





Introduction
A number of renal diseases may be accompanied by malformation syndromes. Oral–facial–digital syndrome type 1 (OFD1) is a rare disorder and involves malformations of the face, oral cavity, hands and feet. It is transmitted as an X-linked dominant condition with lethality in males. Cystic kidney disorders are a recognized feature of OFD1 [1–5]. Caroli's disease is characterized by multifocal segmental dilatation of intrahepatic bile ducts [6,7]. A case of OFD1, associated with cystic renal disease and Caroli's disease, and complicated by end-stage renal failure, is reported here. To our knowledge, Caroli's disease has not been reported previously in OFD1.
Mutation analysis
To test the possible involvement of OFD1, we studied the total genomic DNA by DHPLC analysis on the 23 coding exons that are required to code for the OFD1 transcript. The corresponding polymerase chain reaction (PCR) product was analysed by direct sequencing on both the forward strand and the reverse strand. The primers and conditions used for the mutation analysis have been described earlier [8]. PCRs were carried out on genomic DNA extracted from peripheral blood leukocytes using the Capture Column Kit (Gentra Systems). PCR products were checked on agarose gel and then sequenced on both strands using the ABI Prism Big Dye Terminator Cycle Sequencing Kit (Perkin Elmer) and an ABI 377 automated DNA sequencer. Mutation analysis on the patient was performed by DHPLC using the Wave DNA Fragment Analysis System (Transgenomic, Inc.) according to the manufacturer's instructions. We followed the standard nomenclature to describe the mutation [9]. PCR-amplified products were cloned using the TOPO TA cloning kit (Invitrogen) to separate the mutant and wild-type alleles. Approval from the ethics committee was obtained for the study.
Case
In September 2004, a 20-year-old girl was admitted to our hospital with complaints of malaise and decreased urine volume. Examination of her medical history indicated that oral and facial malformations were noted shortly after her birth. She had therefore undergone oral plastic surgery procedures for cleft palate and cleft tongue. In August 2004, her urine output had decreased to 400 ml/day. On admission, she presented a mildly asymmetrical face, a depressed upper nasal bridge, anteverted nostrils, a thin upper lip, micrognathia, teeth abnormalities, open bite (Figure 1A), a highly arched palate with cleft palate (Figure 1B), and a lobulated tongue (Figure 1C). She had small hands and feet. Both hands had short metacarpals, especially the fourth one, and short phalanges (Figure 2A). The shortness of the phalanges was more conspicuous than the metatarsals in both feet (Figure 2B). Her height was 147 cm and her body mass index, 17 kg/m2. Delayed bone age (age of 18) and borderline mental functioning (IQ test score of 80) were diagnosed. Four generations of the patient's family members were screened for malformations and we were not able to diagnose any family member with somatic malformation, polycystic kidneys or cystic liver diseases. Laboratory analyses provided the following results: haemoglobin 10.4 g/dl, blood urea nitrogen 143 mg/dl and creatinine 8.5 mg/dl. The glomerular filtration rate was estimated at 6 ml/min. Urinalysis showed 10–12 leukocytes and two to three erythrocytes per high-power field and her urine density was 1010. There was no growth of any microbial agent in her urine culture. Abdominal ultrasonography and abdominal magnetic resonance imaging revealed a tubular and cystic dilatation of intrahepatic bile ducts (Figure 3A and B) and bilaterally enlarged kidneys with numerous small cysts in the cortical and medullary sections and without a normal parenchyma (Figure 4A and B). A false tendon in the left ventricular cavity was revealed by echocardiography. Dilatation of extracerebral cerebrospinal fluid in bilateral anterior temporal lobes was also observed in the cranium. Mutation analysis revealed an abnormality in the DNA segment corresponding to exon 9 and the presence of a 5-bp deletion, 837–841 del AAAAG G, leading to a frameshift. This deletion was confirmed after cloning and analysis of the two distinct alleles. Other laboratory parameters were within normal ranges. End-stage renal failure, Caroli's disease and OFD1 were diagnosed. An arteriovenous fistula was performed and the patient was placed on chronic haemodialysis.
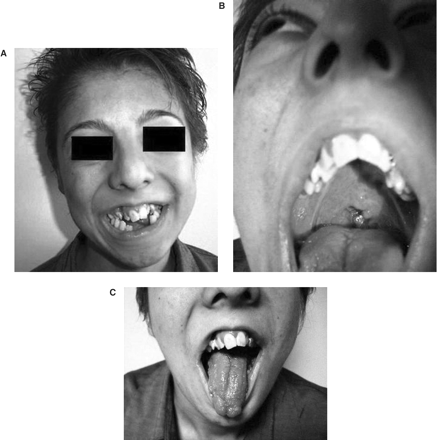
(A) Peculiar face of the patient and tooth abnormalities, (B) cleft palate, (C) lobulated tongue.
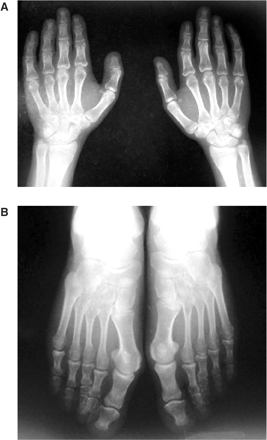
(A) Radiograph of both hands showing short metacarpals and phalanges, (B) radiograph of both feet showing short metatarsals and phalanges. The shortness of the phalanges is more conspicuous than the metatarsals.
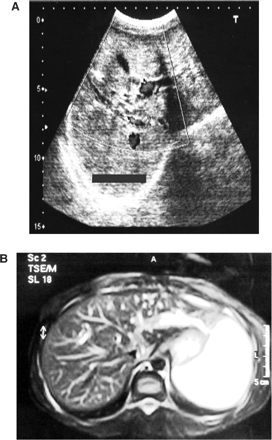
(A) Abdominal ultrasonography revealing tubular and cystic dilatations of intrahepatic bile ducts, (B) magnetic resonance cholangiopancreatography revealing dilated intrahepatic bile ducts with normal extrahepatic bile ducts.
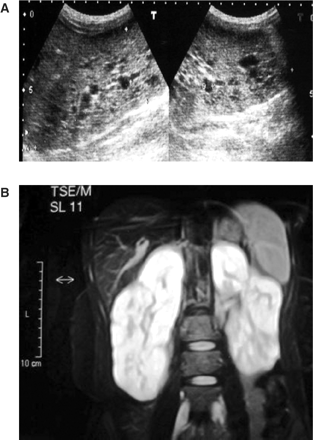
(A) Ultrasonography of kidneys revealing bilateral renal cysts 5–15 mm in size, (B) abdominal MRG revealing bilaterally slightly enlarged kidneys with numerous small cysts in the cortical and medullary sections.
Discussion
Cystic kidney disorders are one of the leading causes of end-stage renal disease. In addition to autosomal dominant polycystic kidney disease (ADPKD), there are other numerous disorders that have renal cysts as a common feature. OFD1 is one such cystic kidney disorder [10]. Renal involvement in OFD1 cases may be as high as 15% and is characterized by multiple cysts [11]. At least nine different forms of oral–facial–digital syndromes have been described, type 1 being the most common. OFD1 occurs approximately in one for every 250 000 live births. It is characterized by malformations of the face, oral cavity and digits with a wide phenotypic variation [1–3]. Usually, renal cystic changes represent a late complication of OFD1 and diagnosis is generally made when renal failure has reached an advanced stage, as in our case [3,5]. End-stage renal disease has been reported in affected females ranging in age from 11 to 70 years [1,4,5]. In our case, the age of the patient was 20 years and her clinical findings were characteristic of OFD1.
The gene responsible for this genetic disorder has been recently identified in the Xp22 region and named OFD1. Expression studies have shown that it is expressed during development and in adult tissues, in all the structures affected by this syndrome [8,12]. In human embryos, OFD1 immunolocalized to the metanephric mesenchyme, oral mucosa, tongue, nasal and cranial cartilage, limb and brain [12,13]. Approximately 75% of OFD1 cases are sporadic and these occur almost exclusively in females [14].
The differential diagnosis of OFD1 with multicystic kidney disease should include other cystic kidney disorders such as ADPKD, tuberous sclerosis and von Hippel-Lindau disease [1,10]. Sometimes OFD1 with multicystic kidneys, as in our case, may be misdiagnosed as ADPKD. It is reported that the cystic kidney disorders associated with OFD1 are different from ADPKD both macroscopically and microscopically [1,2]. In OFD1, multiple cysts are irregularly distributed in the renal parenchyma; the cysts are smaller and more uniform in size in OFD1 than in ADPKD. Histological analysis of OFD1 kidneys demonstrates a predominance of glomerular cysts, whereas this appearance is rare in ADPKD. In contrast to ADPKD, the kidneys are usually normal in size or palpably enlarged and there are minimal changes of renal contour in OFD1. Other distinguishing features are mode of inheritance (the mode of inheritance is X-linked dominant patients with OFD1, in contrast it is autosomal dominant in ADPKD) and the lack of oral, facial and digital abnormalities in ADPKD, tuberous sclerosis and von Hippel-Lindau disease. Furthermore, OFD1 is almost exclusively diagnosed in females because males carrying OFD1 mutations die in utero, usually in the first or second trimester.
Caroli's disease is a rare and complex autosomal recessive congenital disorder characterized by multifocal segmental dilatation of intrahepatic bile ducts. The extrahepatic biliary tree is not affected [6,7]. Caroli's disease predominantly affects females. It is most commonly manifested in childhood and in the second to third decades of life. In our case, we diagnosed that our patient had both OFD1 and Caroli's disease along with renal cystic disease. Caroli's disease can be associated with autosomal recessive polycystic kidney disease and patients may have varying degrees of renal cysts, renal tubular ectasia, interstitial fibrosis and renal failure [6,7]. A rare association with ADPKD has also been reported [15]. Associated cystic dilatation of kidneys is seen in 60–80% of the cases, the most frequent being medullary sponge kidney [10]. Choledhocal cyst, liver cyst, hepatoblastoma, hypertrophic pyloric stenosis and Ehlers-Danlos syndrome may be seen in Caroli's disease [7]. In our case, other syndromes that manifested along with Caroli's disease were excluded by clinical and laboratory findings. The appearance of Caroli's disease can be confused with polycystic liver disease or obstructive bile duct dilatation. Whereas the cystic spaces in Caroli's disease are irregular and communicate with the biliary tree, cysts in polycystic liver disease are rounder and smoother and do not communicate with the bile ducts. Moreover, in Caroli's disease, dilated bile ducts have a random, bizarre pattern and have focal areas of cystic ectasia. This differs in appearance in obstructive bile duct dilatation, where dilatation is most marked centrally, tapers towards the periphery in an organized pattern, and lacks focal areas of cystic dilatation [6,7,15,16].
For now we cannot conclude that there is any strong association between OFD1 and Caroli's disease. In both syndromes there are some cystic changes especially in the kidneys. It may be that the genetic abnormalities in OFD1 and Caroli's disease similarly affect the development of the disorders in the kidney and other organs. Proteins involved in renal cystic disorders in mice and humans have been located to the cilia or basal body [17]. Basal body (a modified centrosome at the base of the primary cilia) localizing proteins are at least partially responsible for kidney pathology in OFD1 [13]. ARPKD patients often develop Caroli's disease and also mutations in cilia-localizing proteins play a role in this situation [18]. Future studies of molecular genetics, mutations in basal body and cilia-localizing proteins, cell biology and biochemical approaches exploring model systems are needed to shed light on the aetiology of Caroli's disease.
In conclusion, the present case is the first description of a patient with both OFD1 and Caroli's disease along with cystic renal disease. Patients diagnosed with OFD1 should also be screened for multifocal segmental dilatation of intrahepatic bile ducts. Careful examination should also be done for oral, facial and digital abnormalities in all female patients presenting with bilateral renal cysts, especially when these cysts are multiple and irregularly distributed in the renal parenchyma.
Conflict of interest statement. None declared.
References
Feather SA, Winyard PJ, Dodd S, Woolf AS. Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease: clinical, radiological and histopathological features of a new kindred.
McLaughlin K, Neilly JB, Fox JG, Boulton-Jones JM. The hypertensive young lady with renal cysts—it is not always polycystic kidney disease.
Sabato A, Fabris A, Oldrizzi L, Montemezzi S, Maschio G. Evaluation of a patient with hypertension and mild renal failure in whom facial and digital abnormalities are noted.
Stoll C, Sauvage P. Long-term follow-up of a girl with oro-facio-digital syndrome type I due to a mutation in the OFD 1 gene.
Coll E, Torra R, Pascual J et al. Sporadic orofaciodigital syndrome type I presenting as end-stage renal disease.
Mrowka C, Adam G, Sieberth HG, Matern S. Caroli's syndrome associated with medullary sponge kidney and nephrocalcinosis.
Dagli U, Atalay F, Sasmaz N, Bostanoglu S, Temucin G, Sahin B. Caroli's disease. 1977–1995 experiences.
Ferrante MI, Giorgio G, Feather SA et al. Identification of the gene for oral-facial-digital type I syndrome.
Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group.
Hildebrandt F, Jungers P, Grunfeld J-P. Medullary cystic and medullary sponge renal disorders. In: Schrier RE, Gottschalk CW, eds.
Romio L, Wright V, Price K et al. OFD1, the gene mutated in oral-facial-digital syndrome type 1, is expressed in the metanephros and in human embryonic renal mesenchymal cells.
Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis.
Feather SA, Woolf AS, Donnai D, Malcolm S, Winter RM. The oral-facial-digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2-Xp22.3.
Mousson C, Rabec M, Cercueil JP, Virot JS, Hillon P, Rifle G. Caroli's disease and autosomal dominant polycystic kidney disease: a rare association?
Madjov R, Chervenkov P, Madjova V, Balev B. Caroli's disease. Report of 5 cases and review of literature.
Zhang Q, Taulman PD, Yoder BK. Cystic kidney diseases: all roads lead to the cilium.
Author notes
1Department of Nephrology, 3Department of Radiology, 4Department of First Internal Medicine, Ataturk Training and Research Hospital, and 5Department of Nephrology, Medical School of Ege University, Izmir, Turkey, 2Department of Medicine, Division of Nephrology, Vanderbilt University School of Medicine, Nashville, TN, USA and 6Telethon Institute of Genetics and Medicine, Naples, Italy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments