





Abstract
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) is a freely available, public archive of human genetic variants and interpretations of their significance to disease, maintained at the National Institutes of Health. Interpretations of the clinical significance of variants are submitted by clinical testing laboratories, research laboratories, expert panels and other groups. ClinVar aggregates data by variant-disease pairs, and by variant (or set of variants). Data aggregated by variant are accessible on the website, in an improved set of variant call format files and as a new comprehensive XML report. ClinVar recently started accepting submissions that are focused primarily on providing phenotypic information for individuals who have had genetic testing. Submissions may come from clinical providers providing their own interpretation of the variant (‘provider interpretation’) or from groups such as patient registries that primarily provide phenotypic information from patients (‘phenotyping only’). ClinVar continues to make improvements to its search and retrieval functions. Several new fields are now indexed for more precise searching, and filters allow the user to narrow down a large set of search results.
INTRODUCTION
ClinVar (1,2) is a freely available, public archive of human genetic variants and interpretations of their significance to disease. It is maintained at the National Center for Biotechnology Information (NCBI), within the National Library of Medicine (NLM) at the National Institutes of Health (NIH). Assertions of the clinical significance of a variant or set of variants are submitted to ClinVar by clinical testing laboratories, research laboratories, locus-specific databases, expert panels and other groups. Submissions include a description of the variant(s); the condition for which the variant was interpreted; the interpretation of the clinical significance of the variant, with the option to provide mode of inheritance; and evidence for that interpretation. ClinVar aggregates submissions based both on the variant and the variant-condition pair, and calculates an aggregate interpretation to indicate whether there is consensus or disagreement among submitters for an interpretation. A review status (https://www.ncbi.nlm.nih.gov/clinvar/docs/review_status/) is assigned to each record to help the user understand what level of review supports the interpretation. Review status is based on submission of the criteria used by the submitter to classify variants, consensus across submitters in the interpretation of the variant and whether an expert panel or practice guideline-providing group has interpreted the variant. The ClinVar dataset may be searched and browsed on the website (https://www.ncbi.nlm.nih.gov/clinvar/) and downloaded on the ftp site (ftp.ncbi.nlm.nih.gov/pub/clinvar/). It is also available programmatically with NCBI’s E-utilities/Entrez direct(https://www.ncbi.nlm.nih.gov/books/NBK25501/).
ClinVar currently holds more than half a million submitted records (https://www.ncbi.nlm.nih.gov/clinvar/submitters/), accounting for >331 000 variants (Figure 1). While most records in ClinVar report germline observations, about 3000 variants include somatic observations. ClinVar includes both sequence variants and structural variants; the database currently includes >15 000 variants >1 kilobase (kb). More than 800 groups from 60 countries submit to ClinVar (Figure 2), including 76 laboratories that submit interpretations from direct clinical testing. Approximately 4700 people use the ClinVar web site each weekday.
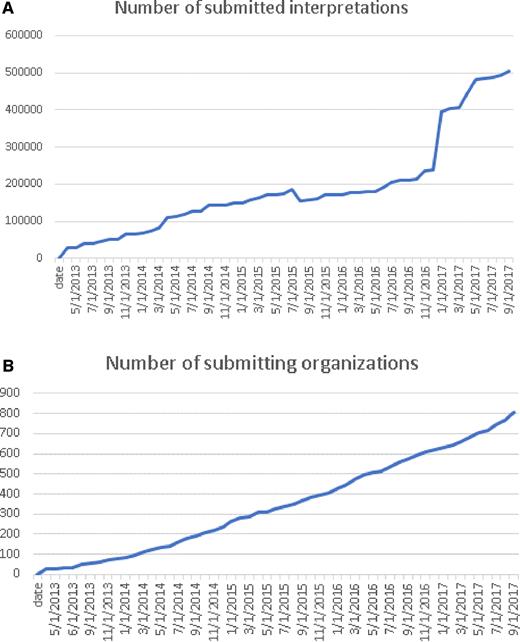
This chart documents the cumulative growth of submissions (A) and organizations that submit to ClinVar (B) since its first public launch in 2013.
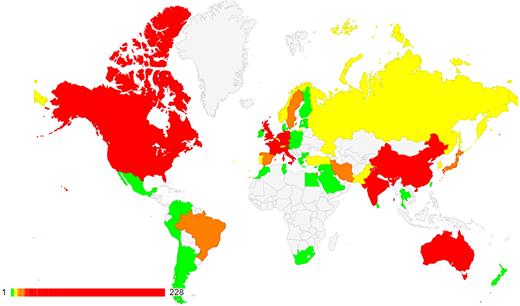
ClinVar holds submissions from >800 organizations, from 60 countries on five continents. See https://www.ncbi.nlm.nih.gov/clinvar/docs/map/ for current counts per country.
NCBI is committed to making ClinVar useful for our users. In this article, we describe recent improvements to ClinVar to make variant-centric data more accessible to enrich the phenotypic content of the database and to make searching the database easier.
IMPROVED ACCESS TO VARIANT-CENTRIC DATA
VCV accession numbers for Variation IDs
The ClinVar Variation ID represents the variant or set of variants that were interpreted (https://www.ncbi.nlm.nih.gov/clinvar/docs/identifiers/#variation). The set of variants that were interpreted may consist of a single variant; multiple variants as a haplotype (in cis); multiple variants as a genotype (in trans) when the individual variants are not interpreted independently; multiple variants where the phase is unknown or multiple variants in different genes. Since its inception, ClinVar has aggregated data for the Variation ID–condition pair and has assigned each pair an RCV accession number (Reference ClinVar). ClinVar now also aggregates data for the Variation ID and assigns an accession number with the prefix VCV (Variation in ClinVar) followed by nine digits. The digits correspond to the Variation ID padded with preceding zeros to make nine digits. The VCV record includes all data for the variant or set of variants, across all diseases reported for the Variation ID. The Variation ID will be retained when VCV accession numbers are added.
For example, Variation ID 96923 represents the variant NM_007294.3:c.4038_4041delAAGA. The accession number for all data aggregated for that variant is VCV000096923, and corresponds to the variation report in the ClinVar web display: https://www.ncbi.nlm.nih.gov/clinvar/variation/96923.
This variant has been reported to ClinVar for two diseases, Breast-ovarian cancer, familial 1 and Hereditary cancer-predisposing syndrome (Figure 3). Thus there are two RCV records for this variant, one for each disease that has been reported:
RCV000083044 for NM_007294.3:c.4038_4041delAAGA and Breast-ovarian cancer, familial 1.
RCV000129276 for NM_007294.3:c.4038_4041delAAGA and Hereditary cancer-predisposing syndrome.
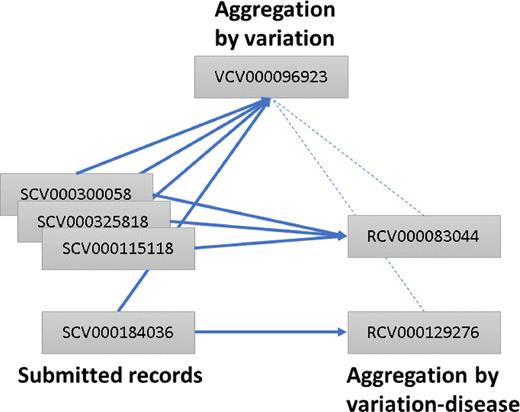
Accessions in ClinVar. Each record submitted to ClinVar is assigned an accession number prefixed with SCV. Submitted records for the same variant and interpreted condition are aggregated into a ‘Reference ClinVar’ record and assigned an accession number prefixed with RCV. Submitted records for the same variant, regardless of disease, are aggregated in a ‘Variation in ClinVar’ record and assigned an accession number prefixed with VCV. VCV records reference the corresponding RCV records and vice versa. Solid lines represent what is aggregated; dotted lines represent what is cross-referenced. In this example, all SCV accessions described a variant that was assigned a Variation ID of 96923 and thus accessioned as VCV00096923. SCV000184036 represents an interpretation relative to a disorder different from that of the others, so it is represented in an RCV distinct from that of the others.
Different levels of aggregation allow the submitter to choose whether to look at all available interpretations and evidence for a variant across all reported diseases or to examine interpretations and evidence for a specific disease. For the ClinVar web display, the variation-level aggregation is the default display; from this page, the variation-disease aggregation can be viewed by clicking the link to ‘see supporting ClinVar records’.
The VCV accession numbers are accessible as part of the variant-centric XML file (see next section). The VCV accession numbers will be versioned so that a history is retained. The VCV version will be incremented whenever a change is made by a submitter to one of the underlying submitted records (SCVs). Versioning will start when the XML file goes into production, anticipated in late 2017. Note that versioning of each VCV record is independent of the release for the corresponding XML product (see below). A VCV record may retain the same version number through many XML releases if the supporting submissions are not updated.
XML for VCV records
The aggregation of data by Variation ID (VCVs) is also represented in a new XML file, named ClinVarVariationRelease, available as a beta release: ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/xml/clinvar_variation/beta/
Full production mode is anticipated to start in late 2017. At that point, the file will be released weekly. The first release of each month will be archived, in parallel to the RCV-centric XML file, ClinVarFullRelease. In addition to the different basis of aggregating data, ClinVarVariationRelease also includes the following novel features relative to ClinVarFullRelease:
ClinVarVariationRelease uses explicit elements to distinguish records for simple alleles, haplotypes and genotypes.
ClinVarVariationRelease uses explicit elements to distinguish between variants that were directly interpreted (InterpretedVariant, e.g. a simple allele submitted with an assertion of clinical significance specific only to that allele) from variants that were interpreted only as part of a haplotype or genotype (IncludedVariant, e.g. a simple allele contributing to the definition of a haplotype, for which the haplotype’s clinical significance was asserted but the simple allele itself was not).
ClinVarVariationRelease represents Human Genome Variation Society (HGVS) (3) expressions for transcripts, and corresponding proteins, as a tuple with molecular consequence.
ClinVarVariationRelease is accompanied by a complementary file of deleted VCV accessions.
ClinVarFullRelease will continue to be provided for users who wish to track data for each Variation ID-condition pair. The two XML files will be generated using the same snapshot of data and will be synchronized with the web display and all reports available on the file transfer protocol (FTP) site. Like ClinVarFullRelease, files for ClinVarVariationRelease are compressed with gzip and have a .gz extension in the file name.
New and improved VCF files
Until October 2017, ClinVar’s files in variant call format (VCF) (4) were organized around the dbSNP (5) rs (reference SNP) number. This meant that in some cases, data for more than one allele were reported on a single row. It also meant that ClinVar variants that were not yet registered in dbSNP were excluded from the file. In addition, each allele may be reported for more than one disease. This resulted in a complex aggregation of data which was complicated to parse. In October 2017, new versions of ClinVar’s VCF files went into production:
ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh37/
ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/
The new files are allele-centric and use the ClinVar Variation ID as the identifier (column 3). This makes it easier to review the data in the VCF file relative to the web display, which is also based on the Variation ID. The files include all variants in ClinVar with a precise genomic location. In other words, variants with imprecise location, such as structural variants identified only by microarray, are not included in the VCF files. Future development will focus on a VCF file for variants with imprecise location. The VCF files also include both variants that were interpreted directly and those that were interpreted only as part of a haplotype or genotype (included variants).
Improvements to the additional information (INFO) tags include:
Several new tags specific to included variants were added (CLNSIGINCL, CLNDNINCL and CLNDISDBINCL). Because these variants have no direct interpretation, the tags indicate the clinical significance and disease for the related haplotypes/genotypes.
The former AF INFO tag was split into three tags, one for each source of allele frequency data: AF_ESP for GO-ESP [https://esp.gs.washington.edu/drupal/]; AF_EXAC for the ExAC Consortium (6); and AF_TGP for the 1000 Genomes Project (7).
A new INFO tag, ALLELEID, reports the Allele ID for the variant (https://www.ncbi.nlm.nih.gov/clinvar/docs/identifiers/#allele).
A new INFO tag, CLNHGVS, reports the top-level genomic HGVS expression for the variant. This may be on an accession for the primary assembly or on an ALT LOCI [https://www.ncbi.nlm.nih.gov/grc]. Note that other HGVS expressions are not included in the VCF file but are found in the tab-delimited file hgvs4variation.txt on the ClinVar ftp site (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/).
A new INFO tag, CLNVI, reports identifiers for the variant in other databases, e.g. OMIM Allelic variant IDs (8).
A new INFO tag, CLNVC, reports the type of variation, e.g. deletion.
A new INFO tag, MC, report the molecular consequence of the variant predicted by NCBI based on the sequence change, including the Sequence Ontology (9) identifier and the molecular consequence term. This tag replaces ASS, DSS, INT, NSF, NSM, NSN, R3, R5, SYN, U3 and U5 in the old format.
A new INFO tag, ORIGIN, reports an integer representing the allele origins that have been observed for the variant and reported to ClinVar. This tag replaces CLNORIGIN in the old format.
Similar to the XML files above, ClinVar’s VCF files are also compressed with gzip and have a .gz extension in the file name.
PHENOTYPE-RICH SUBMISSIONS
Most of the variant interpretations submitted to ClinVar are provided by clinical testing laboratories; however, these laboratories often have little or no knowledge of the clinical features observed in the individual being tested. Consequently, their submissions rarely include any phenotypic data. The patient’s phenotype is often known by the clinician or the patient, but it is not communicated to the testing laboratory. Traditionally, submissions to ClinVar focus on interpretations of variant–disease relationships where the disease is based on established gene–disease relationships rather than the phenotype observed in the individual being tested. ClinVar aims to bridge this gap with submissions that are focused on patient-associated phenotypes. These submissions are distinguished by their collection method (MethodType in XML), either ‘provider interpretation’ or ‘phenotyping only’. Phenotypes may be submitted as Human Phenotype Ontology (HPO) identifiers or terms; terms for clinical features not in HPO may be submitted and are assigned an identifier in MedGen (10).
A clinical provider may submit his or her own interpretation of a variant to ClinVar (distinct from the interpretation from the testing laboratory), including detailed phenotypic information for the patient. The clinician’s interpretation may be based on both the results from the testing laboratory and their own knowledge of the patient’s phenotype. These submissions are considered ‘provider interpretation’ in ClinVar. This type of submission may also include the testing laboratory that identified the variant, the interpretation made by that laboratory and the date that the variant was reported. This information is important because ClinVar may also have an interpretation directly submitted by the testing laboratory. These data are captured to help ClinVar users understand when two submissions may be based on some of the same patient data. It is also useful in case the interpretation that was reported to the clinician is no longer the same as the testing laboratory’s current interpretation. ‘Provider interpretation’ submissions are aggregated in the same way as submissions from clinical testing, research etc. These submissions may provide assertion criteria if available, and their interpretations do contribute to the aggregate interpretation and review status of the variant.
Groups such as patient registries also may have detailed phenotypic data for patients who have received genetic testing, but the registries do not interpret the variant themselves. A registry may submit the variant, the interpreted condition and clinical features observed in patients to ClinVar with collection method ‘phenotyping only’. For this case, there is no interpretation of clinical significance of the variant provided by this submitter, thus these submissions do not contribute to the aggregate interpretation or the review status calculated by ClinVar. Phenotypes or clinical features observed in the patient are required; the submission may also include the testing laboratory that identified the variant, the interpretation made by that laboratory and the date that the variant was reported.
It is important to note that records in ClinVar represent interpretations of variants, not patients. As such, data based on the same patient may be referenced in multiple submitted records. This scenario should not be considered an error; it is analogous to multiple laboratories that reference the same citation in support of their interpretations.
IMPROVEMENTS TO SEARCHING ClinVar
ClinVar uses NCBI’s Entrez search system (5) which provides a great deal of flexibility in searching. Any search term may be used; some recommendations for useful search terms are listed in Table 1. Additional direction on searching and using ClinVar is available at https://www.ncbi.nlm.nih.gov/clinvar/docs/help/.
Recommended types of search terms for ClinVar
| Type of search term | Example |
|---|---|
| gene symbols | PTEN |
| HGVS expressions | NM_000314.4:c.395G>T |
| protein changes | G132V |
| rs numbers | rs121909241 |
| diseases | PTEN hamartoma tumor syndrome |
| clinical features/phenotypes | short stature |
| submitters | NCBI |
| a location on a chromosome for an assembly | 10[chr] AND 89623000:89730000[chrpos37] searches for variants on chromosome 10 between 89623000 and 89730000 based on GRCh37 (chrpos37) |
| Type of search term | Example |
|---|---|
| gene symbols | PTEN |
| HGVS expressions | NM_000314.4:c.395G>T |
| protein changes | G132V |
| rs numbers | rs121909241 |
| diseases | PTEN hamartoma tumor syndrome |
| clinical features/phenotypes | short stature |
| submitters | NCBI |
| a location on a chromosome for an assembly | 10[chr] AND 89623000:89730000[chrpos37] searches for variants on chromosome 10 between 89623000 and 89730000 based on GRCh37 (chrpos37) |
| Type of search term | Example |
|---|---|
| gene symbols | PTEN |
| HGVS expressions | NM_000314.4:c.395G>T |
| protein changes | G132V |
| rs numbers | rs121909241 |
| diseases | PTEN hamartoma tumor syndrome |
| clinical features/phenotypes | short stature |
| submitters | NCBI |
| a location on a chromosome for an assembly | 10[chr] AND 89623000:89730000[chrpos37] searches for variants on chromosome 10 between 89623000 and 89730000 based on GRCh37 (chrpos37) |
| Type of search term | Example |
|---|---|
| gene symbols | PTEN |
| HGVS expressions | NM_000314.4:c.395G>T |
| protein changes | G132V |
| rs numbers | rs121909241 |
| diseases | PTEN hamartoma tumor syndrome |
| clinical features/phenotypes | short stature |
| submitters | NCBI |
| a location on a chromosome for an assembly | 10[chr] AND 89623000:89730000[chrpos37] searches for variants on chromosome 10 between 89623000 and 89730000 based on GRCh37 (chrpos37) |
In addition to searching ClinVar with any search term, users can also perform advanced, focused searches by defining the field in which to look for the query term. For example, clinical significance is indexed as a property of a ClinVar record, so this query: ‘clinsig pathogenic’[Properties] can be used to search for variants that have been reported to be pathogenic. To review all options for indexed fields, consider using the Search Builder tool (https://www.ncbi.nlm.nih.gov/clinvar/advanced). There you can review values for each indexed field, test queries using the fields and save the URL for any query that you want to reuse.
Recent improvements to searching in ClinVar include:
Variants may be searched based on the date that the clinical significance was last evaluated by a ClinVar submitter. This feature was added to make it easier to focus on records that were interpreted within a particular time period. e.g. 2017[Last interpreted] finds all variants where the clinical significance was last evaluated in 2017. This field may also be used as a range to find all variants interpreted after a specific date, e.g. ‘2016/09/10’[Last interpreted]:2017[last interpreted] finds all variants interpreted after 10 September 2016.
Variants in specific genes may be searched by HGNC_IDs (11), as well as by GeneIDs (5), e.g. 1100[hgnc]. This feature was added in part based on discussions with stakeholders indicating that ClinVar should support queries by HGNC identifiers as well as official gene symbols.
Variants reported with specific phenotypes may be searched by HPO (12) IDs, e.g. ‘hp 0004322’[Trait identifier]. This feature was added to make certain that ClinVar could be searched by all trait identifiers, whether for a disease or a clinical feature/phenotype.
Search results for a simple term, such as searching for a gene symbol, can also be focused using the filters on the left side of a search results page. The results can be filtered in several ways including:
Clinical significance. Note that this filter uses the submitted values of clinical significance, not the aggregate value. For example, if you use the filter for ‘Pathogenic’, your search results may include variants with a conflict in interpretation where one or more submissions interpreted the variant as pathogenic, as well as variants where the aggregate clinical significance is pathogenic.
Review status. Filters are available for several of the higher review statuses. There is also a filter to get all records that have ‘at least one star’; in other words, all records where assertion criteria were provided. See https://www.ncbi.nlm.nih.gov/clinvar/docs/variation_report/#review_status for more details.
Allele origin. ClinVar includes interpretations of variants identified in the germline and as somatic events. Note that allele origin refers to an observation of a variant, not the variant itself, so the same variant may have been reported both as germline and as somatic.
Method type. Method type refers to the method that was used to collect the data in each submission, such as clinical testing, research or reporting from the literature only.
Molecular consequence. The molecular consequence of a variant is calculated by NCBI based on the sequence change. For variants in genes with multiple transcripts, there may be more than one predicted molecular consequence.
Variation type. Note that the filter for ‘Deletion’ includes variants submitted as ‘deletion’ and as ‘copy number loss’. Similarly, ‘Duplication’ includes variants submitted as ‘duplication’ and as ‘copy number gain’.
Variant length. This filter is useful for those interested in only small sequence variants or those interested in only large copy number variants (CNV). More than one option in the filter may be selected at a time, e.g. to find all variants larger than 1 megabase (Mb), select both options ‘Between 1 and 5 Mb’ and ‘>5 Mb’.
Variant–gene relationship. This filter distinguishes between variants that affect a single gene, variants that affect multiple genes because the genes overlap on the genome and variants that affect multiple genes because the variant spans the genes. Thus this filter is also useful for those interested in CNVs, which are often large variants that span multiple genes.
Filters are applied per Variation ID, not per submission. However, for some filters (clinical significance and method type), the value that is used for filtering is from submissions for that Variation ID, not an aggregate value. For example, if ‘pathogenic’ and ‘clinical testing’ are selected for clinical significance and method type respectively, the results include Variation IDs where at least one submission reported ‘pathogenic’ and at least one submission is from ‘clinical testing’. But those values are not necessarily from the same submission. Multiple options within a filter may be selected, and the results are the union of those options. For example, when both ‘insertion’ and ‘deletion’ are selected for variant type, the results include both insertion variants and deletion variants, not variants that are both insertions and deletions.
SUMMARY
In its fifth year of operation, ClinVar continues to support our users by providing a publicly available, centralized database for sharing variant interpretations and supporting evidence. Improved access to data aggregated by variant, submissions with phenotypes observed in patients and enhancements to ClinVar’s search function have been provided to meet the needs of ClinVar users. Future challenges include automation of the submission process to allow real-time updates from laboratories and addressing outdated or legacy submissions. ClinVar staff welcome your input on these topics and other aspects of ClinVar. Please contact us at clinvar@ncbi.nlm.nih.gov with your feedback.
ACKNOWLEDGEMENTS
We thank our partners in the ClinGen group, most notably Heidi Rehm, Christa Martin, Steven Harrison, Erin Riggs and Danielle Azzariti, for their continued feedback and guidance to make ClinVar useful for the clinical genetics community.
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
Comments