 68.4% ± 2.9%, and 73.4% ± 1.9%, respectively. The plating efficiencies for untreated SV-HUC-1 cells and SV-HUC-1 cells treated with 20 μM DMSA, 200 μM DMSA, 10 μM DMPS, or 100 μM DMPS were 79.4% ± 4.4%, 73.4% ± 1.9%, 65.7% ± 2.8%, 73.8% ± 2.7%, and 70.0% ± 2.8%, respectively.
DISCUSSION
The present results demonstrate that dithiol compounds at high concentrations decreased, whereas at low concentrations they increased the toxicity of trivalent arsenicals. DMSA has been reported to deplete arsenic in subchronically arsenic exposed rats (Tripathi et al., 1997). A double-blind randomized controlled clinical trial study conducted on a few selected patients from arsenic-affected regions of West Bengal, India, with oral administration of DMSA suggested that DMSA was not effective in producing any clinical or biochemical benefits or any histopathological improvements in skin lesions (Mazumder et al., 1998). Results of this study suggest that DMSA has serious limitations in chelating arsenic from intracellular sites and can only be useful for acute cases of arsenic poisoning. On the other hand, DMPS treatment caused significant improvement in the clinical scores of patients suffering from chronic arsenic toxicity. Increased urinary excretion of arsenic during the period of therapy was the possible cause of this improvement (Mazumder, 2001). DMPS seems to form a complex with MMAIII, which is readily excreted in the urine (Aposhian et al., 2000).
With the oral administration of DMSA or DMPS, concentrations of these dithiol compounds in the human body probably are in the micromolar rather than in the millimolar range. These dithiol compounds at micromolar concentrations effectively enhanced the toxicity of arsenite in our present results. The reasons why dithiols enhance arsenic toxicity are still obscure. However, DTT has been reported to produce H2O2 in a reaction catalyzed by metals, predominately copper. The H2O2 then reacts in a metal-catalyzed Fenton reaction to produce the ultimate toxic species,·OH (Held and Biaglow, 1994; Kachur et al., 1997). The cytotoxicities of various thiols depend on the interplay of the rate of thiol oxidation and the rate of reaction between the thiol and H2O2 produced in the thiol oxidation. If the thiols, e.g., DTT and lipoic acid, react slowly with H2O2, it is toxic, and the toxicity is expressed in a biphasic pattern with no toxicity at high and low thiol concentrations, but increased toxicity at intermediate thiol concentrations (Held and Biaglow, 1994). These phenomena are probably due to higher thiol concentrations possibly having sufficient thiols to consume excessive H2O2 before damage occurs. However, oxidation of thiols with intermediate concentrations produced sufficient H2O2, which could not be completely eliminated by the low levels of thiols, and the excessive H2O2 may have caused toxicity in cells. This explanation can partially be applied to our present results. In the present experiments, neither DTT, DMSA, nor DMPS alone induced nuclear fragmentation (Fig. 1), DNA damage (Fig. 4), micronuclei (Fig. 5), sub-G1 cells (Fig. 6), or reduced colony formation (Fig. 7), either at high or low concentrations. The toxicity of a low concentration of DTT, DMSA, and DMPS was only detected in the presence of As2O3, and reactive oxygen species seemed to be involved in the inverse-hormetic effect of these dithiols. This notion came from the observation that treatment with 2 mM DTT increased but treatment with 50 μM DTT decreased the cellular total antioxidant capacity (Fig. 3), and that 1 and 2 mM DTT increased but 25 and 50 μM DTT decreased the cellular total antioxidant capacity of As2O3-treated NB4 cells (Fig. 3). We feel that more experiments are needed to reveal the mechanism of this inverse-hormetic effect of these dithiols on arsenite toxicity. For example DMPS is known to affect the transportation and metabolism of arsenite (Aposhian et al., 2000; Mazumder, 2001). Low concentration of dithiol may increase whereas high concentration dithiol may decrease the retention of arsenite in cells. Because DMPS is effective in increasing urinary excretion of arsenic (Aposhian et al., 2000; Mazumder, 2001), and antioxidants such as N-acetylcysteine (Huang et al., 1993), pyruvate, and selenium (Samikkannu et al., 2003; Wang et al., 2001) are known to reduce arsenite toxicity, these antioxidants can possibly be used in combination with DMPS for treatment of arsenic poisoning.
References
Aposhian, H. V., Carter, D. E., Hoover, T. D., Hsu, C. A., Maiorino, R. M., and Stine, E. (
Aposhian, H. V., Zheng, B., Aposhian, M. M., Le, X. C., Cebrian, M. E., Cullen, W., Zakharyan, R. A., Ma, M., Dart, R. C., Cheng, Z., et al. (
Barchowsky, A., Roussel, R. R., Klei, L. R., James, P. E., Ganju, N., Smith, K. R., and Dudek, E. J. (
Bau, D. T., Gurr, J. R., and Jan, K. Y. (
Cai, X., Shen, Y. L., Zhu, Q., Jia, P. M., Yu, Y., Zhou, L., Huang, Y., Zhang, J. W., Xiong, S. M., Chen, S. J., et al. (
Cavigelli, M., Li, W. W., Lin, A., Su, B., Yoshioka, K., and Karin, M. (
Chakraborti, P. K., Garabedian, M. J., Yamamoto, K. R., and Simons, S. S., Jr. (
Chen, C. J., Chen, C. W., Wu, M. M., and Kuo, T. L. (
Cullen, W. R., McBride, B. C., Manji, H., Pickett, A. W., and Reglinski, J. (
Delnomdedieu, M., Basti, M. M., Otvos, J. D., and Thomas, D. J. (
Goddard, A. D. (
Gurr, J. R., Bau, D. T., Liu, F., Lynn, S., and Jan, K. Y. (
Gurr, J. R., Liu, F., Lynn, S., and Jan, K. Y. (
Hei, T. K., Liu, S. X., and Waldren, C. (
Held, K. D., and Biaglow, J. E. (
Hu, Y., Su, L., and Snow, E. T. (
Huang, H., Huang, C. F., Wu, D. R., Jinn, C. M., and Jan, K. Y. (
Kachur, A. V., Held, K. D., Koch, C. J., and Biaglow, J. E. (
Liu, F., and Jan, K. Y. (
Lynn, S., Gurr, J. R., Lai, H. T., and Jan, K. Y. (
Lynn, S., Shiung, J. N., Gurr, J. R., and Jan, K. Y. (
Mazumder, D. N. (
Mazumder, D. N., Das Gupta, J., Santra, A., Pal, A., Ghose, A., and Sarkar, S. (
Miller, N. J., Rice-Evans, C., Davies, M. J., Gopinathan, V., and Milner, A. (
Pi, J., Kumagai, Y., Sun, G., Yamauchi, H., Yoshida, T., Iso, H., Endo, A., Yu, L., Yuki, K., Miyauchi, T., et al. (
Rossman, T. G., Uddin, A. N., and Burns, F. J. (
Samikkannu, T., Chen, C. H., Yih, L. H., Wang, A. S., Lin, S. Y., Chen, T. C., and Jan, K. Y. (
Senft, A. P., Dalton, T. P., and Shertzer, H. G. (
Tripathi, N., Kannan, G. M., Pant, B. P., Jaiswal, D. K., Malhotra, P. R., and Flora, S. J. (
Tsai, Y. S., Yang, W. H., Tong, Y. C., Lin, J. S., Pan, C. C., and Tzai, T. S. (
Walton, F. S., Harmon, A. W., Paul, D. S., Drobna, Z., Patel, Y. M., and Styblo, M. (
Wang, A. S., Ramanathan, B., Chien, Y. H., Goparaju, C. M., and Jan, K. Y. (
Wang, T. S., Hsu, T. Y., Chung, C. H., Wang, A. S., Bau, D. T., and Jan, K. Y. (
Wang, T. S., Kuo, C. F., Jan, K. Y., and Huang, H. (
Wang, T. S., Shu, Y. F., Liu, Y. C., Jan, K. Y., and Huang, H. (
Watson, R. W., Redmond, H. P., Wang, J. H., and Bouchier-Hayes, D. (
Author notes
*Institute of Cellular and Organismic Biology, Academia Sinica, Taipei 11529, Taiwan, ROC; and †Hsing Wu College, No. 101, Sec. 1, Fenliao Rd., Linkou Township, Taipei County 24452, Taiwan, ROC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments