





Abstract
The azoxymethane (AOM) model has been widely used to investigate the pathology and genetics of colorectal cancer in rodents. However, there has been wide variation in treatment regimes, making it difficult to compare across studies. Consequently, standardizing AOM treatment and identifying sources of experimental variation would allow better comparisons across studies. In order to establish an optimal dosing regime for detecting experiment-dependent differences in tumorigenesis, we performed a dose curve analysis using AKR/J, SWR/J, and A/J mouse strains previously reported to vary widely in susceptibility to AOM. Although intraperitoneal or subcutaneous administration, but not in utero exposure, resulted in similar levels of tumor induction, significant dose- and strain-dependent effects of AOM were observed. No sex-dependent differences were observed. Increasing the number of treatments uncovered a significant strain-dependent effect on tumor promotion, independent of susceptibility to tumor initiation. Similarly, we used C57BL/6J and DBA/2J intercrosses to demonstrate that small diet modifications can significantly alter AOM-induced tumorigenesis in a background-dependent manner. These results provide experimental support for a standardized AOM treatment and for the importance of controlling both genetic and non-genetic factors when using this model.
Colorectal cancer (CRC) is the second leading cause of cancer deaths in the U.S., with over 150,000 new cases diagnosed each year (American Cancer Society, 2004). Although a small subset of these cases are well-characterized hereditary syndromes, such as familial adenomatous polyposis (FAP) and hereditary non-polyposis colon cancer (HNPCC), the vast majority of CRCs are considered non-familial occurring in individuals with heightened genetic susceptibility as a result of the interaction between multiple genes with low penetrance and environmental exposures (Demant, 2003; DiGiovanni, 1997; Kinzler and Vogelstein, 1996).
The discovery that 1,2-dimethylhydrazine (DMH) and its metabolite azoxymethane (AOM) are specific colon carcinogens paved the way to model non-familial CRC in rodents. Using a wide-variety of dose regimes across different studies, it has been reported that inbred mouse strains vary extensively in their susceptibility to these carcinogens (Diwan and Blackman, 1980; Evans et al., 1974; Nambiar et al., 2003; Papanikolaou et al., 1998). The response of mice to AOM-induced CRC mirrors the occurrence of non-familial CRC in humans, including a preponderance of distal colonic tumors that are pathologically similar to sporadic CRC in the descending (left-sided) human colon (Chang, 1978; Druckrey, 1970). The genetic diversity among inbred strains coupled with similarities of this disease model to human CRC incidence provides relevance for the use of this model to investigate the pathology of non-familial CRC and to identify genetic and non-genetic CRC susceptibility factors. However, because of the wide variety of treatment regimes used in previous studies, interpreting relative susceptibilities of different strains or environmental conditions to AOM-induced CRC across studies remains difficult.
Standardizing the AOM treatment would provide more informative comparisons across studies. In order to establish an optimal dosing regime that would maximize inter-strain differences in tumorigenesis, we assessed AOM-induced tumorigenesis in A/J, SWR/J, and AKR/J mouse strains with varying susceptibility, using a variety of dose regimes. All mice were analyzed six months after the first dose of AOM to reveal significant strain-dependent differences in the effects of AOM, including a role in tumor promotion that has not been previously described. Furthermore, no sex-dependent differences were observed and exposure to AOM in utero was not carcinogenic. However, small dietary modifications revealed strain-dependent effects.
MATERIALS AND METHODS
Mouse strains, husbandry, and genetic crosses.
Founder stocks for A/J, AKR/J, C57BL/6J, DBA/2J, and SWR/J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred in-house. The mice were housed in an AAALAC-approved pathogen-free barrier facility at constant temperature and humidity and on a 12-h light/12-h dark cycle. All mice in the study tested negative for Helicobacter sp. Most mice were group housed by sex with up to five mice per cage in ventilated racks (UNC) or microisolators (Vanderbilt) and provided LabDiet (St. Louis, MO) 5010 maintenance chow containing 5% fat and 24% protein by weight and autoclaved (UNC) or acidified (Vanderbilt) water ad libitum. For diet modification experiments, mice were provided LabDiet 5015 breeder chow containing 11% fat and 21% protein by weight. Approximately equal numbers of males and females were used in all experimental groups.
Female C57BL/6J mice were crossed to male DBA/2J to generate isogenic B6D2F1 hybrids that were subsequently intercrossed to produce a population of B6D2F2 mice. SWAF3 mice were generated by crossing female SWR/J with male A/J to obtain SWAF1 hybrids, followed by two consecutive intercrosses to produce F2 and F3 mice, respectively. The Institutional Animal Care and Use Committee approved all procedures involving mice.
Carcinogen treatment.
A single lot of AOM was obtained from Sigma-Aldrich (St. Louis, MO) in 100 mg isovials and stored at −80°C. Each 100 mg vial was resuspended in 2 ml phosphate-buffered saline (PBS) and individual 250 μl aliquots stored at −80°C until use. A working stock of 1.25 mg/ml AOM was made by diluting individual 250 μl aliquots into 10 ml of saline (0.9% NaCl). Mice, three to five months of age, were treated intraperitoneal (ip) or subcutaneous (sc) with 5, 10, or 20 mg AOM per kg body weight once a week for 2, 4, or 8 weeks as described in the results. Mice dosed at younger than three months of age showed a high sporadic incidence of acute toxicity (data not shown). Prenatal exposures were by maternal ip injection. Age-matched controls were injected with saline.
Tumor histopathology.
Mice were euthanized by CO2 asphyxiation six months after the first AOM dose. Colons, from caecum to rectum, were removed, gently flushed with PBS, and spayed open along the longitudinal axis on Whatmann 3MM filter paper. Tumors 1 mm or larger were counted and measured and their location along the caecal to rectal axis noted. Each colon was loosely rolled from rectum to caecum and held by spearing with a 30-guage needle before fixing overnight in 10% neutral buffered formalin. The fixed colons were dehydrated in ethanol and embedded in paraffin. Six-micron thick cross-sections were cut with a microtome and stained with hematoxylin and eosin to evaluate tumor histopathology.
Statistical analysis.
The nonparametric Mann-Whitney U test was used to analyze all tumor multiplicity and size comparisons. One-sided p values were determined.
RESULTS
Dose Response
The optimal AOM dose for inducing colon tumors, while minimizing potential toxicity, was determined using the A/J strain that was previously reported to be highly susceptible to AOM-induced colon tumors (Nambiar et al., 2003). A/J mice were treated with weekly ip doses of 5, 10, or 20 mg AOM per kg body weight for four consecutive weeks (Fig. 1). Six months after the first AOM dose, mice were analyzed for tumor development. All mice treated with 20 mg/kg of AOM died shortly after the first injection suggesting that AOM is acutely toxic at this dose; there were no premature losses of mice in the other treatment groups. While distal colonic adenomas developed in both the 5 and 10 mg/kg groups, the percentage of tumor-bearing mice (penetrance) was 10-fold higher in the 10 mg/kg group than those treated with 5 mg/kg body weight. Similarly, tumor multiplicity and diameter were significantly higher in the 10 mg/kg treated group, suggesting that AOM was acting as a tumor growth promoter in addition to an initiator. No sex-dependent differences were observed.
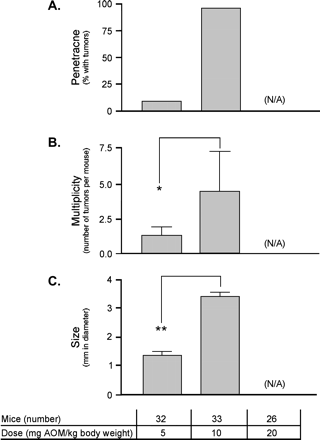
Dose response to AOM. A/J mice were injected ip with 5, 10, or 20 mg AOM per kg body weight for four weeks and analyzed for tumor penetrance (A), multiplicity (B), and size (C). The number of mice used in each treatment group, with approximately equal numbers of males and females, is shown. *p < 0.05; **p < 0.01; (N/A), data not available since all animals died.
Treatment Course and Route of Administration
Having established the maximal tolerated dose for the highly susceptible A/J strain, we next determined the optimal treatment course and route of carcinogen exposure to produce the greatest inter-strain differences in the development of colon tumors. Three strains, AKR/J, SWR/J, and A/J, with reported differences in susceptibility (Nambiar et al., 2003), were administered AOM over three treatment courses and by two routes of administration (Fig. 2). One AKR/J mouse in each of the two 4-week AOM treatment groups developed a single tumor while no tumors developed in untreated controls. The tumors that developed in SWR/J and A/J mice in all treatment groups were located in the distal colon (data not shown). Occasionally extra-colonic lesions were observed, including one uterine tumor in an A/J mouse from the 2-week ip treatment group and rectal tumors in two A/J mice from the 4-week sc group. A rectal tumor was also observed in a single SWR/J mouse from the 8-week treatment course. However, these extra-colonic lesions were not consistently observed suggesting that they are not a major site of AOM carcinogenesis.
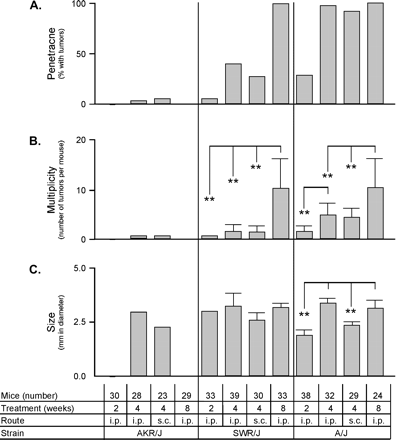
Treatment and route of administration response. Mice were tested using 10 mg AOM per kg body weight with 2, 4, or 8 weekly ip or sc injections. Each treatment regime was analyzed for penetrance (A), multiplicity (B), and size (C). The number of mice used in each treatment group, with approximately equal numbers of males and females, is shown. *p < 0.05; **p < 0.01.
In the 2- and 4-week treatment groups, A/J mice had higher penetrance and tumor multiplicity than SWR/J mice. The difference in tumor susceptibility between SWR/J and A/J was particularly evident in the two 4-week treatment groups. Almost 100% of A/J mice developed colonic tumors in the 4-week treatment groups, while in the less susceptible SWR/J mice, tumor penetrance only reached 100% in the 8-week treatment group suggesting that an 8-week treatment would obscure differences in susceptibility between the SWR/J and A/J strains. Tumor multiplicity was also comparable between A/J and SWR/J mice with the 8-week treatment averaging 8.19 ± 1.4 and 10.3 ± 1.0, respectively. In the 4-week treatment group, the route of administration did not significantly affect tumor penetrance or multiplicity in any of the strains tested. Interestingly, a strain-dependent, route of administration difference in average tumor size was observed. A/J mice in the 4-week sc treatment group developed significantly smaller tumors than the ip group (2.34 ± 0.09 mm versus 3.42 ± 0.13 mm, respectively; p < 0.0001) while there was no difference between sc and ip route of delivery in SWR/J mice. Tumor size also increased in A/J mice between the 2- and 4-week treatment groups (p < 0.0001), but not between the 4- and 8-week treatment groups, suggesting that tumor growth promotion plateaus.
Histopathological Progression
All tumors arising from 2- and 4-week ip treatment groups were pedunculated adenomas (Fig. 3). A/J-derived tumors from the 8-week treatments, although larger, were only slightly more progressed histologically than those from the 4-week treatment groups. However, SWR/J-derived tumors from the 8-week treatment group had a marked increase in tumor progression over that observed in other treatment groups or strains showing that AOM is also a potent strain-dependent tumor promoter. Of the 20 SWR/J tumors characterized histologically from the 8-week treatment group, 4(20%) had crypts with cribriform architecture while 2(10%) showed evidence of local invasion through the basement membrane. Furthermore, many of the SWR/J tumors from the 8-week treatment group had areas totally devoid of characteristic epithelial crypts and inter-glandular stroma, characteristics of carcinoma in situ. None of the A/J or SWR/J 4-week treatment groups demonstrated these characteristics.
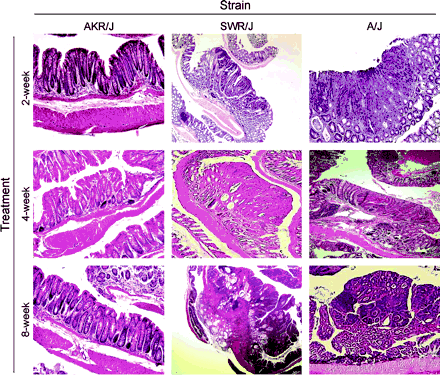
Tumor histology from each treatment group. Representative hematoxylin and eosin stained sections of colons from AKR/J (no tumors), SWR/J and A/J (with tumors) six months after 2, 4, or 8 weekly ip injections of 10 mg AOM per kg body weight.
In Utero Exposure
Mice representing different genetic backgrounds were also tested for the effects of in utero exposure to AOM. AOM was administered ip in one or two doses at 10 mg/kg body weight to pregnant C57BL/6J, DBA/2J, and SWAF3 females between 10 and 18 days of gestation and their offspring necropsied at 6 months of age. Unlike post-natal exposure, no mice exposed pre-natally to AOM developed tumors suggesting that mice are not susceptible to gestational exposure (data not shown).
Diet Modification
To determine the effect of environmental changes on AOM-induced colon tumor development, we tested two commonly available mouse chows on DBA/2J, C57BL/6J, D2B6F1, and D2B6F2 mice using the optimal 4-week ip AOM treatment regime (Fig. 4). Mice were maintained on LabDiet 5010 chow, a standard mouse maintenance chow, or LabDiet 5015 chow, a typical breeder chow with higher fat and lower protein content. Although C57BL/6J mice on the 5015 chow had a 50% lower tumor penetrance than those on the 5010 chow, there were no diet-dependent differences on multiplicity or tumor size in C57BL/6J mice that did develop tumors. DBA/2J and B6D2F1 mice maintained on the 5015 chow showed only a 20–25% lower penetrance compared to those on the 5010 chow. However, unlike C57BL/6J and B6D2F1 mice, DBA/2J had a reduction in multiplicity and tumor size in response to the 5015 chow. Based upon the response of B6D2F1 mice to the diet modification, the lower reduction in tumor penetrance observed in DBA/2J is dominant to the C57BL/6J effect while the lack of diet-induced changes on multiplicity and tumor size observed in the C57BL/6J is dominant to the DBA/2J effect. These results suggest a complex interaction between genetic background and diet on AOM-mediated tumorigenesis. Interestingly, in the segregating F2 generation, tumor penetrance and multiplicity both increased in mice fed 5015 compared to those on 5010, while tumor size decreased. These results are indicative of a polygenic model underlying the genetic background-dependent effects on diet-mediated changes to tumorigenesis. There was no correlation between overall body weight and tumor incidence, and tumor histopathology was not altered by diet (data not shown). Other environmental modifications, like water source (autoclaved versus acidified), bedding (corn cob versus recycled paper), or housing (ventilated versus micro-isolator), had no measurable effect on tumor development in the 4-week ip treated A/J mice (data not shown).
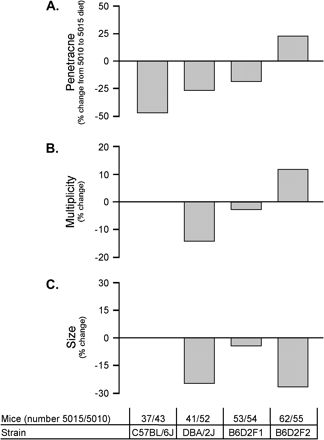
Strain-dependent diet effects on AOM treatment response. All mice were treated with four weekly ip injections of 10 mg AOM per kg body weight. Each strain or cross was analyzed and the percent change in average response from the 5010 to the 5015 diet was determined for penetrance (A), multiplicity (B), and size (C). No difference between the diets would be 0. The number of mice used in each treatment group, with approximately equal numbers of males and females, is shown.
DISCUSSION
The AOM mouse model has been utilized extensively to investigate molecular and genetic aspects of CRC. While, the variety of treatment regimes previously used makes it difficult to compare across studies, a standardized treatment would provide greater consistency for studying host and environmental factors that affect tumorigenesis. Therefore, the purpose of the present study was to identify an optimal AOM treatment to maximize the detection of experimental differences in tumorigenesis, and to provide a standardized protocol that can be used for comparison between studies. Various experimental variables were also investigated to determine their effect on this carcinogen-based CRC model.
Dose curves using the susceptible A/J strain were used to identify 10 mg AOM per kg body weight as the maximal tolerated dose for tumor induction. Although A/J mice are known to be highly susceptible to AOM-induced colon tumorigenesis, mice in the 5 mg/kg treatment group showed a low tumor penetrance, while all mice treated with 20 mg/kg died within a week of receiving the first dose suggesting acute toxicity. This toxicity is similar to fulminant hepatic failure in C57BL/6J mice caused by high doses of AOM (Matkowskyj et al., 1999).
The maximal tolerated dose of AOM was further evaluated to determine the optimal treatment and route of administration to maximize inter-strain differences using A/J, AKR/J, and SWR/J mouse strains that are known to vary in their susceptibility to AOM-induced colon tumorigenesis (Nambiar et al., 2003). Although AKR/J mice were strongly resistant to tumor development, SWR/J and A/J mice showed dose-dependent responses that were relatively independent of the route of administration. Tumor penetrance in A/J mice plateaued at 100% at four weeks of treatment while SWR/J mice did not reach 100% penetrance until eight weeks of treatment. Comparison of tumor multiplicities across treatments showed significant increases in both strains between four and eight weeks. Multiplicity, which was lower at two and four weeks of treatment in SWR/J mice compared to A/J, became equivalent between the strains after eight weeks of treatment. These studies show that four weekly ip treatments with 10 mg AOM per kg body weight is optimal to maximize inter-strain differences for both penetrance and multiplicity of colon tumors. While there may be slight differences in the optimal dose between different laboratory environments, the dose reported here should be generally applicable since we did not detect any effects due to water, bedding, or caging variables.
Although both tumor size and multiplicity increased, tumor morphology changed little in A/J mice with increasing a number of AOM doses; SWR/J mice had similar increases in tumor penetrance. However, unlike A/J, tumors from SWR/J mice did not increase in size but did show greatly increased tumor progression, with signs of local invasion and characteristics of carcinoma in situ, after eight weeks of AOM treatment. This finding shows that AOM is a potent strain-dependent tumor promoter in addition to being a tumor initiator.
Previous studies have shown that administration of the downstream AOM metabolite, methylazoxymethanol (MAM), is a neuroteratogen when administered in utero (Rodier et al., 1991). Although AOM crosses the placenta, our results show that when AOM is administered in utero, offspring remain tumor-free through six months of age. This result is consistent with metabolic studies suggesting that AOM is probably not metabolized to MAM in utero because required oxidation of AOM by the fetal liver does not occur (Miller, 1964; Weisburger, 1971).
In addition to genetic variation, non-genetic factors, like diet, are known to alter AOM-mediated tumorigenesis (Ju et al., 2003; Matsubara et al., 2003). However, most previous dietary studies have been performed on single genetic backgrounds. As more genetically engineered mutants and transgenic models are used to investigate the role of specific genes in CRC, mixed backgrounds become a necessity unless extensive and lengthy backcrossing is performed to generate congenic lines. We used C57BL/6J, DBA/2J, and B6D2 intercrosses as representative backgrounds to investigate potential interactions between genetic background and dietary modifications. Additionally, we used two common diets, a standard maintenance chow (5010) and a breeder chow (5015), that have relatively small compositional differences and as such are also representative of those that might be involved in inadvertent diet mix-ups within animal facilities. Our results showed an interaction between genetic background and diet with C57BL/6J being much more sensitive to the change than DBA/2J when measuring penetrance; this difference is genetically controlled since the DBA/2J effect was dominant based upon the response of B6D2F1 hybrids. Furthermore, this observation was complicated by the fact that multiplicity and tumor size was not changed in C57BL/6J or B6D2F1 mice but was by the DBA/2J background. Unexpectedly, dominance for the effect on multiplicity and tumor size was reverse to that observed for penetrance. The effect in the B6D2F2 mixed population was reverse that observed in the parental strains, indicative of a complex and probably polygenic interaction between the backgrounds.
These studies provide an experimental rationale for a standardized AOM treatment regime that will better support comparisons across studies. Moreover, these results demonstrate that genetic background can significantly modulate the effects of environmental variables necessitating proper experimental control when using the AOM model. Finally, we have discovered that in addition to acting as a tumor initiator, AOM is also a potent and genetic background-dependent tumor promoter.
We thank members of our lab for insightful discussions and suggestions. This work was supported by grants from the National Institutes of Health (CA79869, CA16086, CA106991, ES11391, and ES10126), a National Cancer Institute pilot grant from the Vanderbilt-Ingram Cancer Center (CA68485), and the American Cancer Society (MGO-89341) to D.W.T. A.B. was supported by a National Institutes of Health pre-doctoral fellowship (ES012354). Conflict of interest: none declared.
References
Chang, W. W. (
Demant, P. (
DiGiovanni, J. (
Diwan, B. A., and Blackman, K. E. (
Druckrey, E. (
Evans, J. T., Hauschka, T. S., and Mittelman, A. (
Ju, J., Liu, Y., Hong, J., Huang, M. T., Conney, A. H., and Yang, C. S. (
Kinzler, K. W., and Vogelstein, B. (
Matkowskyj, K. A., Marrero, J. A., Carroll, R. E., Danilkovich, A. V., Green, R. M., and Benya, R. V. (
Matsubara, K., Komatsu, S., Oka, T., and Kato, N. (
Nambiar, P. R., Girnun, G., Lillo, N. A., Guda, K., Whiteley, H. E., and Rosenberg, D. W. (
Papanikolaou, A., Wang, Q. S., Delker, D. A., and Rosenberg, D. W. (
Rodier, P. M., Kates, B., White, W. A., and Muhs, A. (
Author notes
*Curriculum in Toxicology, †Department of Genetics, ‡Department of Cell Biology and Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, Tennessee 37232; §Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, North Carolina 27599; and ¶Center for Environmental Health and Susceptibility and the Lineberger Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments