





Abstract
RPA-coated single-stranded DNA (RPA–ssDNA), a nucleoprotein structure induced by DNA damage, promotes ATR activation and homologous recombination (HR). RPA is hyper-phosphorylated and ubiquitylated after DNA damage. The ubiquitylation of RPA by PRP19 and RFWD3 facilitates ATR activation and HR, but how it is stimulated by DNA damage is still unclear. Here, we show that RFWD3 binds RPA constitutively, whereas PRP19 recognizes RPA after DNA damage. The recruitment of PRP19 by RPA depends on PIKK-mediated RPA phosphorylation and a positively charged pocket in PRP19. An RPA32 mutant lacking phosphorylation sites fails to recruit PRP19 and support RPA ubiquitylation. PRP19 mutants unable to bind RPA or lacking ubiquitin ligase activity also fail to support RPA ubiquitylation and HR. These results suggest that RPA phosphorylation enhances the recruitment of PRP19 to RPA–ssDNA and stimulates RPA ubiquitylation through a process requiring both PRP19 and RFWD3, thereby triggering a phosphorylation-ubiquitylation circuitry that promotes ATR activation and HR.
INTRODUCTION
Cells are constantly challenged by exogenous and endogenous genotoxic stresses that threaten the integrity of their genetic material. DNA replication stress causes the slowing down and stalling of replication forks and is a particularly pervasive source of genomic instability in cancer cells (1,2). Replication stress was recently found to promote chromosomal instability in tumor cells and as such, may constitute an important driver of genome evolution and adaptation during oncogenesis (3). Extensive signaling pathways collectively known as the DNA damage response (DDR) detect a wide variety of DNA lesions including the aberrant replication fork structures generated during replication stress and quickly reprogram the cellular epigenome, transcriptome and proteome to activate cell-cycle checkpoints, repair damaged portions of the genome, stabilize replication forks and ultimately maintain genomic stability (4,5).
During replication stress, the uncoupling of DNA polymerases and replicative helicases at stalled forks produces persistent regions of single-stranded DNA (ssDNA) in the genome (6,7). This ssDNA is rapidly detected and bound by the heterotrimeric ssDNA-binding complex Replication Protein A (RPA; RPA70, RPA32 and RPA14) forming a crucial platform of the DDR: RPA-coated ssDNA (RPA–ssDNA) (8,9). The accumulation of RPA–ssDNA next to dsDNA junctions at blocked forks constitutes a key signal for the recruitment and activation of a number of replication-stress-response proteins including the master checkpoint kinase ataxia telangiectasia-mutated (ATM)- and Rad3-related kinase (ATR). The congregation of multiple factors at primer-template junctions present at stalled forks activates ATR which phosphorylates downstream targets to turn on checkpoints, stabilize blocked forks and facilitate the accurate completion of genome replication (10–13). Stalled forks are actively remodeled by helicases recruited on the RPA–ssDNA platform to form regressed structures that can use homologous recombination (HR)-based mechanisms for immediate restart or which can be nucleolytically processed to yield single-ended DNA double-stranded breaks (DSBs) which may be repaired through alternative recombination-based pathways such as break-induced replication (14,15). In addition to its fundamental role in DDR signaling and replication fork protection, RPA–ssDNA is also a central player during normal DNA replication and in most repair pathways including mismatch repair (MSH), nucleotide excision repair (NER) and HR (16–18).
To regulate its many functions in genome maintenance, the RPA–ssDNA platform is extensively modified during the cell-cycle and in response to genotoxic agents (8,19,20). For instance, RPA32 is phosphorylated by cyclin-dependent kinases (CDKs) on serine residues 23 and 29 during S-phase (S23) and G2/M (S23 and S29) and this has been shown to promote cell-cycle progression in human cells (21–23). Moreover, phosphorylation of these CDK sites is also enhanced by DNA damage which concomitantly causes the N-terminus of RPA32 to undergo hyper-phosphorylation at numerous additional serine and threonine residues through the conjugated activity of the three master phosphoinositide-3-kinase-related kinases (PIKKs) of the DDR: ATR, ATM and DNAPK (24–30). Constitutive hyper-phosphorylation of RPA32 has been shown to impede the association of the RPA complex with active replication centers (31). This led to the suggestion that RPA32 phosphorylation by PIKKs reallocates its activity from DNA replication to DNA damage signaling and repair functions. Accordingly, phosphorylation of RPA has been found to stimulate DNA repair and promote the protection and recovery of replication forks following genotoxic insults (22,26,29,32,33). Furthermore, there is evidence that RPA phosphorylation stimulates checkpoint activation and maintenance (26,33). Finally, RPA32 phosphorylation and its subsequent dephosphorylation are important for the regulation of the HR repair pathway (25,34,35). More recently, an HR-promoting role for DNA damage-induced SUMOylation of RPA70 has also been proposed (19).
We and others have recently shown that DNA damage induces RPA ubiquitylation (36–38). During replication stress, the PRP19 E3 ubiquitin ligase, which normally functions in RNA maturation, associates with RPA and stimulates its ubiquitylation (39–41,37). Our data and that of another research group established that PRP19 functions as a ubiquitin ligase on the RPA–ssDNA platform to promote ATR checkpoint activation and replication fork repair (37,42). PRP19 also participates in HR but whether it functions as a ubiquitin ligase on the RPA–ssDNA platform to promote this repair pathway remains undisclosed (43). Another ubiquitin ligase, RFWD3 was also shown to promote RPA complex ubiquitylation and to be required for replication fork restart and HR as well (38). How these two ubiquitin ligases work together to promote the RPA-centered DDR has not been addressed.
Here, we have investigated the regulation of RPA ubiquitylation in response to damage. We report that whereas RFWD3 is constitutively associated with the RPA complex, PRP19 binds to the RPA complex after DNA damage. We show that RPA32 phosphorylation stimulates both its interaction with PRP19 and its ubiquitylation in response to damage. Recruitment of PRP19 to sites of UV laser microirradiation also depends on RPA32 phosphorylation. We identify a positively charged pocket in the PRP19 WD40 repeat domain required for its interaction with RPA. Finally, we demonstrate that the damage-stimulated recognition of RPA by PRP19 and its ubiquitin ligase activity are both required for optimal RPA ubiquitylation, checkpoint activation and the repair of DSBs through HR. Our work describes a novel phosphorylation–ubiquitylation cascade occurring on RPA–ssDNA that drives ATR activation and DNA repair through the HR pathway and thus provides a greater molecular understanding of the role of RPA modification in genome maintenance.
MATERIALS AND METHODS
Cell culture
HeLa, HEK293T and U2OS DR-GFP cells were grown in Dulbecco's modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics.
Antibodies
The following antibodies were used in this study: PRP19 (ab27692, 1:400 IF, 1:1000 WB), ATM pS1981 (ab81292, 1:1000 WB), DNAPKcs pS2056 (ab18192, 1:1000 WB) and RPA32 pT21 (ab109394, 1:5000 WB) antibodies were from Abcam, CHK1 (SC-8404, 1:1000 WB), Myc (SC-40, 1:1000 WB) and HA (SC-7392, 1:1000 WB) antibodies were from Santa Cruz; RPA32 antibodies (MA1–26418, 1:500 IF, 1:1000 WB) were from Thermo, ATM (A300–299A, 1:1000 WB), RPA32 pS4/8 (A300–245A, 1:1000 WB), RPA32 pS33 (A300–246A, 1:5000 WB), RFWD3 (A301–397A, 1:250 IF, 1:1000 WB) antibodies were from Bethyl; ubiquitin antibodies were from Covance (MMS-257P, 1:1000 WB); CDC5L antibodies were from BD-Bioscience (612362, 1:1000 WB); anti-FLAG antibodies were from Sigma (F1804, 1:800 IF, 1:1000 WB); CHK1 pS345 (2348, 1:1000 WB), GAPDH (5174, 1:1000 WB), DNAPKcs (12311, 1:1000 WB) and tubulin (2144, 1:1000 WB) antibodies were from Cell Signaling.
Molecular cloning and cell line generation
The pDEST myc Ala10 RPA32 construct was obtained by PCR amplification and BP cloning into a Gateway Entry vector pDONR221 followed by LR recombination into a pDEST myc Gateway Destination vector. The template used for cloning was pET11d-RPA32 Ala10 (kind gift from Dr Marc Wold, University of Iowa) which encodes a RPA32 construct in which the following residues are mutated to alanines: S4, S8, S11, S12, S13, T21, S23, S29, S33 and S39. For generating the stable HA-RPA32 WT and HA-RPA32 Ala10 cell lines, pDONR221 RPA32 WT or RPA32 Ala10 were LR recombined into a pHAGE EF1α 3XHA-tag destination vector. Viruses were prepared and infection and selection of stable HeLa cell populations stably expressing the RPA32 constructs were performed using puromycin as a selection reagent. For the DR-GFP homologous recombination assays and RPA ubiquitylation complementation experiments, entry vectors containing WT, mPocket2, Δ-Ubox, Y405A and 3Xmut (C5A, V17E, Y33A (U-box defective)) PRP19 constructs were recombined into a pHAGE EF1α 3XHA-tag Destination vector. Viruses were produced and infection was performed to isolate stable clones of U2OS DR-GFP and HEK293T cells for the HR and ubiquitylation assays respectively.
RNA interference
The following siRNAs were used in this study. Ctl, GGGUAUCGACGAUUACAAAdTdT, PRP19 #1, 5′-GGGAUCUGGCGAAGCUU AAGAACUU-3′, PRP19 3′UTR, 5′-UGUAAGCAGUGAUCUAGUUUCAUUA-3′, RFWD3, 5′-GGACCUACUUGCAAACUAUdTdT-3′ (44) and RPA32 3′UTR, 5′-AGUCAGAGGAGACAUUUGAUAGAUG-3′ (Stealth siRNA from Thermo Fisher Scientific/Life Technologies). Cells were transfected with siRNAs using the Lipofectamine RNAimax reagent as per manufacturer's instructions (Life Technologies).
Protein complex purification
To capture SFB-tagged RFWD3 or PRP19 and its derivatives, cells were lysed in NETN buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% Igepal) containing protease and phosphatase inhibitors and incubated with streptavidin-coated magnetic beads (Life Technologies). Beads were retrieved with magnets and washed three times with NETN buffer before the captured proteins were eluted with SDS sample buffer.
Isolation of ubiquitylated proteins
Cells were transfected with pCDNA3.1 His6-ubiquitin (kind gift from Dr Jianping Jin, University of Texas Medical School at Houston) or pCDNA4T/O Strep-HA ubiquitin (kind gift from Dr Niels Mailand, University of Copenhagen). Cells expressing His6-ubiquitin were lysed in guanidium-HCl lysis buffer (6 M Guanidium HCl, 20 mM Tris–HCl pH 8.0, 0.5 M NaCl, 5% glycerol, 25 mM Imidazole). Following sonication, Ni-NTA denaturing pulldown was carried out directly in guanidium–HCl buffer using Ni-NTA agarose resin (Invitrogen). Precipitates were washed twice in guanidium–HCl buffer containing 0.1% Tween-20, twice in buffer B (guanidium–HCl buffer 1:4 in buffer C) and twice in buffer C (25 mM Tris–HCl pH 6.8, 150 mM NaCl, 25 mM Imidazole, 10 mM NEM, 5% glycerol, 0.1% Tween-20) before resuspension in Laemlli buffer. Cells expressing Strep-HA-tagged ubiquitin were processed as described previously (45). For the complementation experiments, HEK293T cells were infected with lentiviruses expressing HA-tagged PRP19 constructs. Populations of cells expressing the various constructs were selected via puromycin resistance carried by the lentiviral vector. Isolated clones from these populations were used to carry out the RPA ubiquitylation complementation experiments.
DR-GFP homologous recombination assays
U2OS DR-GFP cells (46) (a kind gift from Dr Maria Jasin, Memorial Sloan Kettering Cancer Center) were transfected with Ctl or PRP19-targeting siRNA and 24 h later plasmids expressing the I-Sce-I nuclease and mCherry were co-transfected. Forty eight hours later, the efficiency of homologous recombination was evaluated as the % of mCherry-positive cells that were also GFP positive using a BD FACS Jazz Cell Sorter (BD Biosciences). For the complementation experiments, U2OS DR-GFP cells were infected with lentiviruses expressing HA-tagged PRP19 constructs. Populations of cells expressing the various constructs were selected via puromycin resistance carried by the lentiviral vector. These cells were transfected with a 3′UTR targeting PRP19 siRNA to monitor the capacity of the various mutants to support HR.
UV-laser microirradiation
Cells were treated with 10 μM BrdU 24 h prior to the microirradiation. Before microirradiation, media was replaced with DMEM without phenol red. Microirradiation was performed using a 355 nm UV laser on an Arcturus Veritas Laser Capture Microscope (Life technologies) at the Advanced Tissue Resource Center of the Harvard NeuroDiscovery Center or with a 355 nm UV laser on MMI Cell Cut Plus Laser Capture microscope (Molecular Machines & Industries AG) at the Université de Sherbrooke.
Determination of the electrostatic potential of the PRP19 WD40 domain
The surface electrostatic potential of human PRP19 (pdb 4LG8) was generated using the PDB2PQR web server (47) and the adaptive Poisson–Boltzmann solver (APBS) (48) and were subsequently represented using PyMol (http://www.pymol.org/).
RESULTS
PRP19 interacts with RPA and RFWD3 after DNA damage
In response to DNA damage, the RPA complex is poly-ubiquitylated (36–38). We recently reported that depletion of the PRP19 E3 ubiquitin ligase causes a substantial decrease in RPA32 ubiquitylation in response to DNA damage (37). More recently, RFWD3, another E3 ubiquitin ligase previously implicated in the elaboration of the RPA–ssDNA-based DDR was also shown to stimulate damage-induced RPA complex ubiquitylation in HeLa cells (38). In agreement with a role for both of these ligases in this process, we found that depletion of PRP19 or RFWD3 in HEK293T cells strongly impedes camptothecin (CPT)-induced RPA70 ubiquitylation without significant changes in ubiquitin expression levels (Figure 1A). These results suggest that while both PRP19 and RFWD3 are required for DNA damage-induced RPA ubiquitylation, neither ligase is sufficient in vivo. Thus, the two E3 ligases must work in concert in a DNA damage-induced manner.
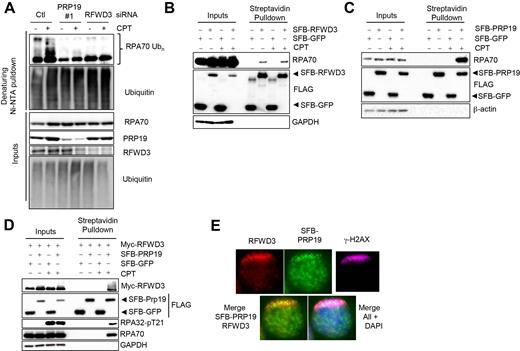
PRP19 assembles with RFWD3 on RPA–ssDNA in response to DNA damage and promotes RPA ubiquitylation. (A) PRP19 and RFWD3 depletion perturb DNA damage-induced RPA ubiquitylation. Cells were transfected with siRNAs targeting either PRP19 or RFWD3 and a vector expressing His6-tagged ubiquitin, treated or not with CPT and lysed under denaturing conditions. Ni-NTA pulldown was performed to isolate ubiquitylated proteins. (B) Cells were transfected with SFB- (S-protein, FLAG, streptavidin-binding peptide) GFP or SFB-RFWD3 vectors and streptavidin pulldown of SFB-tagged proteins in untreated or CPT-treated cells was performed. The indicated proteins were immunoblotted. (C) Cells were transfected with SFB-GFP or SFB-PRP19 vectors and streptavidin pulldown of SFB-tagged proteins isolated from untreated or CPT-treated cells was performed. The indicated proteins were immunoblotted. (D) Cells were transfected with SFB-GFP or SFB-PRP19 and myc-RFWD3 vectors. Streptavidin pulldown of SFB-tagged proteins isolated from untreated or CPT-treated cells was performed and the indicated proteins were immunoblotted. (E) HeLa cells transfected with an SFB-PRP19 vector and pre-sensitized with BrdU were UV laser microirradiated. Immunofluorescence against endogenous γ-H2AX, RFWD3 and FLAG epitope was subsequently performed to monitor RFWD3 and PRP19 accrual at damage sites.
The mechanism that triggers RPA ubiquitylation in response to DNA damage remains unclear. The regulation of the interactions between ubiquitin ligases and their substrates is a prevalent control mechanism for stimulus-induced ubiquitylation (49). Interestingly, although the RFWD3–RPA interaction is constitutively found in unstressed cells and is not affected by DNA damage (Figure 1B and (44,50)), the interaction between the PRP19 E3 ubiquitin ligase and RPA is greatly stimulated by CPT treatment (Figure 1C). Moreover, RFWD3 and PRP19 were found as part of the same complex only upon DNA damage induction and colocalized as punctate foci in UV laser microirradiation stripes suggesting that they might function together to stimulate damage-induced ubiquitylation by congregating on the RPA–ssDNA platform (Figure 1D and E). These results also indicate that molecular events must exist to trigger the RPA–PRP19 interaction and/or the ability of RFWD3 to induce RPA ubiquitylation in response to damage.
RPA hyper-phosphorylation correlates with increased PRP19 binding and RPA ubiquitylation
Regulatory relationships between different PTMs such as phosphorylation and ubiquitylation play an important part in the precise control of signaling pathways (51). We thus set out to investigate the relationship between phosphorylation and ubiquitylation of the RPA complex. The RPA70 and RPA32 subunits of the RPA complex are both ubiquitylated in response to CPT treatment (Figure 2A and Supplementary Figure S1A) (37,38). Furthermore, RPA32 ubiquitylation upon CPT treatment is also accompanied by a strong induction in RPA32 hyper-phosphorylation. To further investigate the genotoxic circumstances that might lead to RPA complex ubiquitylation, we tested the ability of different DNA damaging agents to induce this modification (Figure 2B and Supplementary Figure S1B). In agreement with previous results, we found that CPT (1 μM), ultraviolet radiation (UV, 50 J/m2) and hydroxyurea (HU, 4 mM) are potent inducers of RPA ubiquitylation, whereas ionizing radiation (IR, 10 γ) did not induce this modification (38). Interestingly, whereas CPT, UV and HU strongly induce hyper-phosphorylation of RPA, IR is comparatively a much weaker inducer of RPA32 N-terminal phosphorylation (Figure 2B). The correlation between RPA32 phosphorylation and ubiquitylation led us to examine the kinetics of each modification. Whereas RPA32 hyper-phosphorylation could be readily detected as soon as an hour after CPT addition, the increase in mono and poly-ubiquitylation of RPA32 and RPA70 could only be detected 2 h after CPT treatment and kept increasing at subsequent timepoints (Figure 2C, Supplementary Figure S1C and data not shown). To address whether ubiquitylated RPA was also phosphorylated, we used site-specific antibodies to detect phosphorylation of RPA32 at residues S4/8, S33 and T21 in ubiquitylated proteins isolated from CPT-treated cells. Whereas phosphorylation of RPA32 at each of the sites could be detected after 1 h, ubiquitylated phospho-RPA32 started increasing after 2 h of treatment indicating that at least some species of RPA32 are phosphorylated and ubiquitylated at the same time (Figure 2D). Interestingly, the damage-induced interaction of PRP19 with RPA is also detected after 2 h of CPT treatment, which correlates with the kinetics of RPA ubiquitylation (Figure 2E). Moreover, we found that PRP19 exhibits a strong preference for the hyper-phosphorylated form of RPA suggesting that this modification might help tether PRP19 to the RPA–ssDNA platform (compare P-RPA32/RPA32 ratios in input and PRP19 pulldown in Figure 2E). Collectively, these results show that RPA hyper-phosphorylation correlates with increased PRP19 binding and RPA ubiquitylation.
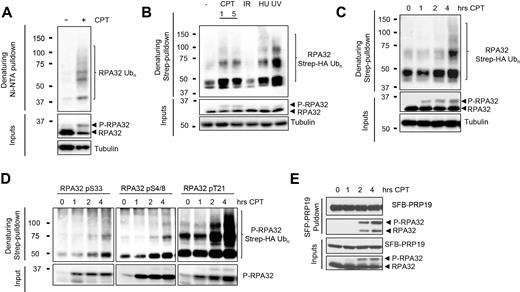
RPA32 is phosphorylated and ubiquitylated in response to DNA damage that targets active replication forks. (A) Cells were transfected with a vector expressing His6-tagged ubiquitin and lysed under denaturing conditions. Ni-NTA pulldown was performed to isolate ubiquitylated proteins. The indicated proteins were detected with specific antibodies. (B) Cells were transfected with a vector expressing Strep-HA ubiquitin and treated with 1 or 5 μM CPT, 10 γ IR, 4 mM HU or 50 J/m2 UV for 4 h. Ubiquitylated proteins were isolated by Strep-Tactin pulldown under denaturing conditions. (C) Cells transfected as in (B) were treated with 1 μM CPT for the indicated times and total RPA32 or (D) phosphorylated RPA32 species were detected using specific antibodies after Strep-Tactin pulldown. (E) A stable HEK293T cell line expressing SFB-PRP19 was treated with CPT 1 μM for the indicated times. SFB-PRP19 and its interactors were isolated using streptavidin-associated beads.
PIKKs promote RPA hyper-phosphorylation, PRP19 binding and RPA ubiquitylation
Because the RPA32 N-terminus is targeted by the PIKKs ATM, ATR and DNA-PK in response to damage, we examined the importance of each of these kinases for RPA phosphorylation and ubiquitylation using specific inhibitors (26,27,52,53). As shown in Figure 3A and Supplementary Figure S2A, inhibition of a single kinase led to weak decreases in RPA32 and RPA70 ubiquitylation with the most pronounced decrease observed with the single ATR inhibition. It is worth noting that to robustly detect RPA ubiquitylation, 4 h of CPT treatment were necessary. This prolonged treatment results in only a weak inhibition of RPA hyper-phosphorylation levels when ATM, ATR or DNA-PK inhibitors are used individually (Figure 3A). However, even after 4 h of CPT treatment, inhibition of all three kinases at the same time completely abrogated both damage-stimulated RPA phosphorylation and ubiquitylation. Supplementary Figure S2B shows the effective inhibition of each kinase when using VE-821, KU55933 and NU7026 to inhibit ATR, ATM and DNA-PK respectively. Concomitantly, whereas the inhibition of individual kinases partially inhibited the damage-induced PRP19-RPA interaction, inhibition of all three kinases at the same time completely abrogated the formation of this complex after damage (Figure 3B). In agreement with the partial impairment of the PRP19-RPA interaction, inhibition of single kinases slightly reduced the recruitment of PRP19 to RPA-labeled sites of UV-laser microirradiation (Figure 3C and D). Strikingly, inhibition of all 3 kinases led to a complete abrogation of PRP19 accumulation at laser stripes. Instead of being actively recruited, PRP19 was excluded and formed an anti-stripe at the site of UV-laser damage when microirradiation was performed in the presence of ATM, ATR and DNA-PK inhibitors (White arrow, Figure 3C bottom panel). Such anti-stripes are reminiscent of the behavior of other RNA processing proteins at sites of UV damage and might be attributable to the downregulation of transcription at sites of DNA damage (54–57). Therefore, PRP19 is likely released from transcription sites in the damaged areas, and subsequently recruited to damage sites by phospho-RPA.
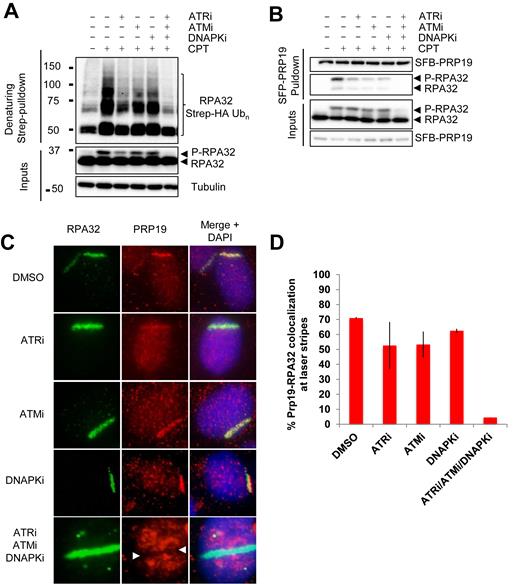
RPA32 ubiquitylation is regulated by PI3K-like kinases. (A) Cells were transfected with a vector expressing Strep-HA-tagged ubiquitin, pre-treated for 1 h with VE-821 (ATRi, 10 μM), KU55933 (ATMi, 10 uM), NU7441 (DNAPKi, 2 μM) or a combination of all three inhibitors, treated with 1 μM CPT for 4 h and lysed under denaturing conditions. Strep-Tactin pulldown was performed to isolate ubiquitylated proteins. (B) Cell transfection with an SFB-PRP19 vector were treated as in A and streptavidin pulldown was performed to isolate PRP19 along with its interactors. Controls for the efficiency of the inhibitor treatments are provided in (S2A). (C and D) U2OS cells were pre-sensitized with 10 μM BrdU, treated with the indicated inhibitors along with DRB for 1 h and damaged by UV-laser microirradiation. Recruitment of PRP19 and RPA32 was monitored 2 h after damage by immunofluorescence.
RPA32 phosphorylation is required for PRP19 binding and RPA ubiquitylation
PIKKs phosphorylate multiple substrates upon DNA damage (55,58,59). To ascertain whether RPA phosphorylation might specifically promote its interaction with PRP19 and its ubiquitylation, we tested whether a form of RPA32 lacking multiple phosphorylation sites (S4, S8, S11–13, T21, S23, S29, S33, T39) at its N-terminus could interact with PRP19 in response to DNA damage. This mutant was selected because it cannot be phosphorylated upon DNA damage but is still able to bind ssDNA and destabilize DNA helixes as well as the WT version in vitro (60).
We first engineered cell lines expressing WT or Ala10 RPA32 mutants at a level similar to endogenous RPA32 and tested whether impairment of RPA32 phosphorylation might impact its interaction with PRP19. Upon CPT treatment, WT RPA32 interacted strongly with PRP19 upon DNA damage, but the Ala10 mutant, although expressed similarly as WT and able to relocate to sites of DNA damage (Figure 4B and C), was unable to productively interact with PRP19 upon CPT treatment (Figure 4A). Contrastingly, RFWD3 interacted equally well in the presence or absence of DNA damage with both WT and the Ala10 mutant, indicating that the phosphorylation status of the RPA32 N-terminus has no effect on its constitutive interaction with RFWD3 (Supplementary Figure S3).
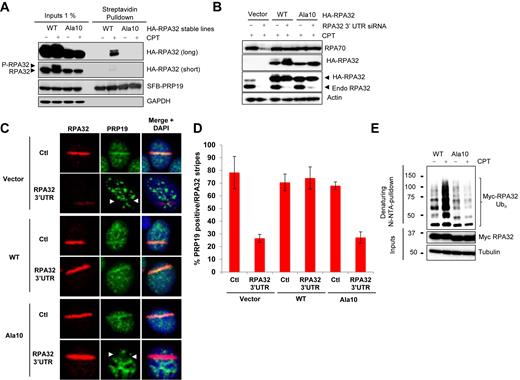
DNA-damage-induced RPA phosphorylation promotes its ubiquitylation. (A) HeLa cell lines stably expressing HA-tagged WT or Ala10 RPA32 mutants were transfected with a vector expressing SFB-PRP19. Cells were then treated with CPT 1 μM for 4 h, lysed and SFB-PRP19 and its interactors were isolated using streptavidin-associated beads. (B) Stable HeLa cell lines expressing the indicated HA-tagged RPA32 constructs were transfected with an siRNA targeting the 3′ untranslated region for the RPA32 mRNA. 72 h later, cells were treated with 1 μM CPT for 2 h, lysed and the indicated proteins were detected using specific antibodies. (C) Alternatively, cells transfected as in (B) were microirradiated and processed for immunofluorescence to examine PRP19 recruitment to laser stripes. (D) Histogram representing the recruitment of endogenous PRP19 to RPA32 stripes after laser microirradiation. The error bars correspond to biological triplicate experiments. At least 100 microirradiated cells were examined for each replicate. (E) Cells were transfected with vectors expressing His6-tagged ubiquitin and myc-tagged WT or Ala10 RPA32 mutants and treated with 1 μM CPT for 3 h. Ubiquitylated proteins were isolated by denaturing Ni-NTA pulldown.
To further examine the importance of RPA32 phosphorylation for PRP19 relocalization to RPA–ssDNA upon DNA damage, we knocked-down endogenous RPA32 in cell lines expressing HA-tagged WT or Ala10 RPA32 mutants using a 3′UTR-targeted siRNA (Figure 4B). As previously described, knockdown of endogenous RPA32 destabilized RPA70 (Figure 4B, first 2 lanes). Expression of HA-tagged WT or Ala10 RPA32 rescued RPA70 levels indicating that both of these constructs form stable RPA complexes. Next, we monitored the recruitment of PRP19 at UV-laser stripes in cell lines where endogenous RPA32 was replaced by WT or Ala10 RPA32. As shown in Figure 4C, RPA32 knockdown in non-complemented cells dramatically decreased the recruitment of PRP19 to sites of DNA damage. In fact, instead of PRP19 recruitment, anti-stripes of PRP19 could be observed at most RPA32 positive stripes (Figure 4C, upper panels and D). Of note, the intensity of RPA32 stripes in non-complemented cells was much weaker than in control siRNA-treated cells indicating an efficient knockdown of endogenous RPA32. In cells carrying WT RPA32-HA, the level of PRP19 recruitment remained very high despite the knockdown of endogenous RPA32 (Figure 4C, middle panels and D). However, in cells expressing only the Ala10 mutant, the recruitment of PRP19 was strongly decreased. Moreover, anti-stripes were present in most RPA32 positive stripes (Figure 4C, lower panel, D). These data strongly suggest that PRP19 recruitment onto RPA–ssDNA requires RPA32 hyper-phosphorylation.
Finally, we tested whether RPA phosphorylation promotes its ubiquitylation. As shown in Figure 4E, whereas the ubiquitylation of WT RPA32 was greatly induced by CPT treatment, the ubiquitylation of Ala10 RPA32 was unresponsive to DNA damage. Because the Ala10 mutant still interacts constitutively with RFWD3 but cannot be ubiquitylated efficiently, our data suggests that the RFWD3–RPA interaction is not sufficient for maximal damage-induced RPA ubiquitylation. It is plausible that PIKK-mediated RPA hyper-phosphorylation promotes its interaction with PRP19, which may function in concert with RFWD3 to induce RPA ubiquitylation during replication stress. In a non-mutually exclusive scenario, hyper-phosphorylation of RPA may render RPA a better substrate for RFWD3 and/or PRP19, contributing to the damage-induced RPA ubiquitylation.
A positively charged surface on the PRP19 WD40 repeat domain mediates its damage-induced interaction with RPA
Because RPA phosphorylation is required for its interaction with PRP19 at sites of damage, we looked for positively charged surfaces on the WD40 repeat domain of PRP19, which is essential for its interaction with RPA (37). We reasoned that these electropositive patches might mediate PRP19 damage-induced interaction with the phosphorylated RPA complex. Using the available crystallographic structure of human PRP19 WD40 repeat domain (Structural Genomics Consortium, PDB:4LG8), we were able to identify two surface accessible basic pockets on the WD40 surface that may potentially accommodate negatively charged peptides (Figure 5A and B). We mutated positively charged lysine and arginine residues in each pocket to alanines in order to decrease their electrostatic potential and evaluated the ability of these mutants to interact with RPA in a damage-inducible manner. Although mPocket1 (K244A K265A K266A) still interacted with the RPA complex as well as WT, mutating two residues in Pocket 2 completely abolished the ability of PRP19 to interact with the RPA complex in response to damage (Figure 5C). Altogether, these data imply that following RPA32 phosphorylation, PRP19 is recruited to damaged replication forks via a basic pocket in its WD40 domain where it may cooperate with RFWD3 to ubiquitylate RPA and regulate damage signaling and repair.
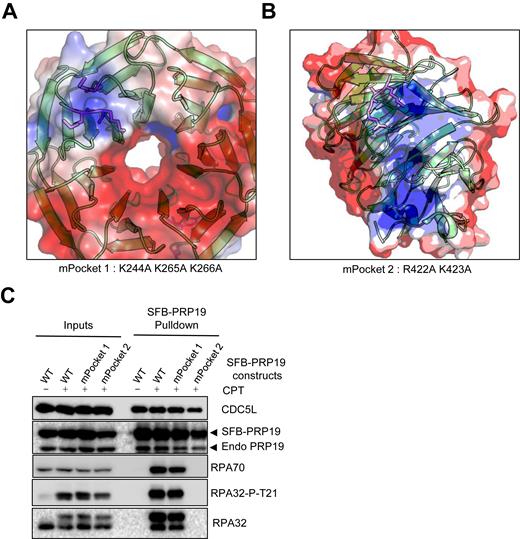
The PRP19 WD40 domain contains an electropositive surface that mediates its damage-induced interaction with the RPA complex. (A and B) Electrostatic surface potential of the WD40 repeats domain of human PRP19 identifies two major electropositive surfaces (blue). The mutated positively charged residues in the mPocket1 and mPocket2 PRP19 constructs are shown in magenta. (C) Cells were transfected with WT SFB-PRP19 or the indicated mutants and treated with 1 μM CPT 24 h later. SFB-PRP19 constructs and their interactors were isolated using streptavidin-associated beads and immunoblotted with the indicated antibodies.
The RPA-binding surface and ubiquitin ligase activity of PRP19 are required for damage-induced RPA ubiquitylation
We next addressed the roles of the PRP19 RPA binding pocket and its U-box E3 ubiquitin ligase domain for RPA ubiquitylation. First, we monitored the impact of PRP19 depletion using a 3′UTR-targeted siRNA on RPA32 and RPA70 ubiquitylation upon DNA damage. As seen in Figure 6A and similarly to another PRP19-targeting siRNA (Figure 1A), PRP19 depletion strongly abrogated both RPA32 and RPA70 ubiquitylation upon CPT treatment. We could also see a similar decrease of HU-induced RPA ubiquitylation following PRP19 depletion (Supplementary Figure S4A). Next, we obtained stable cell lines expressing siRNA-resistant HA-tagged WT and mutant PRP19. Using the WT PRP19 cell line, we could rescue RPA32 and RPA70 CPT-induced ubiquitylation, thereby demonstrating that PRP19 is necessary for RPA ubiquitylation. We further tested whether the ability of PRP19 to interact with RPA and its ubiquitin ligase activity of PRP19 are required for RPA ubiquitylation. Using stable cell lines expressing mPocket2 PRP19 or a PRP19 completely lacking its U-box E3 ubiquitin ligase domain (ΔU-box), we determined that both of these PRP19 mutants were unable to support optimal RPA complex damage-induced ubiquitylation (Figure 6B). Thus, our data clearly demonstrate that both the ubiquitin ligase activity of PRP19 and its damage-inducible interaction with RPA are required for optimal RPA complex ubiquitylation during replication stress.
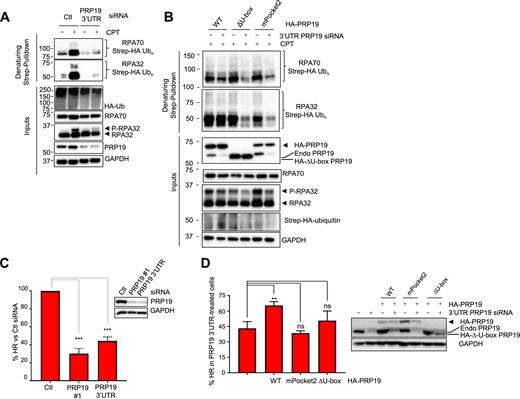
The PRP19 E3 ubiquitin ligase activity and its electropositive RPA-binding pocket are required for RPA ubiquitylation and homologous recombination. (A) Cells were transfected with Ctl or PRP19 3′UTR-targeted siRNA and 24 h later with a vector expressing Strep-HA ubiquitin. Twenty four hours later, cells were treated with 1 μM CPT for 3 h and lysed under denaturing conditions. Strep-Tactin pulldown was performed to isolate ubiquitylated proteins. (B) Cells expressing the indicated HA-PRP19 contructs were transfected with Ctl or PRP19 3′UTR-targeted siRNA and a Strep-HA ubiquitin vector and exposed to CPT as in A. Ubiquitylated proteins were isolated and the indicated proteins were detected by immunoblotting. (C) U2OS DR-GFP cells were transfected with the indicated siRNAs and subsequently co-transfected with mCherry and I-Sce-I expressing plasmids. Homologous recombination efficiency is measured as the % of GFP- and mCherry-positive cells to normalize for transfection efficiency and HR efficiency is plotted relatively to Ctl siRNA transfected cells. The graph represents the mean obtained from three independent experiments (***P < 0.001, Student's t-test). (D) U2OS DR-GFP cells stably expressing the indicated HA-PRP19 constructs were transfected with Ctl or PRP19 3′UTR siRNAs and subsequently co-transfected with mCherry and I-Sce-I expressing plasmids. The graphs represents the % HR in PRP19 3′UTR siRNA-transfected cells relative to Ctl siRNA-transfected cells for each cell line and were compiled from three independent experiments (**P < 0.01, ns = non-significant, Student's t-test).
PRP19 functions as an E3 ubiquitin ligase on RPA–ssDNA to promote HR
RFWD3 and PRP19 were both shown previously to promote DSB repair through HR but whether PRP19 functions as a ubiquitin ligase on RPA–ssDNA to promote this DSB repair pathway has not been thoroughly addressed (38,43,61). To monitor HR by gene conversion, we used the well-established DR-GFP system to determine whether depletion of PRP19 with two independent siRNAs would impede this repair pathway (46). In agreement with previous results, we observed a 60–70% decrease in gene conversion efficiency upon PRP19 depletion. Next, we performed complementation experiments using a stable cell line expressing an HA-tagged WT PRP19 at near endogenous levels. In this cell line, we used the PRP19 3′UTR-targeting siRNA to specifically deplete endogenous PRP19 and were able to partially but reproducibly rescue the HR defect (Supplementary Figure S4B). One possible explanation for this incomplete rescue may reside in the fact that N-terminal tagging of PRP19 with HA epitopes might perturb its ability to ubiquitylate substrates as the tag is adjacent to its U-box. In agreement with this, RPA ubiquitylation is also partially rescued by WT PRP19 (Figure 6B). In addition, we further tested whether the ability to interact with RPA and the ubiquitin ligase activity of PRP19 are required for its function in HR. Using stable cell lines expressing mPocket2 PRP19 mutant or PRP19 completely lacking its U-box E3 ligase domain (ΔU-box), we were unable to rescue the HR defect. Moreover, the Y405A PRP19 mutant which we have previously shown is unable to productively interact with RPA upon damage and a triple point mutant (3Xmut, C5A/V17E/Y33A) in conserved residues of the U-box domain of PRP19 were also unable to significantly rescue the HR defect caused by PRP19 depletion (Supplementary Figure S4C) (62,63). Of note, both the 3Xmut construct and the mPocket2 mutants were defective for ATR activation in response to CPT (Supplementary Figure S4D,E). Altogether, these data strongly argue that PRP19 must be recruited by RPA–ssDNA and function as an E3 ligase to promote full ATR activation and the HR repair pathway.
DISCUSSION
A phosphorylation-ubiquitylation cascade on the RPA–ssDNA platform
The DDR is regulated by a complex array of PTMs which modify existing proteins to reorient their activities towards genome maintenance at the sites of DNA damage (64,65). These PTMs occur on two main nucleoprotein structures that orchestrate the DDR: chromatin and RPA–ssDNA. Several seminal papers published in the last few years have elegantly shown that on the chromatin flanking DSBs, phosphorylation of the histone variant H2A.X triggers a series of events leading to the recruitment of the RNF8 and RNF168 ubiquitin ligases which work together to regulate repair pathway choice at DSBs (64,66).
We now show that a similar phosphorylation–ubiquitylation PTM cascade occurs on RPA–ssDNA to promote ATR signaling and HR. Our work supports a working model whereby RPA phosphorylation through the combined efforts of ATM, ATR and DNA-PK promotes the recruitment of PRP19 which works together with RFWD3 to ubiquitylate RPA and potentially additional substrates (Supplementary Figure S6). Prior to DNA damage, RFWD3 is associated with RPA32 but our data show that it cannot efficiently polyubiquitylate the RPA complex without (1) RPA32 phosphorylation and (2) PRP19 tethering to RPA–ssDNA. Additionally, phosphorylation of RFWD3 by ATM and ATR may stimulate its ubiquitin ligase activity (67). The requirement of these conditions for optimal RPA ubiquitylation may limit the extent of this modification to prevent erroneous ATR activation and engagement of the HR pathway during normal DNA replication.
How might PRP19 and RFWD3 work together to promote full RPA ubiquitylation? RFWD3 depletion does not appear to affect the recruitment of PRP19 on RPA–ssDNA produced by UV laser microirradiation (Supplementary Figure S5A and B). Conversely, depletion of PRP19 or RFWD3 did not significantly impact each other's cellular levels (Supplementary Figure S5B–D). Interestingly, RFWD3 has been shown to collaborate with the MDM2 ubiquitin ligase to promote p53 ubiquitylation (68). In that context, RFWD3 alone is unable to ubiquitylate p53 but when combined with MDM2, it strongly enhances p53 ubiquitylation. It is plausible that a similar collaboration between RFWD3 and PRP19 supports optimal RPA ubiquitylation. It is also possible that RFWD3 and PRP19 might target different lysine residues on the RPA subunits which when combined allow robust ubiquitylation of the complex. Further work will be required to sort out these hypotheses. In yeast, there is no obvious ortholog of RFWD3, Prp19 however is highly conserved and is also known as Pso4 for PSOralen sensitive 4 as it was identified in a genetic screen for sensitivity to this crosslinking agent. Similarly to PRP19, Prp19/Pso4 functions in interstrand crosslink repair and mitotic recombination (69,70). It will be interesting to determine whether Prp19/Pso4 also associates with and ubiquitylates yeast RPA in response to damage.
Interestingly, both RFWD3 and PRP19 also promote RPA phosphorylation (37,38,50). Mutation of specific RPA32 ubiquitylation sites or impairment of its association with RFWD3 also dampen RPA phosphorylation (38). These prior data along with the evidence provided here that RPA phosphorylation tethers PRP19, which together with RFWD3 stimulates RPA ubiquitylation, cement the existence of a feed-forward loop on RPA–ssDNA. Similarly to the propagation of phosphorylation and ubiquitylation across chromatin adjacent to DSBs by the γ-H2AX-RNF8-RNF168 DDR axis, the ability of both PRP19 and RFWD3 to promote RPA phosphorylation and ubiquitylation may support the simultaneous spreading of these modifications to extensive stretches of RPA–ssDNA at stalled replication forks or upon resection of DSB to ensure robust damage signaling and repair via HR (37,38,42).
PRP19 promotes RPA phosphorylation via ATR activation by acting as a ubiquitin ligase on RPA–ssDNA (Supplementary Figure S4D,E and (37,42,71)). More specifically, PRP19 promotes phosphorylation of RPA32 S33 by ATR which further stimulates RPA hyper-phosphorylation at additional sites by ATM and DNA-PK kinases (22,29). We showed previously that the ATR-ATRIP complex has affinity for K63-linked ubiquitin chains and that ATR-ATRIP recruitment to damage sites is decreased upon PRP19 downregulation. K63-linked chains along with other forms of non-degradative ubiquitin moieties are conjugated to the RPA complex suggesting that RPA ubiquitylation could promote its phosphorylation by enhancing ATR activation and perhaps the accessibility of phosphorylation sites to PIKKs (38). Concomitantly, phosphorylated RPA might also be a better substrate for ubiquitylation, which would also enhance the robustness of the feed-forward loop. The PRP19-XAB2 complex was also recently shown to regulate DSB resection (61). It is possible that some of the decrease in ATR activation and RPA ubiquitylation caused by PRP19 depletion is due to a defect in DSB resection. However, since PRP19 can ubiquitylate RPA directly in vitro and because PRP19 complex depletion also decreases RPA ubiquitylation and phosphorylation in response to short HU treatment which does not cause DSBs and resection (Supplementary Figure S4A and (71)), we favour the possibility that PRP19 acts directly on the RPA complex to promote its ubiquitylation and phosphorylation via ATR activation (37).
In the case of RFWD3, while its depletion can impede both CHK1 and RPA32 phosphorylation in certain cell types, the decrease in CHK1 phosphorylation appears to be more context-specific than for PRP19 (38,44,50). As such it is worth noting that proteins such as NBS1 and the recently described ATR activator ETAA1 may promote the phosphorylation of RPA32 by ATR but are not critically important for CHK1-targeting which is more sensitive to TOPBP1 depletion (72–75). It is possible that RFWD3 and PRP19 participate in ATR activation under complementary circumstances with PRP19 functioning both at the double-stranded/single-stranded DNA junction to mediate ATR activation and CHK1 phosphorylation and on RPA–ssDNA to promote RPA phosphorylation. Notably, a direct interaction between the CDC5L component of the PRP19 complex and ATR was shown to promote canonical ATR signaling through CHK1 phosphorylation and might contribute to the observed differences between PRP19 and RFWD3 depletion on ATR activity (71). Constrastingly, RFWD3 would function mostly on RPA–ssDNA itself, hence the greater impact of its depletion on RPA phosphorylation.
Our data clearly shows that PRP19 preferentially interacts with the hyper-phosphorylated form of the RPA complex and that damage-induced RPA phosphorylation is required for its ubiquitylation and interaction with PRP19 in vivo. There are a number of ways through which RPA phosphorylation might trigger its interaction with PRP19. One possibility is that PRP19 might directly recognize phosphorylated RPA itself similar to the recently described enhanced interaction between PALB2 and phosphorylated RPA in vitro (32). However, we were unable to detect a direct interaction between the WD40 repeat domain of PRP19 and RPA32 N-terminus phosphopeptides (data not shown) suggesting that while phosphorylation of RPA promotes the PRP19-RPA interaction in vivo, it is not sufficient to recapitulate the full PRP19-RPA interaction in vitro. This is in agreement with the fact that mutation of the RPA70 N-terminus also impedes the RPA-PRP19 interaction (37). The BCAS2 subunit of the core PRP19 complex may also contact RPA directly, again suggesting a multipronged PRP19-RPA interaction in vivo (42). Alternatively, RPA phosphorylation may promote the removal of proteins that might compete with PRP19 for RPA binding. For instance, RPA phosphorylation impairs its interaction with the MRN complex (76).
RPA ubiquitylation and HR
Both PRP19 and RFWD3 promote DNA repair through HR (Figure 6C and D and Supplementary Figure S4B and C and (38,43)). A previous paper concluded that PRP19 itself but not its ubiquitin ligase activity are required for HR. However, the mutant used to complement the HR defect in this previous report contained a small 3 amino acid deletion V17, S18, P19 in the middle of the UBOX domain (62,63). Because no data was presented to demonstrate the non-functionality of this mutant it is possible that it still has sufficient ubiquitin ligase activity to support HR. Here, we provide 2 additional PRP19 ubiquitin ligase-deficient mutants; one in which the complete U-box domain has been removed (ΔU-box) making it impossible for PRP19 to recruit E2 partners and another (3Xmut) in which we mutated three highly conserved residues equivalent to functionally important amino acids in Saccharomyces cerevisiae Prp19 C5A (C3), V17E (L15) and Y33A (Y31). Each of these mutations is unable to rescue the viability defect of the Prp19 mutant yeasts indicating that the ubiquitin ligase activity is obliterated in individual mutants (62,63). We also show that two independent RPA interaction point mutants of PRP19 are unable to support efficient HR. Thus, our data strongly supports a model in which the PRP19 ubiquitin ligase activity on RPA–ssDNA is important for HR and ATR activation in mammalian cells.
At this point, it remains to be determined how PRP19 and RFWD3-mediated ubiquitylation on the RPA–ssDNA platform promotes HR. Indeed, the presence of PRP19 and RFWD3 on the RPA–ssDNA platform puts them in the vicinity of multiple genome maintenance factors that could become ubiquitylation substrates during the DDR (8). Recently, RFWD3 was shown participate in the ubiquitylation of both RPA and RAD51 in response to mitomycin C treatments (67). Additionally, a mutation in a Fanconi anemia patient that impairs the interaction of RFWD3 with RPA interferes with RPA and RAD51 ubiquitylation. Ubiquitylation of RPA and RAD51 appears to promote their clearance from interstrand crosslinking damage sites which paves the way for latter steps in the HR pathway (67,77). Interestingly, PRP19 depletion also increases the retention time of RAD51 foci during HU-induced replication stress (37). It will be interesting to determine whether PRP19 and RFWD3 have overlapping as well as unique ubiquitylation targets on RPA–ssDNA and determine how this plays into DDR activation and DNA repair by HR.
In conclusion, our work shows for the first time that RPA phosphorylation acts to bring together the RFWD3 and PRP19 ubiquitin ligases on RPA–ssDNA, which promote ATR activation and HR. We provide a mechanism that explains the DNA damage regulation of RPA ubiquitylation and show that DNA damage signals propagate on both the chromatin and RPA–ssDNA platforms through similar phosphorylation and ubiquitylation-based feed-forward loops, highlighting fundamental organizational principles of the DDR.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Dr Marc Wold, Dr Xiaohua Wu, Dr Niels Mailand, Dr Jianping Jin, Dr Caroline Saucier, Dr Andrew E.H. Elia, Dr Stephen J. Elledge and Dr Maria Jasin for the generous gift of reagents. We thank Dr Hai Dang Nguyen for reagent characterization. We also want to thank Daniel Garneau and Dominique Jean for technical support during the microscopy and FACS experiments.
FUNDING
Discovery Grant from the Natural Sciences and Engineering Research Council of Canada [Discovery grant 5026 to A.M]; Next-Generation of Scientists Scholarship from the Cancer Research Society [21531 to A.M]; Project Grant from the Canadian Institutes of Health [Project Grant 376288 to A.M.]; National Institutes of Health [GM076388 and CA197779 to L.Z]. Funding for open access charge: Next-Generation of Scientists Scholarship; NIH Grant.
Conflict of interest statement. None declared.
REFERENCES
Author notes
These authors contributed equally to this work as first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments