





Abstract
Web 3DNA (w3DNA) 2.0 is a significantly enhanced version of the widely used w3DNA server for the analysis, visualization, and modeling of 3D nucleic-acid-containing structures. Since its initial release in 2009, the w3DNA server has continuously served the community by making commonly-used features of the 3DNA suite of command-line programs readily accessible. However, due to the lack of updates, w3DNA has clearly shown its age in terms of modern web technologies and it has long lagged behind further developments of 3DNA per se. The w3DNA 2.0 server presented here overcomes all known shortcomings of w3DNA while maintaining its battle-tested characteristics. Technically, w3DNA 2.0 implements a simple and intuitive interface (with sensible defaults) for increased usability, and it complies with HTML5 web standards for broad accessibility. Featurewise, w3DNA 2.0 employs the most recent version of 3DNA, enhanced with many new functionalities, including: the automatic handling of modified nucleotides; a set of ‘simple’ base-pair and step parameters for qualitative characterization of non-Watson–Crick double-helical structures; new structural parameters that integrate the rigid base plane and the backbone phosphate group, the two nucleic acid components most reliably determined with X-ray crystallography; in silico base mutations that preserve the backbone geometry; and a notably improved module for building models of single-stranded RNA, double-helical DNA, Pauling triplex, G-quadruplex, or DNA structures ‘decorated’ with proteins. The w3DNA 2.0 server is freely available, without registration, at http://web.x3dna.org.
INTRODUCTION
In addition to carrying genetic information, nucleic acids contain structural, regulatory, and enzymatic signals related to their cellular functions. Understanding the structural features of DNA and RNA at the atomic level is crucial for deciphering the ‘codes’ of gene expression/regulation, and for designing new therapeutics and nanomaterials. Experimentally determined 3D structural information about nucleic acids is available from the Protein Data Bank (PDB) (1) and the dedicated Nucleic Acid Database (NDB) (2). Exploration of 3D nucleic acid structures involves three closely related components: analysis, visualization, and modeling of molecular conformation, dynamics, and interactions. 3DNA (3–5) is an integrated suite of computer programs with capabilities in each of these categories. Over the years, 3DNA has been widely used, and adopted in many bioinformatics resources, including the NDB.
3DNA, along with Curves+ (6), is one of the most popular software products for the analysis of DNA/RNA double-helical structures. Moreover, by adopting the same standard base reference frame (7), the two programs yield similar numerical values for the rigid-body parameters used to describe the 3D arrangements of the complementary bases within Watson–Crick (WC) base pairs and the steps taken by successive WC base pairs (bp) along double-helical structures (8). 3DNA and Curves+, however, diverge from one another in the analysis of non-WC pairs, and they provide different characterizations of groove dimensions and helical distortions. 3DNA also stands out among the most popular nucleic-acid model building tools, using local bp and step parameters for customized construction of arbitrary nucleic acid structures, and a ‘fiber’ (9) component for easy generation of over fifty types of uniform helical structures. By contrast, the Nucleic Acid Builder (NAB) (10) employs a combination of rigid-body transformations and distance geometry to generate helical and unusual, non-helical DNA and RNA structures. Like 3DNA, the NUPARM/NUCGEN software (11) and the updated RNAHelix modeling program (12) allow for the analysis and construction of sequence-dependent nucleic acid structures in terms of local bp parameters. NUPARM, however, calculates bp parameters differently from 3DNA (5,13), which is unique for the rigorous reversibility of its analysis/rebuilding routines. In addition, 3DNA offers a characteristic base-block representation of a nucleic acid structure that is both simple and informative for visualization. These schematic images are used by both the PDB and the NDB. Please refer to Supplemental Data S5 for a list of related external resources.
The web 3DNA (w3DNA) server (14) makes commonly-used features of the 3DNA suite of command-line programs readily accessible. Other web servers for nucleic acid structures include the Curves+ web server (15) for analysis, 3D-DART (16), and the ‘make-na’ server (10), built on top of NAB, for modeling (Supplemental Data S5). Nevertheless, w3DNA stands out for its convenience in use, and a rich set of features for analysis, rebuilding, and visualization. Since its initial release in 2009, w3DNA has continuously served the community with virtually no changes. At the time of this writing, the server has been cited over 230 times (including 34 times in 2018), according to Google Scholar. Recent citations to w3DNA include: calculations of the DNA helical axis and twist angles (17), determination of the conformational parameters of DNA bound to PfoI and other CCGG-family restriction endonucleases (18), and generation of initial atomistic duplex and triplex models (19,20) for molecular dynamics simulations.
Due to the lack of updates, however, w3DNA has clearly shown its age in terms of modern web technologies, and it has long lagged behind further developments of 3DNA per se (3,21). For example, w3DNA uses a fixed list of modified nucleotides mapped to their canonical counterparts (e.g. 5-methylcytidine [5MC] to ‘c’, expressed as a lower-case letter to distinguish it from the standard ‘C’). The list was compiled from an analysis of all entries in the NDB around 2009, covering the most common cases. However, each new modified nucleotide demands a manual update of the list, and the addition of new cases to the list has become a frequent request at the 3DNA Forum (http://forum.x3dna.org). Another well-known issue is the analysis of RNA or non-canonical DNA structures that contain non-WC pairs (Supplementary Data S1). The existing set of local bp and step parameters in 3DNA is mathematically rigorous, allowing for an unambiguous characterization of any pair of interacting bases and serving as input for exact model building. For non-WC pairs, however, the 3DNA local bp parameters sometimes become cryptic, and the local step parameters may seem to be nonsensical. Obviously, the w3DNA server needs a major overhaul to take better advantage of 3DNA and modern web technologies, and to remain useful.
The w3DNA 2.0 server (Figure 1) presented here overcomes all known shortcomings of w3DNA while maintaining its battle-tested characteristics. The new web server provides a modern interactive user interface for the analysis, visualization, and modeling of nucleic-acid-containing structures. Technically, w3DNA 2.0 implements a simple and intuitive interface (with sensible defaults) for increased usability, and it complies with HTML5 web standards for broad accessibility. As a result, even novice or occasional users can quickly get started using a browser (such as Chrome, Safari, or Firefox) on desktops, tablets, or smartphones. Featurewise, w3DNA 2.0 employs the most recent version of 3DNA which is enhanced with many new functionalities, including: (i) the automatic handling of modified nucleotides, which streamlines the analysis process; (ii) a set of ‘simple’ bp and step parameters for qualitative characterization of non-WC double-helical structures (Figure 1B, and Supplementary Data S1); (iii) new structural parameters that integrate the rigid base plane and the backbone phosphate group, the two nucleic acid components most reliably determined with X-ray crystallography (Figure 2 and Supplementary Data S3); (iv) high resolution images of the base-block schematic representation of each nucleic acid structure (cf. with those in the PDB and NDB) and interactive visualization of the same with JSmol (22); (v) restraint optimization of backbone geometry (23) of models from customized rebuilding (Figure 1D and E); (vi) in silico base mutations that preserve the backbone geometry (Figure 3E and F) and (vii) a notably improved module for building models of single-stranded RNA, duplex DNA, G-quadruplex, and DNA structures ‘decorated’ with proteins (Figures 1D, 1E, and 3B-F).
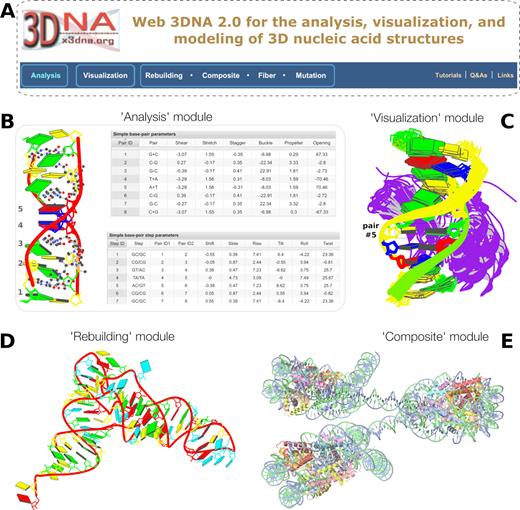
Summary of web 3DNA 2.0. (A) The homepage, highlighting the three major components of the server (boxed) and links to key resources. (B) Excerpt from the ‘Analysis’ of a drug–DNA complex (PDB entry 1xvk) (38), showing a base-block image and tabulations of the ‘simple’ base-pair and step parameters. (C) Schematic ‘Visualization’ of an ensemble of NMR structures of a protein–DNA complex (PDB entry 2moe) (31). The models are aligned locally in the reference frame of the fifth base pair, with its minor-groove edge (colored black) facing the viewer. The protein, colored purple, binds in the major groove of the DNA. (D) An example of ‘Rebuilding’ a model from the local base-pair and step parameters obtained from an ‘Analysis’ of a tRNA structure (PDB entry: 1fir) (33). (E) ‘Composite’ model of a DNA ‘decorated’ with proteins. Here a nucleosome (PDB entry: 4xzq) (34) is used as a template to construct a three-nucleosome, chromatin-like structure. Color code for base rectangular blocks: A, red; C, yellow; G, green; T, blue; U, cyan. (B–D) were generated automatically via the 3DNA ‘blocview’ program, which calls MolScript (24) and Raster3D (25); (E) was rendered using JSmol (22). The annotations were created using Inkscape (https://inkscape.org).
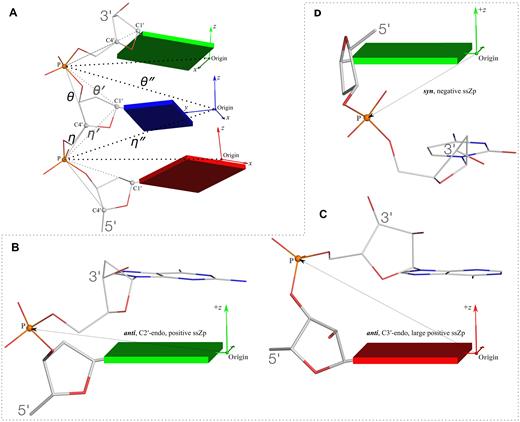
New structural parameters that connect base and backbone atoms. (A) A pair of virtual torsion angles (η″ and θ″) that are based on the positions of the phosphorus atoms (P) and the origins of the intervening bases in their standard base reference frames (7). Two closely related forms of virtual backbone torsion angles are depicted for comparison: the classic version (η and θ) defined by the P and C4′ atoms (27), and a more recent variant (η′ and θ′) based on the P and C1′ atoms (28). Here, an ApTpG trinucleotide from a B-DNA fiber model is used for illustration, with base reference frames attached. Note that the standard base frames of purines and pyrimidines are symmetrical with respect to the dyad of an idealized Watson–Crick base pair, and thus independent of base identity (21): the base origin is accordingly more displaced from the atoms of T (a pyrimidine) than those of A or G (purines). (B–D) Single-stranded phosphate displacement, ssZp, in representative helical structures showing: (B) a small, positive number for a GpG step from a B-DNA fiber model where a C2′-endo sugar attaches to a base in an anti conformation (ssZp = +1.84 Å); (C) a large, positive value for an ApA step from an RNA fiber model where a C3′-endo sugar attaches to a base in an anti conformation (ssZp = +4.38 Å); (D) a negative value for a GpC step from a Z-DNA fiber model where the G adopts a syn conformation (ssZp = −1.74 Å). Color code for base blocks: A, red; G, green; T, blue. The illustrations were generated with DSSR (21) and PyMOL (https://pymol.org).
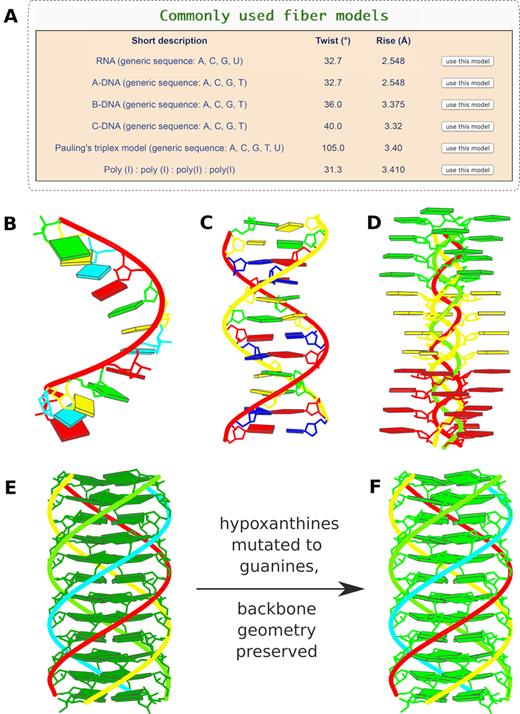
Commonly-used fiber models and in silico base mutations. (A) Six commonly used models highlighted in the ‘Fiber’ module: single-stranded RNA, double-helical A-, B-, and C-form DNA, the Pauling triplex model (32), and the parallel polyI:polyI:polyI:polyI quadruplex. (B) Single-stranded RNA fiber model of base sequence AUCGAUCGAUCG. (C) Double-helical B-DNA fiber model with sequence ATCGATCGATCG on the leading strand. (D) Pauling triplex model with each strand of sequence AAAACCCCGGGG. (E) parallel polyI:polyI:polyI:polyI quadruplex model with 12 layers of hydrogen-bonded hypoxanthine tetrads. Models in (B-E) were generated using the default settings on the w3DNA 2.0 server, each taking just two mouse clicks. (F) All hypoxanthine bases along the poly I chains mutated to guanine via the ‘Mutation’ module, leading to a parallel G-quadruplex. Color code for base blocks: A, red; C, yellow; G, green; T, blue; U, cyan; I, dark green.
Indeed, during its nearly two decades of evolution, 3DNA has accumulated more features than a typical user may care to employ (Figures 1-3) and a non-expert user can get lost in the details of the numerous conformational parameters (e.g. ‘global’ versus ‘local’ versus ‘simple’). To help users become oriented with the interface, the w3DNA 2.0 server contains a complete set of tutorials (Supplementary Data S4), and each feature has a help icon linked to the corresponding tutorial section. The Supplementary Data also provide concrete examples of new structural parameters (Sections S1 and S3) and sample input files for the ‘Rebuilding’ module (Section S2). The website is further enhanced with pictorial illustrations and annotations of pertinent parameters in the ‘Analysis’ module. Mathematical details of the core analysis/rebuilding algorithms can be found in the initial 3DNA publication (5) and references cited therein. Since 3DNA version 2.4 (which backs the web server) is open source, interested users can examine the source code of any feature. Moreover, all user questions are always welcome and promptly addressed on the 3DNA Forum. For example, one entry on the FAQ (frequently-asked-questions) section of the 3DNA Forum is titled ‘How to calculate a DNA bending angle?’ Generally speaking, the curvature of a complicated double helix in 3D is hard to quantify and difficult to visualize. For a duplex with two fragments that are relatively straight, one can fit a least-squared helical axis to each and then derive an angle between the two as an estimate of the magnitude of bending, and an additional phase angle for the direction of bending (S. Harvey, personal communications, November to December 2015).
Overall, the new server has a streamlined interface, is simple to use, and comes with an unmatched set of features. It has the capabilities to serve a huge userbase, for both research and education. Since its public release in December 2018, w3DNA 2.0 has been accessed by an average of ∼150 unique IP addresses each month from users around the world, and visitor numbers keep increasing. The w3DNA 2.0 server is freely available, without registration, at http://web.x3dna.org.
MATERIALS AND METHODS
Datasets
To facilitate the analysis of nucleic-acid-containing structures from the PDB, a very common user demand, we constructed a database of those entries for w3DNA 2.0. The current database is populated by all PDB entries (with metadata) from the 6 March 2019 release that contain the 3D coordinates, in traditional PDB format, of at least one nucleotide. Gigantic structures, such as the ribosome, that are available only in PDBx/mmCIF format are thus excluded from the database. Each selected entry has been analyzed with 3DNA version 2.4 (3–5) to obtain a large set of structural parameters describing base morphology and backbone geometry. The resulting datasets are deposited in tables managed by a MySQL database engine. Additionally, several text files containing various structural parameters and five schematic images of base-block representations from diverse perspectives have been stored in a folder linked to each structure.
Users can also upload a PDB-formatted coordinate file of a nucleic acid structure to the server. Except for a lack of metadata (such as experimental methodology or links to external resources), the analysis pipeline and the output interface are similar to those for a PDB entry.
Web server
The w3DNA 2.0 web server is hosted at Columbia University, and runs on Ubuntu Linux (16.04.6 LTS) with 128 GB memory and a 20 TB hard drive. Other significant software packages used include: Apache (version 2.4.18, https://httpd.apache.org), PHP (version 7.2.15, http://www.php.net), MySQL (version 5.7.25, https://www.mysql.com), JSmol (version 14.29.4) (22) for the interactive display of 3D models, and Phenix (23) (version 1.14) for the base-restraint optimization of backbone geometry. The base-block schematic images were generated by calling MolScript (24) and Raster3D (25). The client-side user interface was implemented using HTML5, the CodeIgniter PHP framework (https://www.codeigniter.com/), and JavaScript libraries, including jQuery (http://jquery.com). The server has a 20 MB size limit for uploaded coordinate files. For DNA/RNA structures with <350 nucleotides, it normally takes ∼20 s for analysis, and less than 1 min for rebuilding with backbone optimization (or ∼5 s without optimization).
The w3DNA 2.0 server conducts computations synchronously in the backend via a combination of embedded 3DNA software (version 2.4) and other components glued together by ad hoc scripts. Once a user has provided a PDB structure identifier (id) or base sequence (along with other options, depending upon the chosen module; see Figure 1A), the backend scripts first validate the user input (e.g. a 4-letter PDB id, or A/C/G/T for a DNA base sequence). Subsequently, the server conveys appropriate 3DNA commands for analysis, modeling, or image plotting.
RESULTS
The w3DNA 2.0 server includes six modules (Figure 1A): ‘Analysis’, ‘Visualization’, ‘Rebuilding’, ‘Composite’, ‘Fiber’, and ‘Mutation’, the last four of which fall under the category of model building. Each module has practical default settings, so users only have to click a few buttons to see the main features.
The ‘Analysis’ module
3DNA (5) identifies nucleotides including modified ones, WC or non-WC pairs, co-planar base multiplets, double-helical fragments, and calculates a large set of bp and backbone parameters. The analysis component of w3DNA 2.0 accepts a user-provided PDB id or an uploaded, PDB-formatted coordinate file for structural characterization. For a PDB entry, the server searches the database tables for pre-populated metadata and fetches pre-stored schematic images and text files. For a user-uploaded file, the analysis pipeline is performed anew based solely on the ATOM/HETATM coordinates, with no consideration of metadata that may exist in the coordinate file.
Modified nucleotides
The original w3DNA server (14) used a fixed list of modified nucleotides mapped to their canonical counterparts. This mapping process is needed to derive base reference frames for the calculation of bp parameters. For example, 5-methylcytidine, named 5MC in the PDB, is mapped to ‘c’. The modified nucleotide is expressed as a lower-case letter to distinguish it from the standard nucleotide. The list (with entries like ‘5MC c’ stored in a file called ‘baselist.dat’) was compiled from an analysis of all entries in the NDB around 2009 and covered all modified nucleotides known experimentally at the time. Each new modified nucleotide, however, demands a manual update of the list. This is cumbersome for server maintenance, and causes inconvenience to end users. Over the years, we have continuously revised the algorithm for the identification of modified nucleotides and the list is no longer necessary (21). In w3DNA 2.0, we have adopted a middle-ground approach: the ‘baselist.dat’ file is checked first to allow for control of the mapping process. If a modified nucleotide is not found in the list, it is assigned automatically.
Output sections
The output page includes five sections: (i) a structural summary (available for PDB entries) including structure title, primary citation, experimental method, and links to external resources; (ii) schematic base-block representations with informative color coding in five perpendicular perspectives (see example in Figure 1B and below); (iii) a JSmol window for interactive visualization; (iv) files of raw parameters, including the main output file which contains a detailed description of 3DNA-derived conformational parameters, the local bp/step parameters file that can be used for rebuilding (see below), and the (virtual) torsion angles file that quantifies backbone geometries and spatial connections between the bases and backbone (see below); (v) nine interactive Grid-View tables embracing bp/step parameters and torsion angles. See Figure 1B for two examples of such tables, which can be sorted interactively and downloaded in CSV (comma-separated values) format for further processing.
‘Simple’ parameters for non-WC pairs
The spatial arrangement of the two bases in a pair can be rigorously quantified by six rigid-body parameters—three translations (Shear, Stretch, and Stagger) and three rotations (Buckle, Propeller, and Opening). Similarly, taking each bp as a rigid body, a set of six step parameters (Shift, Slide, Rise for translations, Tilt, Roll and Twist for rotations) can be used to describe the relative position and orientation of neighboring bps. The numerical values of these local bp and step parameters depend upon the choice of reference frames (13). For WC pairs and DNA/RNA duplexes consisting of these canonical pairs, local 3DNA parameters make ‘intuitive’ sense (e.g. a Rise of around 3.4 Å and a Twist of 32–36°). For non-WC pairs, however, the 3DNA local bp parameters can sometimes become cryptic, and the local step parameters may seem to be nonsensical (for example, null or negative Rise values, as reported on the 3DNA Forum; see Supplemental Data S1).
In 2015, Richardson contributed two articles to the Computational Crystallography Newsletter (CCN, vol. 6, pp. 28–31 and pp. 47–53) on the importance of bp non-planarity at biologically significant positions in high-resolution nucleic acid structures (e.g. functional binding sites) and the need to account correctly for this non-planarity in deriving models of DNA and RNA based on low-resolution data. There, the bp non-planarity is quantified by two angular parameters, Buckle and Propeller. Inspired by her work, we developed a set of six ‘simple’ bp parameters (CCN, 2016, vol. 7, pp. 6–9), which are complete and more intuitive for qualitative characterization of non-WC pairs, and have already been used in the literature for the characterization of G-tetrads in G-quadruplexes (26). We have subsequently extended the description to a new set of six ‘simple’ step parameters.
Figure 1B shows an example of the ‘simple’ bp and step parameters for PDB entry 1xvk, a complex of DNA with echinomycin which contains two A+T Hoogsteen base pairs (nos. 4 and 5). This echinomycin-(GCGTACGC)2 complex has a single DNA strand as the asymmetric unit. The biological unit is contained in a multi-model MODEL/ENDMDL ensemble of two models, each corresponding to one chain of the duplex. Due to the perfect symmetry and opposing directions of the bp reference frames, the central TA/TA step (no. 4, with two Hoogsteen pairs) has a local Rise of zero, and a Twist of 180 degrees, values that do not make ‘intuitive’ sense (Figure 1B, base-block image). On the other hand, the corresponding ‘simple’ parameter description of step no. 4 has a Rise of 3.1 Å, and a Twist of 26°, which are more intuitively reasonable.
Overall, bp geometry can be described in more than one way. The existing set of six local bp and step parameters in 3DNA is mathematically rigorous, allowing for an unambiguous characterization of any pair of interacting bases and serving as input for exact model building. The new sets of six ‘simple’ bp and six ‘simple’ step parameters provide a more intuitive interpretation of structural variations in non-WC-containing structures. The term ‘simple’ is used to distinguish the new set of parameters from the traditional 3DNA local parameters. It should be emphasized that the ‘simple’ parameters are for structural description only, and are not suitable as input for the 3DNA ‘rebuild’ program. The two types of parameters complement one another and serve different audiences and/or purposes. Numerical values of both are readily obtained with 3DNA. See Supplemental Data S1 for details.
New parameters connecting base and backbone atoms
In w3DNA 2.0, we have also introduced two new structural parameters that integrate the rigid base plane and the backbone phosphate group, the two nucleic acid components most reliably determined with X-ray crystallography. These new parameters take advantage of the standard base reference frame (7), which is symmetrical with respect to the dyad of an idealized Watson–Crick base pair, and independent of base identity.
Using the origins of the base reference frames and the positions of the phosphorus atoms (P), we define a pair of virtual angles (Figure 2A): η″ by origin(i–1)-P(i)-origin(i)-P(i+1); θ″ by P(i)-origin(i)-P(i+1)-origin(i+1). The classic virtual backbone torsion angles (η and θ) are based on the simplification of the sugar-phosphate backbone by Olson (27) in terms of the P and C4′ atoms. Subsequent interpretations of low-resolution crystallographic density maps by Pyle and co-worker (28) led to the use of a variant angle pair (η′ and θ′) based on the P and C1′ atoms. The C1′ atom was chosen based on its more reliable location in X-ray crystal structures, due to its covalent linkage to the base. Following this line, we reason that the base ‘origin’, which is defined by a least-squares fitting procedure using the whole rigid base plane, is even more reliably located.
Inspired by the finding of Richardson et al. (29) on the correlation between the sugar puckering and the perpendicular distance between the bridging P atom and the preceding C1′–N1/9 glycosidic bond, we extended the 3DNA double-helical parameter Zp (30) to a single-stranded analog (ssZp). As shown in Figure 2B–D, ssZp is defined as the z-coordinate of the bridging P atom expressed in the standard reference frame of the preceding base. This parameter is sensitive to both sugar puckering (C2′-endo versus C3′-endo) and base conformation (syn versus anti): for a C2′-endo sugar attached to an anti base as in B-form DNA, ssZp is a small, positive number; for a C3′-endo sugar attached to an anti base as in A-form DNA and RNA, ssZp is large and positive; for a base in a syn conformation as the guanines in Z-form DNA and in certain G-quadruplexes, ssZp is negative. The potential advantage of ssZp over the perpendicular distance using C1′–N1/9 is similar to that for the virtual angles mentioned above.
The ‘Visualization’ module
The visualization module can be used to examine a single structure or a multi-model ensemble, such as a series of NMR-derived or computer-simulated structures, in schematic base-block representations, with backbone ribbons and protein cartoons (Figure 1C). If the entry contains a single structure, block images are automatically created to show the model from five perpendicular perspectives. Each image has a corresponding downloadable link and is enlarged with mouse hovering. If the entry corresponds to an ensemble of structures, users can specify the range of models for display and choose a bp for local alignment. Figure 1C shows an ensemble of 10 solution NMR structures of a protein–DNA complex from PDB entry 2moe (31). Here the structures are aligned on the fifth bp, containing one of the two methyl-cytosines recognized on its major-groove edge by the protein. The exposed and unrecognized minor-groove edge of the aligned base pairs is at once apparent from the shading automatically introduced by the software.
The rectangular base-block representation is simple and informative (Figures 1–3). With color-coding, the base identity, pairing geometry, and stacking interactions are immediately obvious in small to mid-sized nucleic acid structures. It is worth noting that this schematic representation has been adopted by both the PDB and the NDB, and propagated into other online bioinformatics resources. In w3DNA 2.0, we have significantly increased the resolution of these block images, which look much crisper and are of publication quality.
Stacking diagrams (5) of all the bp steps in a double-helical structure can also be generated (not shown, see Supplementary Data S4.2). The view is taken down the z-axis of the middle frame used in calculating step parameters. As for base-block images, we have increased the resolution of the images for stacking diagrams in the updated server and provided options for the labeling of nucleotides.
The modeling modules
The w3DNA 2.0 server contains four modeling modules that enable users to create new 3D nucleic acid structures or perform base mutations of existing ones. The structures are generated by a combination of ad hoc scripts and 3DNA modeling routines with user-specified bp parameters and templates. The choices include five model-building types: (i) customized single-stranded RNA or double-helical DNA structures with user-specific base sequences and rigid-body parameters (Supplemental Data S2); (ii) mixed DNA double-helical structures comprised of canonical A-, B-, and C-type DNA fragments; (iii) protein-decorated DNA structures with user-defined binding sites and templates of known DNA-protein complexes; (iv) 56 uniform fiber-diffraction models (9), containing single-stranded RNA, different helical forms of DNA, hybrid DNA/RNA helices, triplexes (32), and quadruplexes; (v) models with bases mutated at user-specified locations while preserving backbone geometry.
All resulting 3D models share the same output interface: (i) a schematic base-block representation in the principal-axis frame; (ii) a coordinate file in PDB format, which can be downloaded for further study and (iii) a JSmol window for interactive visualization.
Rebuilding
To build a customized DNA/RNA model, the user is required to prepare a text file containing the base sequence and rigid-body parameters of the desired structure. Two example parameter files (Supplemental Data S2) can be found on the customized modeling section, where clicking a sample-file link auto-fills the corresponding content in the text area. Users can also upload a parameter file in valid format. In practice, the input file is most conveniently generated by running the ‘Analysis’ module on a starting structure, and then modifying the resultant parameter file as needed. After selecting the desired backbone conformation and choosing whether to perform backbone geometry optimization, users can click the ‘Build Model’ button to trigger the modeling pipeline.
Figure 1D shows a tRNA model built from the local bp and step parameters (Supplemental Data S2) obtained from an analysis of PDB entry 1fir (33), the crystal structure of HIV reverse-transcription primer tRNA(Lys,3). Here, the 15 modified bases have been replaced by their standard counterparts. The initial model rebuilt using 3DNA employs a standard RNA helical backbone (with C3′-endo sugar puckering) attached to each base. The resultant backbone, however, is oftentimes distorted, without proper covalent linkages between neighboring nucleotides. Users have the option to optimize this approximate backbone connection, using Phenix (23), while keeping the base atoms fixed. The 3DNA-Phenix combination leads to a model where the base geometry strictly follows the parameters prescribed in the user-specified file, and the backbone is regularized with improved stereochemistry and a ‘smooth’ appearance in ribbon representation.
Building a mixed, naturally curved DNA structure with A-, B- and C-form components is also available in this section. After choosing the number of segments from a pull-down menu, users can type in the base sequence, an optional repeating number, and choose the helical form(s) of DNA.
Composite
To construct a protein-decorated DNA model, users need to specify the number of protein-binding sites, the base sequence, and the helical form (B- or A-type DNA). For each binding site, users then assign a binding position along the DNA model and a protein–DNA complex template (via a PDB id). The binding position corresponds to the middle of the DNA in the protein–DNA complex. Different binding sites can use the templates of different protein–DNA complexes. The server performs checks to avoid the overlap of neighboring binding positions.
From a starting B- or A-form DNA model, the (different) binding proteins may distort the straight double helix into a complicated 3D shape, depending on the configuration of the DNA template in each protein–DNA complex. This ‘composite’ modeling is an advanced topic and involves many components of the 3DNA suite and some ad hoc scripts. Figure 1E shows such a ‘composite’ model of DNA ‘decorated’ with proteins. Here the X-ray crystal structure of a nucleosome from PDB entry 4xzq (34) is used as a template to construct a three-nucleosome, chromatin-like model. Specifically, the nucleosomal DNA in 4xzq has 146 bp. Approximated to 150 bp, the middle position is at 75. Leaving 10 bp at both ends, and two linkers of 25 bp each in the middle, we need a DNA of 520 bp (2 × 10 + 3 × 150 + 2 × 25) to wrap around three nucleosomes. The three protein-binding sites are thus centered at base positions 85, 260, and 435 of the 520-bp DNA, i.e., 175-bp dyad spacing between successive nucleosomes. The model presented here assigns a B-form fiber model to the four protein-free DNA regions (two linkers plus both ends), each with a poly dA sequence in the leading strand.
Fiber
We have extended the regular DNA/RNA helical models (5) that can be constructed, most based on early fiber diffraction studies of Arnott and co-workers (9), to a total of 56. New additions include a single-stranded RNA model of generic sequence (A, C, G and U), derived from the A-form DNA fiber model (9) and the historic Pauling triplex model (32), an unusual structure with exposed bases consistent with the response of DNA to extreme stretching and over-winding (35).
Based on user feedback, we have optimized the interface for constructing fiber models (Figure 3A). The six most commonly used models are highlighted at the top of this section. Now users can easily build regular models of single-stranded RNA, double-helical A-, B- or C-DNA, and the Pauling triplex, with user-specified base sequences. On the other hand, the polyI:polyI:polyI:polyI quadruplex is specific to hypoxanthine. Figure 3B–E were generated with default settings, each taking just two-button clicks (first select a model, and then press the ‘Build’ button) and a few seconds waiting time.
Mutation
A new functionality of w3DNA 2.0 is the in silico base mutations of nucleic acid structures, by calling the command-line program ‘mutate_bases’. Notably, the mutation process preserves both the geometry of the sugar-phosphate backbone and the base reference frame (position and orientation). As a result, re-analyzing the mutated model gives the same bp and step parameters as those of the original structure. The 3DNA ‘mutate_bases’ program has already been employed in the literature for inference of statistical protein–DNA potentials (36) and homology modeling of the Escherichia coli FMN aptamer (37).
The starting structure for mutations can be specified by a PDB id or uploaded from a coordinate file. Alternatively, models constructed by other w3DNA 2.0 modeling modules mentioned above can be directly used for base mutations. Figure 3F shows an example of the mutation of all the hypoxanthines of the polyI:polyI:polyI:polyI quadruplex (Figure 3E) to guanine, leading to a parallel G-quadruplex.
CONCLUSION
The new w3DNA 2.0 web server provides a modern interactive user interface for the analysis, visualization, and modeling of 3D nucleic acid structures, including their complexes with proteins and other ligands. The server embeds popular and new features of the 3DNA suite of command-line programs via a simple and intuitive web interface, with a built-in set of sensible defaults. As a result, even novice or occasional users can quickly get started and benefit from the many features that 3DNA has to offer. Specifically, w3DNA 2.0 allows users: (i) to gather information and gain insight about DNA/RNA conformational features, via the ‘Analysis’ module; (ii) to create high-resolution base-block schematic images that are both simple and highly revealing, via the ‘Visualization’ module and (iii) to construct 3D models that can be used in various computational applications, such as atomic-level simulations of DNA/RNA structures or their complexes with proteins, via the ‘Modeling’ modules. Furthermore, user questions are always welcome and promptly addressed on the 3DNA Forum.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank the user community for using w3DNA and providing us with feedback, and Yin Yin Lu for proofreading the manuscript. We appreciate the comments and suggestions of the three anonymous reviewers who helped improve and clarify this manuscript and the website.
FUNDING
National Institutes of Health (NIH) [R01GM096889 to X.J.L., R01GM034809 to W.K.O.]. Funding for open access charge: NIH [R01GM096889].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
Comments