





Abstract
Despite recent advances in human genetics, model organisms are indispensable for human disease research. Most human disease pathways are evolutionally conserved among other species, where they may phenocopy the human condition or be associated with seemingly unrelated phenotypes. Much of the known gene-to-phenotype association information is distributed across diverse databases, growing rapidly due to new experimental techniques. Accessible bioinformatics tools will therefore facilitate translation of discoveries from model organisms into human disease biology. Here, we present a web-based discovery tool for human disease studies, MORPHIN (model organisms projected on a human integrated gene network), which prioritizes the most relevant human diseases for a given set of model organism genes, potentially highlighting new model systems for human diseases and providing context to model organism studies. Conceptually, MORPHIN investigates human diseases by an orthology-based projection of a set of model organism genes onto a genome-scale human gene network. MORPHIN then prioritizes human diseases by relevance to the projected model organism genes using two distinct methods: a conventional overlap-based gene set enrichment analysis and a network-based measure of closeness between the query and disease gene sets capable of detecting associations undetectable by the conventional overlap-based methods. MORPHIN is freely accessible at http://www.inetbio.org/morphin.
INTRODUCTION
Most human disease pathways are evolutionarily conserved with other organisms. For example, the nematode worm Caenorhabditis elegans, which is relatively distant in phylogeny from humans, is used as a model system to study human Parkinson's disease (1). Despite limited functional mimicry of some human diseases (2) and recent advances in patient-based disease genetics due to genome-wide association studies (GWAS) and disease genome sequencing, non-human model organisms remain indispensable in human disease research, because (i) disease-associated DNA variants typically explain only a small proportion of disease heritability; (ii) detailed molecular mechanisms of disease processes often cannot be studied directly in humans for ethical reasons (3). While model organisms will remain critical for human disease research into the future, the functional relevance of pathways conserved between humans and other species can sometimes be nonobvious (4), hampering identification of new human disease models in other, more experimentally tractable organisms. The identification of human disease-relevant pathways conserved in model organisms will allow new opportunities for studying diseases, disease genes and drug candidates, and often allow for genetic manipulations of the disease phenotype to illuminate molecular mechanisms of disease progression. Therefore, bioinformatics tools that can efficiently translate the biology of model organisms into new insights about human diseases are critical to connect model organism observations to human disease research.
Here, we present a new web-based tool for prioritizing the human diseases most relevant to a given set of model organism genes. MORPHIN (model organisms projected on a human integrated gene network) harnesses model organisms to investigate human disease by performing an orthology-based projection of model organism pathway genes onto a human integrated functional gene network (HumanNet (5)). Genome-scale functional gene networks have proven useful in prioritizing novel candidate genes for phenotypes in diverse species, including for human diseases (6). A typical use case for MORPHIN is as follows: suppose a user obtained a list of worm (C. elegans) genes involved in, for example, dauer induction, and wants to identify the human diseases most relevant to the worm dauer induction pathway. The user can submit the worm genes for dauer induction to the MORPHIN server, which then returns the following results: (i) human orthologs of the worm query genes, (ii) a list of human diseases significantly associated with the worm dauer induction pathway, based upon gene set enrichment or network-based closeness, (iii) gene networks from HumanNet between human orthologs of the worm genes for dauer induction and the associated human disease genes, (iv) a list of prioritized human orthologs of worm genes most relevant to each associated human disease. The user can actively link the model organism pathway to the most relevant human diseases, and thus potentially identify new disease models and find novel candidate genes for those human diseases.
MORPHIN substantially differs from other tools for mapping model-to-disease pathway associations, which are based on either intersection of orthologous genes (e.g. Phenologs (4), KOBAS (7)) or semantic similarity (e.g. PhenomeNET (8), PhenoDigm (9)). For example, MORPHIN employing the Fisher's exact test is equivalent to searching for phenologs for the model organism gene set. However, MORPHIN additionally uses not only overlap but also functional links between orthologous genes from pathways of model species and human to measure their association, by projecting orthologous genes of the model species on a human gene network, enabling detection of functional association between pathways with no overlap (10). This extra detection powered by a network algorithm, RIDDLE (Reflective Diffusion and Local Extension), may be particularly beneficial if annotation of a model pathway or a human disease is largely incomplete. Moreover, gene functional links within and between pathways allow prioritization of the model pathway genes for a disease, and may provide new molecular insights about pathogenesis.
DESCRIPTION OF MORPHIN
Overall design of MORPHIN server
The overall design of MORPHIN is summarized in Figure 1. Once a user submits a set of model organism query genes, MORPHIN performs the following analyses. First, MORPHIN identifies human orthologs of the submitted model organism query genes using the INPARANOID algorithm (11). MORPHIN then measures associations between human disease pathways and the query genes, calculated by overlap-based Fisher's exact test and network-based RIDDLE algorithm, and returns all significantly associated human diseases. Third, MORPHIN visualizes gene networks depicting the functional couplings between the query genes and human disease genes. Finally, MORPHIN returns a list of query genes prioritized by relevance to each of the associated human diseases. Figure 2 shows representative screenshots of the MORPHIN web interface.
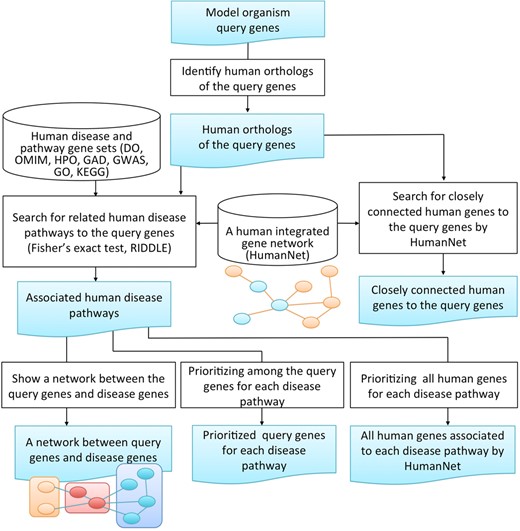
The overall design of the MORPHIN web server. Once a user submits a set of query genes for one of nine supported model organisms, MORPHIN performs the following analyses: MORPHIN first identifies human orthologs of the submitted model organism genes using INPARANOID (11). MORPHIN then searches for related human disease pathways to the query genes using Fisher's exact test (12) and RIDDLE (10). Third, for each significantly associated human disease pathways, MORPHIN displays the gene network between the query genes and disease genes by HumanNet links. Finally, MORPHIN prioritizes the query genes for the relevant human disease.

Screenshots of MORPHIN resulting from a query using six worm genes modulating dauer induction. (A) A table of human orthologs for the submitted six worm genes. The third column shows the in-paralog confidence score for each ortholog. (B) A table of significantly associated human disease pathways ranked by the Fisher's exact test. The P-value represents the statistical significance by Fisher's exact test; the q-value represents the adjusted significance for multiple hypotheses test;
Available model organisms and identification of human orthologs
The MORPHIN web server currently supports nine model organisms: C. elegans (worm), Danio rerio (zebrafish), Dictyostelium discoideum (social ameba), Drosophila melanogaster (fruit fly), Mus musculus (mouse), Rattus norvegicus (rat), Saccharomyces cerevisiae (budding yeast), Schizosaccharomyces pombe (fission yeast) and Xenopus laevis (African clawed frog). The protein sequences of human, zebrafish, mouse, rat were downloaded on 25 February, 4 December, 9 September 2013 from the NCBI Reference Sequence (RefSeq) database (http://www.ncbi.nlm.nih.gov/refseq) (13), worm WS239 from Wormbase (http://www.wormbase.org) (14), social ameba on 28 January 2014 from DictyBase (http://dictybase.org) (15), fruit fly release 5.54 from FlyBase (http://flybase.org) (16), budding yeast on 25 November 2013 from Saccharomyces Genome Database (http://www.yeastgenome.org) (17), fission yeast on 28 January 2014 from PomBase (http://www.pombase.org) (18) and African clawed frog on 28 January from UniProt (http://www.uniprot.org) (19). Users can submit query genes using gene names or unique systematic IDs, although we generally recommend using unique systematic IDs. Details of supported gene IDs are available on the MORPHIN tutorial page.
To identify human orthologs for the model organism query genes, MORPHIN employs the INPARANOID 4.1 standalone algorithm (http://inparanoid.sbc.su.se) (11), which allows multiple human orthologs for a given query gene by considering not only the best ortholog but also its functionally similar paralogs (in-paralogs) (20). This algorithm achieves a balance between sensitivity and specificity in identifying orthologs across two species (21,,22) by distinguishing in-paralogs duplicated after speciation from out-paralogs duplicated before speciation. Users can specify the desired organism and in-paralog score threshold. This score indicates the relative similarity to the two-way best-hit orthologs and ranges from 0 to 1, where 1 indicates the maximum likelihood of orthology. The suggested default threshold is 0, which maximizes sensitivity at the expense of specificity. However, the in-paralog score threshold can be increased so as to use only the most confident orthologs in subsequent analyses. Notably, where gene expansions have occurred, INPARANOID allows for multiple human orthologs for each model organism gene, each associated with its own in-paralog score (Figure 2A).
MORPHIN shows GO annotations for each human ortholog of the query genes, from which we may quickly capture functional properties of query genes in a human context. For more extensive functional characterization of the query genes in human contexts, MORPHIN also returns a list of the human genes closely connected to the query genes in HumanNet along with their GO annotations. Such annotations supplement the group-wise analysis search for functionally associated human disease pathways.
Identification of human diseases associated with the model organism pathway
MORPHIN returns those human diseases significantly associated with the query gene set as determined by two algorithms: gene set enrichment by Fisher's exact test (12) (Figure 2B) and RIDDLE, a measure of network proximity between two gene sets (10) (Figure 2C). Fisher's exact test is a classic overlap-based enrichment analysis, which measures the statistical significance of the observed overlap between two gene sets. When adjusted for multiple hypotheses (assessing significance as a q-value) (23), this method is generally considered to be statistically robust. However, this test requires the presence of common member genes between two gene sets. Considering that current annotations for many pathways are still largely incomplete, we might thus anticipate associations between pairs of gene sets to be missed due to failure to observe the overlap. To overcome this limitation, we previously developed a network-based measure of association between two gene sets, RIDDLE (10). RIDDLE determines functional closeness between two gene sets using a set-wise distance on an integrated functional human gene network, HumanNet (5). Because RIDDLE measures association based upon network connections between two gene sets, not their overlap in gene content, it can also detect relationships between gene sets which have no overlap, thus increasing its power to identify relevant human diseases.
While Fisher's exact test cannot detect associations between gene sets with no overlap, it showed slightly better performance for sets with overlap in a previous study using simulated test data (10). In addition, we anticipate that the two methods may be somewhat complementary since they employ entirely different methodologies. Therefore, MORPHIN provides the search results from both methods in two tables of candidate human diseases, one ranking pathways with q-value < 0.1 by Fisher's exact test and the other ranking pathways with FDR (false discovery rate) < 0.01 by RIDDLE (limited to the top 1000 pathways).
MORPHIN currently tests gene sets from seven databases of human pathways, including five databases cataloging human disease genes: (i) Disease Ontology (http://disease-ontology.org downloaded on 4 April 2014) (24), (ii) Genetic Association Database (GAD, http://geneticassociationdb.nih.gov, downloaded on 14 December 2013) (25), (iii) Genome-Wide Association Study Catalog (GWAS Catalog, http://geneticassociationdb.nih.gov, downloaded on 5 December 2013) (26), (iv) Human Phenotype Ontology (HPO, http://www.human-phenotype-ontology.org/ downloaded on 7 April 2014) (27), (v) Online Mendelian Inheritance in Man (OMIM, http://omim.org, downloaded on 4 December 2013) (28) and two databases for pathways or biological processes: (i) Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp, downloaded on 18 December 2013) (29) and Gene Ontology biological processes (GOBP, http://www.geneontology.org, downloaded on 17 December 2013) (30). Particularly, GOBP annotations are provided with various types of evidence including exclusively computational annotation (inferred from electronic annotation). To achieve high specificity in MORPHIN analysis, we used only highly reliable annotation with experimental or literature evidence: inferred from direct assay, expression pattern, genetic interaction, mutant phenotype, physical interaction and traceable author statement. Users also need to be aware of potentially inaccurate gene-to-disease associations by GWAS due to its mapping strategy, based on a gene's physical proximity to functional genetic variation affecting diseases.
Network visualization of human disease pathways
Network representations of human disease genes associated with model organism query genes can help interpret the associations and better prioritize genes of interest. MORPHIN provides network visualizations for each candidate human disease significantly associated with the model organism query genes. Clicking on the network icon beside each human pathway name opens a web-based network view (Figure 2D) displayed using Cytoscape Web, an interactive network browser (http://cytoscapeweb.cytoscape.org/) (31). The Cytoscape Web browser requires Flash Player to be installed on the local client machine. (Note that visualizing particularly large networks may be problematic depending on the performance of the local client machine.)
Genes are grouped into three categories, represented as boxes: query genes (orange nodes), disease genes (blue nodes) and overlap genes between two gene sets (red genes), and are linked by HumanNet functional associations. MORPHIN shows both group-level connections (black edges) and gene-level connections (blue edges) (Figure 2D). The RIDDLE algorithm can find connections between a group of query genes and a group of disease genes with no overlapping genes (see Figure 3 for examples). Sometimes two groups are connected in the absence of gene-level connections between them. This is possible because RIDDLE measures closeness between two groups of genes using not only direct connections but also indirect ones. Clicking on a network link shows detailed information about that link including its supporting evidence and confidence scores (LLS, log likelihood score) (32,,33). Clicking on a node provides detailed information for that gene including its name, category (query gene, disease gene or overlap gene) and total connection score to the disease genes, represented as a weighted sum of LLS as calculated in (33).
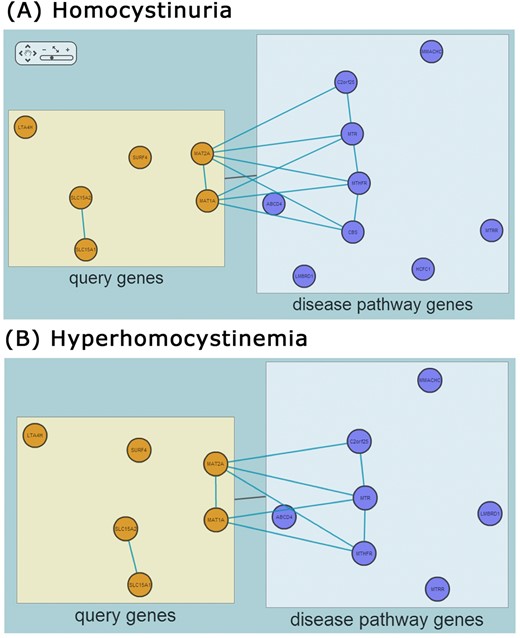
Networks between human orthologs of the 11 worm genes linked to an increased number of fat associated organelles (query genes) and human homocystinuria (A) or hyperhomocysteinemia (B) genes (disease pathway genes). Despite no overlap between query genes and disease pathway genes (there is no box for overlap genes) RIDDLE detected statistically significant association between them by using HumanNet-based connections between genes from the two gene sets.
Prioritization of queried model organism genes for a disease
Although a group of query genes from a model organism is found to be significantly associated with a human disease, individual query genes may not be equally relevant to the disease. Hence, prioritizing the query genes for the disease by functional relevance will be useful for follow-up functional studies focusing on key candidate genes. Query genes that overlap with the human disease genes might be considered to be top candidates, which will be listed in the first table. The rest of the query genes are prioritized for relevance to the disease based on network connectivity scores to the disease genes and listed in the second table (Figure 2E). For more reliable candidate selection, MORPHIN employs two complementary ranking systems: group ranks and global ranks. Group ranks are assigned among the group of query genes, while global ranks are assigned for all genes in HumanNet. Both group and global ranks of each query gene are presented in the table, allowing the user to assess both relevance within the query gene set and relevance relative to all other human genes. A query gene highly ranked among its group as well as among all human genes may be a particularly strong candidate for the disease. Lastly, MORPHIN returns all human genes prioritized for the disease by total connectivity scores to the disease genes in the third table for users who want to see disease candidate genes other than the query genes.
EVALUATION OF MORPHIN
In order to evaluate the capability of MORPHIN for disease model discovery, we assessed its power to prioritize gene-to-disease relationships annotated by two distinct methods: manual curation and semantic similarity. To construct validation sets of gene–disease relationships, we downloaded 149 and 88 manually curated gene-OMIM sets from Wormbase (14) and Mouse Genome Informatics (34), respectively, on 14 April 2014. For fly and zebrafish, such annotations by manual curation are not available. We therefore compiled 68 and 152 gene-OMIM sets for fly and zebrafish, respectively, from PhenomeNet (8), which is a cross-species phenotype ontology network based on semantic similarity (selecting pairs with similarity score > 0.1).
In all four species, MORPHIN, with the default in-paralog score threshold, outperformed the conventional Fisher's exact test in prioritizing reference gene-OMIM sets (Figure 4). MORPHIN that employs not only the overlap-based Fisher's exact test but also the network-based RIDDLE algorithm significantly improved identification of the reference gene-OMIM sets over the use of using Fisher's exact test only in mouse, worm, fly and zebrafish; 3.4 percentage points(pp), 12.1pp, 8.8pp and 3.3pp more matches in the top 10 ranks, respectively. The network-based method was critical particularly for fly and zebrafish, in which the overlap-based Fisher's exact test could identify none or only a few reference gene-OMIM relationships. The different degree of contribution of RIDDLE across the four species may be attributable to the differences in cross-species phenotype association approaches. Manual curation generally uses literature information, which mostly contains experimental data, and the majority of traditional disease models have been established from orthology-based hypotheses. Therefore, gene–disease models by manual curation are likely to contain orthologous overlaps between model species and human. In contrast, semantic approaches of gene–disease modeling do not consider genetic components of testing phenotypes, allowing associations between phenotypes of model species and human diseases even in the absence of orthologous genes in common.
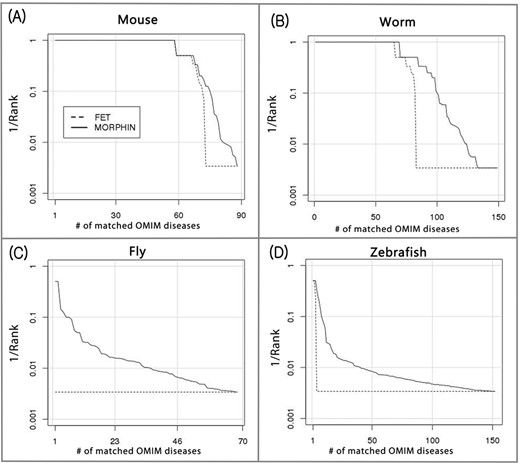
MORPHIN improves identification of human diseases associated with four model organism pathways. The reciprocal of the rank of the matching subset of reference gene-OMIM disease pairs is shown for Fisher's exact test (FET) and MORPHIN in mouse (A), worm (B), fly (C) and zebrafish (D). MORPHIN identified 85.2%, 67.1%, 8.8% and 5.3% of the reference gene-OMIM pairs, while the conventional FET method identified 81.8%, 55.0%, 0.0% and 2.0% in the top 10 ranks for mouse, worm, fly and zebrafish, respectively. The RIDDLE algorithm improved the performance of MORPHIN by 3.4pp, 12.1pp, 8.8pp and 3.3pp in the four species, respectively.
CASE STUDIES
To demonstrate application of MORPHIN in identification of new disease models, here we present two case studies using worm genes annotated according to the worm phenotype ontology (WPO) (35). First, MORPHIN shows a pre-computed example using six worm genes modulating dauer induction (WPO:0001539; F52D10.3, C54D1.3, Y110A7A.10, F52B5.5, R13H8.1, F55A3.3), which is known as an animal model of human diabetes (36). Both Fisher's exact test and RIDDLE identified diabetes-related terms among the top ranked human diseases (Figure 2B and C). Fisher's exact test identified ‘Type II diabetes mellitus (KEGG)’ in the fourth rank and RIDDLE identified ‘Pregnancy in Diabetics (GAD)’ in the third rank and ‘Type II diabetes mellitus (KEGG)’ in the 16th rank. Dauer formation is known to be regulated by insulin signaling pathways in worm (37). RIDDLE also identified two insulin-related biological processes, ‘cellular response to insulin stimulus (GOBP)’ and ‘insulin-like growth factor receptor signaling pathways (GOBP)’, tied in the fourth rank, while Fisher's exact test returns the most relevant term ‘insulin signaling pathway (KEGG)’ as the 41st rank.
Another example is a set of 11 worm genes associated with an increased number of fat associated bodies (WPO:0001888; F54C9.7, C54H2.5, C17G10.5, R11H6.1, C02A12.4, F01G10.3, ZK622.3, F01G10.2, K04E7.2, ZC416.6, C49F5.1). Interestingly, MORPHIN returned pathway or disease terms related to homocysteine metabolism. Homocysteine is an intermediate in methionine metabolism and acts as a methyl donor. High levels of homocysteine in blood and urine confer risk of cardiovascular diseases (38). Animal studies have suggested that a high fat diet is associated with an elevated homocysteine level (39). The Fisher's exact test identified the association of the query genes with ‘Cysteine and methionine metabolism (KEGG)’ but not with human disease terms. In contrast, RIDDLE identified not only the related pathway ‘homocysteine metabolic process (GOBP)’ but also the related diseases ‘Homocystinuria (HPO)’ and ‘Hyperhomocysteinemia (HPO)’ as third and fifth ranks, respectively, suggesting some of the query genes may be relevant to defects of homocysteine metabolism. There are no overlapping genes between the query genes and each of the two diseases (Figure 3), explaining why only the network-based RIDDLE method was effective at identifying the association with such diseases, which now suggest potential future experimental directions.
CONCLUSIONS
MORPHIN identifies human diseases most relevant to model organism pathway genes using highly sensitive and statistically robust methods incorporating a human gene network. For each significant human disease association, MORPHIN also prioritizes the query genes for disease relevance and visualizes the gene network comprised of query and disease genes. MORPHIN thus facilitates connecting model organism discoveries with relevant human diseases by (i) identifying potential new model systems for disease research, (ii) prioritizing new disease gene candidates for follow-up studies and (iii) using network data and visualization to promote insight about underlying biology of human disease. MORPHIN will be updated as significant changes in annotations for human disease pathways and new versions of HumanNet become available in the future.
FUNDING
National Research Foundation of Korea (2010–0017649, 2012M3A9B4028641, 2012M3A9C7050151 to I.L.); National Institutes of Health, National Science Foundation, Cancer Prevention Research Institute of Texas, U.S. Army Research (58343–MA); Welch (F-1515) Foundation to E.M.M.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments