




Abstract
Myotonic dystrophy type 1 (DM1) is an inherited dominant muscular dystrophy caused by expanded CTG·CAG triplet repeats in the 3′ untranslated region of the DMPK1 gene, which produces a toxic gain-of-function CUG RNA. It has been shown that the severity of disease symptoms, age of onset and progression are related to the length of the triplet repeats. However, the mechanism(s) of CTG·CAG triplet-repeat instability is not fully understood. Herein, induced pluripotent stem cells (iPSCs) were generated from DM1 and Huntington's disease patient fibroblasts. We isolated 41 iPSC clones from DM1 fibroblasts, all showing different CTG·CAG repeat lengths, thus demonstrating somatic instability within the initial fibroblast population. During propagation of the iPSCs, the repeats expanded in a manner analogous to the expansion seen in somatic cells from DM1 patients. The correlation between repeat length and expansion rate identified the interval between 57 and 126 repeats as being an important length threshold where expansion rates dramatically increased. Moreover, longer repeats showed faster triplet-repeat expansion. However, the overall tendency of triplet repeats to expand ceased on differentiation into differentiated embryoid body or neurospheres. The mismatch repair components MSH2, MSH3 and MSH6 were highly expressed in iPSCs compared with fibroblasts, and only occupied the DMPK1 gene harboring longer CTG·CAG triplet repeats. In addition, shRNA silencing of MSH2 impeded CTG·CAG triplet-repeat expansion. The information gained from these studies provides new insight into a general mechanism of triplet-repeat expansion in iPSCs.
INTRODUCTION
More than 30 hereditary diseases in humans are caused by expansion of a simple triplet-repeat sequences in genomic DNA, such as CTG·CAG (and the reverse orientation, CAG·CTG), GAA·TTC and CGG·CCG (1,2). These expanded triplet-repeat sequences are unstable and frequently change in length during intergenerational transmission and within somatic cells. For example, myotonic dystrophy type 1 (DM1) patients may have between 50 and 5000 triplet repeats in their pathogenic allele, while Huntington's disease (HD) patients harbor between 38 and 180 repeats (3). Why some repeats expand more than others still remains an important unresolved question. In addition, the intrinsic expansion tendency of different triplet repeats is also not well understood.
DM1 is an inherited autosomal dominant muscular dystrophy caused by expanded CTG·CAG triplet repeats in the 3′-untranslated region (UTR) of the DMPK1 gene, which produces a toxic gain-of-function CUG RNA (4,5). DM1 patients may have between 50 and 5000 CTG·CAG triplet repeats in their pathogenic allele, while normal alleles have between 6 and 34 repeats (6). HD is an inherited neurodegenerative disorder with an expansion of CAG·CTG triplet repeats to more than 35–40 repeats–but rarely above 60–in the Huntingtin (HTT) gene (3,7,8). Studies have shown that the severity of disease symptoms, age of onset and progression are related to the size of the CTG/CAG triplet-repeat expansion in both DM1 and HD (9). Since the CTG/CAG triplet-repeat expansion mutation was identified (4,5), triplet-repeat expansion has been studied extensively in bacteria (10,11), yeast (12–14), mouse (15–17), mammalian cells (8,18–22), human cell extracts (23–25) and patient tissue (6,26–28), but the molecular mechanisms of expansion remain poorly understood. These systems differ substantially from humans and from each other in terms of triplet-repeat size, DNA replication rate, the cell type in which triplet-repeat expansion occurs, and in terms of chromatin structure. Therefore, studies in CTG·CAG triplet-repeat expansion diseases are still limited by the availability of an appropriate cell model in which to study the mechanism of CTG·CAG triplet-repeat instability in the human genome.
In our previous studies (29,30), we showed that GAA·TTC triplet repeats are highly unstable in Freidreich ataxia (FRDA) induced pluripotent stem cells (iPSCs) and that their expansion involves the highly active mismatch repair system. Our findings provided the first human cellular model to study the mechanisms of GAA·TTC triplet-repeat expansion in the context of the endogenous cellular FXN gene in human cells. However, that study did not address the generality of the findings to other trinucleotide repeat diseases. To determine whether the triplet-repeat instability observed in FRDA iPSCs is unique to GAA·TTC repeats or represents a more general phenomenon, we derived both HD and DM1 iPSCs. We found that CTG·CAG triplet-repeat expansion also occurred in DM1 iPSCs, but not in differentiated embryoid bodies (D-EB) or neurospheres (NS), and that the rate of expansion is dependent on the length of the repeats. Similar to other reports (31), the relatively small CAG·CTG triplet repeats in the HTT gene do not expand in HD iPSCs. We also find that the CTG·CAG triplet repeats in DM1 fibroblasts are heterogeneous in length, but when individual iPSC clones are examined, these latter cells have unique expanded alleles, thus providing a series of clones with different repeat numbers but an otherwise identical genetic background. These cells should prove useful for further analysis of DM1 pathophysiology.
RESULTS
Derivation of iPSCs from DM1 and HD patient fibroblasts
Primary fibroblasts from two DM1 patients (GM03991 and GM06076) and one HD patient (GM04285) were reprogrammed by four-transcriptional factor over-expression (OCT4, SOX2, Klf4 and c-Myc). We observed no difference in the efficiency of DM1 and HD fibroblast reprogramming compared with unaffected or FRDA fibroblasts (29,30). iPSC colonies with ESC morphology were selected and expanded. Analysis by qRT polymerase chain reaction (PCR) (Fig. 1A and B) and immunostaining (Fig. 1C) showed that our DM1 and HD iPSC lines expressed genes indicative of pluripotency. iPSC-derived EB expressed markers of endoderm, mesoderm and ectoderm (Fig. 1D). The iPSCs were also differentiated in vivo into teratomas that manifested elements of all three embryonic germ layers (Fig. 1E). Therefore, these DM1 and HD iPSCs met the most stringent criteria for pluripotency.
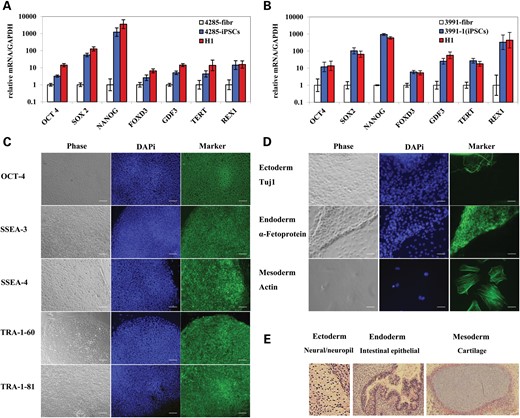
Characterization of HD and DM1 iPSCs. HD and DM1 iPSCs show similar expression of pluripotency genes as H1 hESCs. (A) HD (GM04285) fibroblasts, iPSCs and H1 hESCs. (B) DM1 (GM03991) fibroblasts, iPSCs (clone 1) and H1 hESCs. mRNA levels are normalized to GAPDH. Error bars indicate SEM of duplicate measurements. (C) Staining of pluripotency markers in DM1 (GM03991) iPSCs (clone 1). Phase contrast (gray); nuclear staining (blue); pluripotency markers staining (green). Tra-1-60 and Tra-1-81, surface markers; SSEA-3 and SSEA-4, stage-specific embryonic antigens; OCT4, transcription factor. Scale bars represent 0.1 mm. (D) Staining of three germ layer markers in DM1 (GM03991) iPSCs (clone 1) EB. Phase contrast (gray); nuclear staining (blue); three germ layer markers staining (green). Tuj1, ectoderm marker; a-Fetoprotein, endoderm marker; actin, mesoderm marker. Scale bars represent 0.05 mm. (E) Representative in vivo differentiation assay for assessing the pluripotency of HD iPSCs. Teratoma tissue sections were stained with hematoxylin and eosin and imaged by brightfield microscopy. Differentiated tissues representing ectoderm (neural/neuropil tissue), endoderm (intestinal epithelia) and mesoderm (cartilage) are shown.
Somatic instability of CTG·CAG triplet repeats in primary fibroblasts
It is known that expanded CTG·CAG triplet repeats in DM1-associated alleles are genetically highly unstable and undergo changes in repeat-length in both the germline and soma (32). Small-pool polymerase chain reaction (SP-PCR) has been used to detect the rarer mutant molecules (26). This method requires PCR amplification of the triplet-repeat sequence in multiple small pools of input genomic DNA, containing on the order of 0.5 to 200 genomic equivalents, followed by Southern blot analysis (33). SP-PCR can detect rare molecules comprising <10% of the total population. However, SP-PCR is a less effective technique when investigating triplet-repeat instability over time.
Similar to our findings in FRDA (29,30), DM1 patient-derived iPSCs could be an ideal model to study triplet-repeat instability. During reprogramming, each fibroblast that captures all four-transcription factors can grow to one iPSC colony, each with a unique triplet-repeat length. Thus, the isolation of different iPSC clones with different repeat lengths represents the somatic instability in the starting fibroblasts population. The analysis of repeat length can be done by conventional PCR, giving the advantage over SP-PCR of a robust signal and greater reproducibility.
Genomic DNA from DM1 fibroblasts (GM03991) produced at least four different bands with conventional PCR (Fig. 2). The PCR band corresponding to ∼20 CTG·CAG triplet repeats comes from the normal allele (DM1 is a dominant disorder). Bands with ∼120, 500 and 600 CTG·CAG triplet repeats are amplified from the expanded alleles and show triplet-repeat somatic instability in DM1 fibroblasts. In total, 41 DM1 GM03991-derived iPSC clones were isolated after reprogramming. PCR from all 41 iPSC clones showed the presence of the normal allele, as did the fibroblasts, and of expanded alleles of different repeat lengths (Fig. 2). The majority of iPSC clones (26 clones, 63.4%) had ∼120 CTG·CAG triplet repeats. Seven iPSC clones (17.1%) had a much longer triplet repeat (∼1300), which was not detected in the fibroblasts, possibly because of poor PCR efficiency in amplifying repeats that represent only a small percentage of the population. There were three iPSC clones (7.3%) with ∼600 CTG·CAG triplet repeats and two iPSC clones (4.9%) with ∼500 CTG·CAG triplet repeats, repeat numbers that were also seen in fibroblasts. A slight triplet-repeat length difference between the iPSCs and fibroblasts was observed due to triplet-repeat expansion in iPSCs, which will be addressed below. We also obtained one iPSC clone (2.4%) with ∼100 CTG·CAG triplet repeats and another (2.4%) with ∼200 CTG·CAG triplet repeats, representing repeat numbers that were not detected in fibroblasts. In addition, we noted that one iPSC clone (iPSC clone #30, 2.4%) had both ∼120 and ∼1300 CTG·CAG triplet repeats, which could come from two fibroblasts during reprogramming, resulting in a mixed iPSC clone. Thus, our 41 DM1 iPSC clones with different repeat lengths clearly demonstrate somatic instability within the parent fibroblast population.
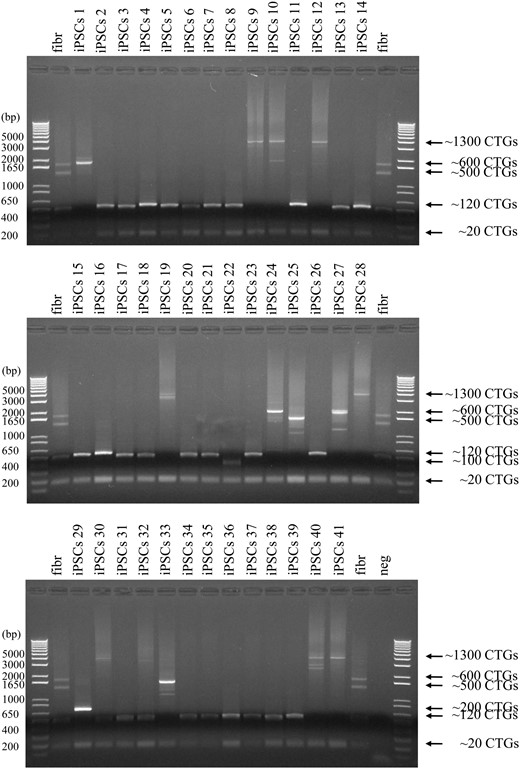
Somatic CTG·CAG triplet-repeat instability in GM03991fibroblasts. PCR amplification of CTG·CAG triplet repeats from GM03991 fibroblasts (fibr) and 41 iPSC clones derived from GM03991 fibroblasts. Arrows indicate the size of the normal allele (∼20 CTG·CAG repeats) and the expanded repeats.
Length-dependent triplet-repeat expansion in DM1 iPSCs
In our previous work, we found that the GAA·TTC triplet repeats expand on propagation of FRDA iPSCs (30). This finding makes iPSCs an ideal system to study the accumulation rate of the GAA·TTC triplet repeats. It was also observed that GAA·TTC repeats at two unrelated genetic loci (2q36, 16 repeats; 4q31.1, 30 repeats) (34) remained unchanged between iPSCs and their three corresponding donor fibroblasts (29) even though these loci were at Alu elements similar to FXN GAA·TTC triplet repeats. It seemed that either triplet-repeat length or repeat loci played an important role in the repeat expansion in iPSCs. Using a human reporter cell model, Ditch et al. (35) found that an uninterrupted GAA·TTC triplet-repeat sequence displayed an overall tendency toward progressive expansion as in FRDA iPSCs and the interval between 88 and 176 triplet repeats was an important length threshold where expansion rates dramatically increased. Our iPSCs with different CTG·CAG repeat lengths are therefore an ideal model to study the relationship between repeat length and expansion rate at the same genomic locus.
The CTG·CAG repeat expansion rate in iPSCs was indeed repeat length dependent. In HD iPSCs, the 46 CAG·CTG triplet repeats were stable for 12 passages (∼12 weeks) (Fig. 3A). In DM1 GM06076 iPSCs, the 57 CTG·CAG triplet repeats were also stable for 16 passages (Fig. 3B). The CTG·CAG triplet repeats in DM1 GM03991 iPSC clone 3 (126 repeats) expanded with an expansion rate of ∼1 repeat per passage (Fig. 3C). CTG·CAG triplet repeats in DM1 GM03991 iPSC clone 1 (773 repeats) expanded more rapidly, with an expansion rate of ∼22 repeats per passage (Fig. 3D). Importantly, CTG·CAG triplet-repeat expansion only occurred on the pathogenic allele, not the normal allele. Using our HD and DM1 iPSC models, we identified the interval between 57 and 126 repeats as being an important length threshold where expansion rates dramatically increased. Moreover, longer repeats showed faster triplet-repeat expansion.
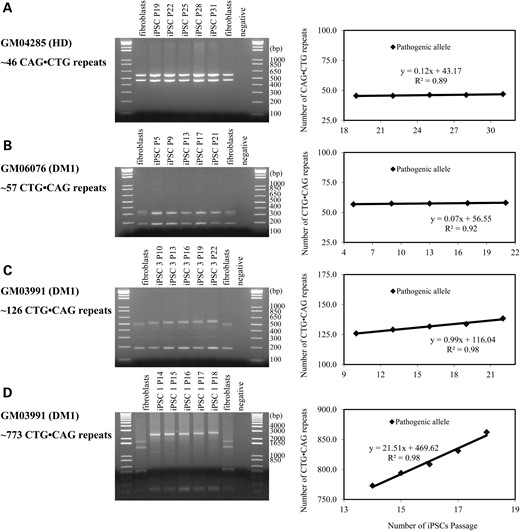
CAG·CTG and CTG·CAG triplet-repeat expansion in iPSCs over time. (A) PCR amplification of CAG·CTG repeats from HD fibroblasts GM04285 and iPSCs at different passages (left panel), and measured expansion rates (right panel); (B) same as in (A) for DM1 fibroblasts GM06076 and corresponding iPSC clone 3; (C) Same as in (A) for DM1 fibroblasts GM03991 and corresponding iPSC clone 3; (D) same as in (A) for DM1 fibroblasts GM03991 and corresponding iPSC clone 1.
Length-dependent recruitment of mismatch repair components on CTG·CAG triplet repeats
Our previous work has shown that the mismatch repair system is involved in GAA·TTC triplet-repeat instability in FRDA iPSCs (29,30). Mismatch repair components MSH2, MSH3 and MSH6 were all up-regulated in FRDA iPSCs compared with the parent fibroblasts. These increased protein levels are not related to the FRDA phenotype, as unaffected GM08333 iPSCs and H1 ES cells showed a similar up-regulation of MSH2, MSH3 and MSH6 proteins, compared with fibroblasts (30). As expected, gene expression analysis of MSH2 and MSH6 showed a significant up-regulation of their corresponding mRNAs in HD and all three DM1 iPSCs compared with fibroblasts, and similar to the expression in H1 ES cells (Fig. 4A–D). However, MSH3 gene expression shows no clear difference in iPSCs and H1 ES cells compared with fibroblasts (Fig. 4A–D). This up-regulation in iPSCs is consistent with results in DM1-derived human embryonic stem cells (hESCs), which show significantly higher expression levels of mismatch repair genes in undifferentiated cells compared with differentiated stages (8).
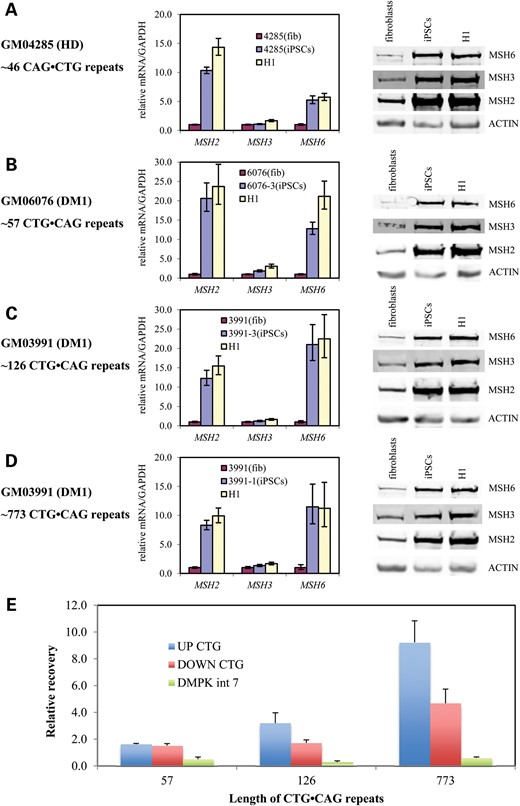
Mismatch repair mRNA and protein are highly expressed in iPSCs, but only occupied the DMPK1 gene harboring longer CTG·CAG triplet repeat. MSH2, MSH3 and MSH6 mRNA and protein levels in (A) HD fibroblasts (GM04285), iPSCs and H1 hESCs, (B) DM1 fibroblasts (GM06076), iPSC clone 3 and H1 hESCs, (C) DM1 fibroblasts (GM03991), iPSC clone 3 and H1 hESCs, (D) DM1 fibroblasts (GM06076), iPSC clone 1 and H1 hESCs. mRNA levels were normalized to GAPDH mRNA level and the amount of actin protein was used as a loading control for western blotting. Error bars indicate SEM of triplicate measurements. (E) Chromatin immunoprecipitation showing MSH2 occupancy on the DMPK1 gene intron 7 (DMPK int 7), upstream (UP CTG) and downstream (DOWN CTG) of the CTG·CAG triplet-repeat expansion. DNA recovery is relative to the recovery of FXN intron 2 DNA.
Since the protein levels of MSH2, MSH3 and MSH6 are dependent not only on the level of their respective mRNA, but also upon MutSα and MutSβ complex stability (36), we monitored the levels of MSH2, MSH3 and MSH6 in HD and DM1 fibroblasts, in HD and DM1 iPSCs and in H1 ES cells. Indeed, expression of MSH2, MSH3 and MSH6 proteins was up-regulated in HD and all three DM1 iPSCs compared with the parent fibroblasts and similar to the expression in H1 ES cells (Fig. 4A–D). The protein levels of each of the MMR components MSH2, MSH3 and MSH6 appear to be similarly up-regulated in the iPSCs and ES cells. Moreover, while the amounts of each of the MMR proteins clearly increase, the ratio of MSH3 to MSH6 (and hence the ratio of MutSβ to MutSα) does not significantly change between fibroblasts and iPSCs.
Our previous ChIP assays (29) showed that, we can detect occupancy of MSH2 downstream of the FXN GAA·TTC triplet repeats in FRDA iPSCs and not in an unaffected iPSC line. Since DM1 iPSCs have a similar mismatch repair component up-regulation profile, we next explored the occupancy of MSH2 upstream and downstream of CTG·CAG triplet repeats in DM1 iPSCs with different repeat lengths. For these ChIP experiments, after cross-linking, genomic DNA was sonicated to an average length of ∼3 kb. In DM1 GM06076 iPSCs with 57 CTG·CAG triplet repeats, there was no clear binding of MSH2 either upstream or downstream of CTG·CAG triplet repeats (Fig. 4E). In contrast, we observe occupancy of MSH2 upstream of 126 CTG·CAG triplet repeats in DM1 GM03991 iPSC clone 3, and both upstream and downstream of the ∼773 CTG·CAG repeats in DM1 GM03991 iPSC clone 1 (Fig. 4E). In addition, the ChIP assay did not show binding of MSH2 in DMPK intron 7 (DMPK int 7), which is ∼5 kb away from the repeats (Fig. 4E). The increased occupancy of MSH2 on longer CTG·CAG repeats both upstream and downstream correlates with the observed repeat expansion rate at the longer alleles.
The mismatch repair component MSH2 is involved in CTG·CAG triplet-repeat expansion
Several studies have pointed to the involvement of the mismatch repair pathway in triplet-repeat instability (16,30,37,38). In addition, our previous work has shown that the mismatch repair pathway is involved in GAA·TTC triplet-repeat expansion in FRDA iPSCs. However, in vitro and in vivo data also show that CTG·CAG triplet-repeat instability involves oxidative DNA damage and base excision repair (BER) (25,39,40). To gain insight into the mechanism of CTG·CAG triplet-repeat expansion in iPSCs, we down-regulated the key component MSH2 by means of lentiviral shRNA knockdown in DM1 iPSCs. shRNA-mediated knockdown of MSH2 had no effect on MSH3 and MSH6 mRNA levels (Fig. 5A), while the level of all three proteins clearly decreased (Fig. 5B and C). This is not surprising as it is known that the protein levels of MSH2, MSH3 and MSH6 are dependent upon MutSα and MutSβ complex stability (36). As CTG·CAG triplet-repeat expansion occurs only on the pathogenic allele, we measured changes in repeat length on the expanded allele after MSH2 gene knockdown (Fig. 5D–G). iPSC genomic DNA was collected four to eight passages after MSH2 knockdown. PCR analysis showed that the expanded alleles accumulated ∼22 CTG·CAG triplet repeats at every passage in scrambled control iPSCs (Fig. 5D and E), but only ∼4.4 CTG·CAG triplet repeats in MSH2 knockdown iPSCs (Fig. 5F and G). We conclude that the down-regulation of MSH2 impedes CTG·CAG triplet-repeat expansion in DM1 iPSCs and further implicates the mismatch repair enzymes in CTG·CAG triplet-repeat instability.
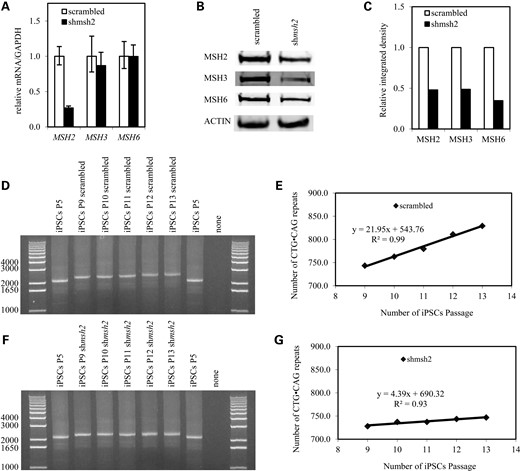
CTG·CAG triplet-repeat expansion involves the mismatch repair component MSH2. (A) MSH2, MSH3 and MSH6 mRNA levels in DM1 iPSCs (GM03991 iPSC clone 24) expressing MSH2-targeting shRNA. mRNA levels are normalized to GAPDH. Error bars indicate SEM of triplet measurements. (B) Western blot of MSH2, MSH3 and MSH6 protein in DM1 iPSCs expressing MSH2-targeting shRNA; (C) densitometric analysis of (B); data are normalized to actin signal; (D) PCR of CTG·CAG triplet repeats in iPSCs before transduction (P5), four passages (P9) to eight passages (P13) after transduction with scrambled shRNA (scrambled); (E) CTG·CAG triplet-repeat expansion rate (over passage) in scrambled iPSCs. The rate of CTG·CAG triplet-repeat expansion in the pathogenic allele was analyzed. (F) PCR of CTG·CAG triplet repeats in iPSCs before silencing (P5), four passages (P9) to eight passages (P13) after transduction with shRNA against MSH2 (shMSH2); (G) CTG·CAG triplet-repeat expansion rate (over passage) in shMSH2 iPSCs. The rate of CTG·CAG triplet-repeat expansion in the pathogenic allele was analyzed.
CTG·CAG triplet-repeat expansion occurs in DM1 iPSCs, but not in differentiated cells
We and others have shown that triplet-repeat expansion in iPSCs or hES cells ceases upon differentiation (8,30,41). Herein, we monitored the CTG·CAG triplet-repeat expansion in DM1 EB, D-EB and NS. EB were generated as previously reported and cultured in ESC medium without basic FGF (42). Genomic DNA was collected after 2, 4 and 6 weeks of EB formation. PCR analysis showed that the CTG·CAG triplet-repeat expansion rate in EB was very similar to that in iPSCs (Fig. 6A1, A2, B1, B2). EB, consisting of ectodermal, mesodermal and endodermal tissues, recapitulate many aspects of cell differentiation during early mammalian embryogenesis and differentiate into derivatives of all three germ layers (43). Further differentiation of EB to the three germ layers was achieved by changing the EB media to differentiated EB media. The overall tendency of CTG·CAG triplet repeats was to cease expansion and become stable within 6 weeks of becoming further differentiated, although a small degree of expansion in the first 2 weeks of differentiation was observed (Fig. 6).
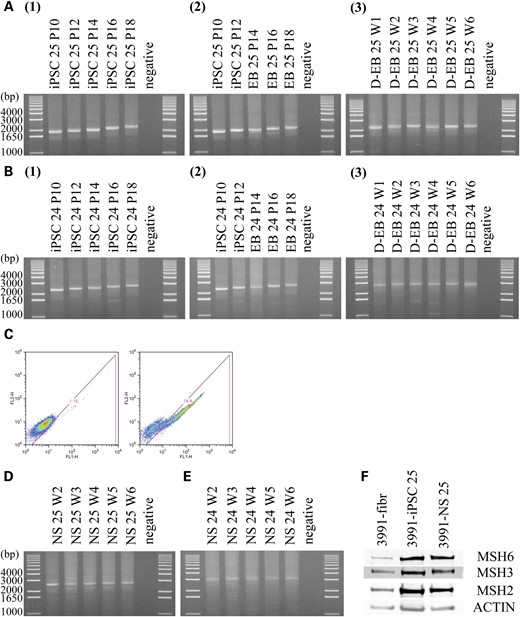
CTG·CAG triplet-repeat expansion occurs in DM1 iPSCs, but not in differentiated cells. (A) Gel analysis of PCR products obtained with genomic DNA extracted from DM1 iPSC clone 25 (A1), EB (A2) and differentiated EB (A3) at different passages (P) or weeks (W). (B) Same as in (A) for iPSC clone 24. (C) Flow cytometry analysis of NS incubated with anti-nestin antibody (right panel) or negative control (left panel). (D) Gel analysis of PCR products obtained with genomic DNA extracted from DM1 neurosphere (NS) clone 25 at different weeks (W). (E) Same as in (D) for DM1 neurosphere (NS) clone 24. (F) MSH2, MSH3 and MSH6 protein levels in GM03991 fibroblasts, iPSCs (clone 25) and NS (clone 25) as measured by western blotting. Actin levels were used as a loading control.
In vitro differentiation of DM1 iPSCs to NS was performed as previously described (30,44–47). Flow cytometry showed that the neurosphere population is 75% positive for the neural stem cell marker protein nestin (Fig. 6C). CTG·CAG triplet repeats stop expanding once differentiated into NS, showing stability at week two to six post neurosphere formation (Fig. 6D and E).
We and others have shown that in FRDA iPSCs and DM1 hESCs, the mismatch repair gene expression levels in differentiated stages are significantly lower than undifferentiated cells (8,30). However, a recent report shows that in FRDA iPSCs the levels of MSH2 and MSH6 stay high after differentiation (41). Similarly, we do not detect significantly lower levels of mismatch repair components in DM1 NS compared with iPSCs (Fig. 6F).
DISCUSSION
While CTG·CAG repeat instability has been observed in human DM1 embryonic stem cell lines (8,19), no reports on CTG·CAG instability in DM1 iPSCs have been presented. We therefore sought to investigate whether our findings of repeat expansion in FRDA iPSCs (29,30) would be recapitulated in other triplet-repeat disorders. We previously reported that upon generation of FRDA iPSCs, the GAA·TTC repeats in the FXN gene expand in a manner analogous to intergenerational repeat expansion observed in families affected with this disease (29,30). GAA·TTC repeats in the FXN gene continue to expand on propagation of the iPSCs, and such expansion is dependent on the levels of the mismatch repair enzymes. Our findings are in contrast to other reports on the derivation of FRDA iPSCs, where both expansions and contractions of the repeats have been observed in FRDA iPSCs, relative to the starting fibroblasts (41,48). This difference could be due to either differences in the lengths of the repeats in the parent fibroblasts or to different methods employed for iPSC generation and analysis of the resulting clones (29,30,41,48).
DM1 is an inherited dominant muscular dystrophy caused by the expanded CTG·CAG triplet repeats in the 3′-UTR of the DMPK1 gene. Transcripts from the mutant allele contain an expanded CUG repeat and are retained in nuclear foci along with splicing factors of the muscleblind-like (MNBL) protein family (49,50). Muscleblind sequestration leads to misregulated alternative splicing and other changes in the muscle transcriptome (50). In addition, at least 17 neurological diseases have similar CTG·CAG triplet-repeat expansions. In most of these disorders, the disease severity is related to the length of the repeat expansion. At present, no therapeutic approach exists to prevent or slow the triplet-repeat expansion and thereby reduce disease severity or delay disease onset (51). CTG·CAG triplet-repeat expansion has been studied extensively in bacteria (10,11), yeast (12–14), mouse (15–17), mammalian cells (8,18–22), human cell extracts (23–25) and patient tissue (6,26–28), while intrinsic expansion tendency of triplet repeats still remain enigmatic (3). In this study, we first generated iPSCs from DM1 patient fibroblasts and detected CTG·CAG triplet repeats in each iPSC clone with conventional PCR. Homogeneous lengths of CTG·CAG triplet repeats in each iPSC clone allow for a convenient system to study the mechanism(s) of repeat expansion.
CTG·CAG triplet repeats exhibit somatic instability in DM1 fibroblasts. Somatic instability of CTG·CAG triplet repeats in patient tissues has been reported based on SP-PCR (26,27). Individual repeat length was detected in SP-PCR with a few genomic DNA molecules by DNA sequence dilution. The PCR signal is then monitored by southern blot analysis. This method is time consuming with a relatively low quality result. Fibroblast reprogramming makes it possible to measure the triplet-repeat length in individual fibroblasts, as each iPSC clone comes from one single fibroblast. This is also one reasonable explanation for the observation of triplet-repeat expansions and contractions during iPSC derivation reported by other groups (48,52,53). For example, on generation of fragile X syndrome iPSCs, CGG·CCG repeat contractions and expansions were observed (52,53). Based on our findings, we surmise that the triplet-repeat expansions and contractions observed by others during iPSC derivation represent somatic instability in the parent fibroblasts (52,53).
Triplet-repeat expansion is length dependent. Accumulating data from bacteria (10,11), yeast (12–14), mouse (15–17) and mammalian cell (8,18–22) models reveal that somatic gene mosaicism and triplet-repeat expansion occur as the result of multiple small changes. Ditch et al. (35) have reported that the interval between 88 and 176 GAA·TTC triplet repeats is an important length threshold where expansion rates dramatically increase in their mammalian cell model. Here, we found that the interval between 57 and 126 CTG·CAG triplet repeats as being an important length threshold where expansion rates dramatically increased in DM1 iPSCs. Xia et al. (54) have reported that the 44 CAG·CTG triplet repeats in the ATXN2 gene, which is responsible for spinocerebellar ataxia type 2 (SCA2), are stable throughout iPSC reprogramming and neural differentiation. One explanation for this result is that 44 CAG·CTG triplet repeats in SCA2 is below the length threshold for expansion. Similarly, the CAG·CTG triplet repeats in HD iPSCs remain stable (this study and (31)). Although cells harboring these short repeats manifest clear disease phenotypes, iPSCs do not recapitulate the repeat instability observed in HD patients and in mouse models (40). Understanding the threshold of triplet-repeat expansion in the human genome will help design better cell and mouse models to study the disease phenotype.
Generation of expanded CTG·CAG triplet repeats in DM1 iPSCs involve the mismatch repair pathway. Although it has been proposed that formation of non-B DNA structures by triplet repeats is required for expansion (55,56), the molecular mechanisms of expansion remain poorly understood. Previous studies using bacteria (10,11), yeast (12–14), mouse (15–17) and mammalian cell (8,18–22) models indicated that triplet-repeat expansion occurs not only during DNA replication (57,58) but also during DNA repair (10,57,59–61), transcription (62,63) or recombination (64,65). Recent studies suggested that CTG·CAG triplet-repeat expansion occurs during the repair of DNA lesions produced by oxidative stress (39). In particular, the oxidized guanine base 8-oxoguanine within sequences containing CAG·CTG repeats may induce formation of pro-expansion intermediates through strand slippage during DNA BER. BER may be directly involved in somatic triplet-repeat expansion, because knockout of the Ogg1 gene abolished age-dependent neuronal CAG·CTG triplet-repeat expansion in a HD mouse model (40). However, in mice, the mismatch repair protein complex MSH2–MSH3 is essential for CAG·CTG triplet-repeat expansion in both germ cells (61) and somatic cells (60). Herein, we found that the mismatch repair component MSH2 binds expanded CTG·CAG triplet repeats at the DMPK1 locus in DM1 iPSCs. Knockdown of MSH2 in these cells clearly decreased the CTG·CAG triplet-repeat expansion rate. We previously reported that knockdown of MSH6 blocks expansion of the GAA·TTC repeats in the frataxin gene in Friedreich's ataxia iPSCs (30) and we would suspect that such knockdown in DM1 iPSCs would have a similar effect. We have been unable to effectively knockdown MSH3 in iPSCs; however, another group has shown the participation of MutSβ in repeat expansion in a GAA·TTC repeat reporter cell line (66).
Our data support the involvement of the mismatch repair pathway in CTG·CAG triplet-repeat expansion in DM1 iPSCs. Unlike FRDA iPSC, where differentiation to NS resulted in down-regulation of mismatch repair components (MSH2, MSH3 and MSH6), neurosphere differentiation from DM1 iPSCs did not show a similar down-regulation of these proteins (Fig. 6F) (8,30,41). The basis for this difference is unclear. It is also still not clear how the high fidelity DNA mismatch repair pathway participates in triplet-repeat expansion. Nonetheless, the finding that high levels of mismatch repair proteins in iPSCs lead to repeat expansion provides a useful resource to generate differentiated cells with varying numbers of repeats. Such cells will be of use in elucidating the pathophysiology of trinucleotide expansion diseases.
Our results are in agreement with a model wherein expanded CTG·CAG triplet-repeat alleles adopt non-B DNA structures, which in turn promote further expansion by recruitment of the mismatch repair enzymes. However, the exact mechanisms involved in MMR recruitment remain to be identified. Our observations open a new avenue for investigating and designing new compounds, which have the ability to target CTG·CAG triplet repeats and interfere recruitment of MMR, to block triplet-repeat instability. It has been shown that the severity of disease symptoms, age of onset and progression are related to the size of CTG·CAG triplet repeats (9). Preventing triplet-repeat expansion raises the hope that the severity of pathophysiology might be reduced or its onset delayed, thereby widening the therapeutic window for this deadly disease (3).
MATERIALS AND METHODS
Cell culture
Fibroblasts (GM03991, GM06076, GM04285) were obtained from Coriell Institute (Camden, NJ, USA). Fibroblasts, ESCs/iPSCs, NS, HEK293T, Phoenix and Plat-A cells were cultured as previously described (30,67). EB were cultured in ESC medium without basic FGF (EB media). D-EB were grown with 10% FBS in DMEM, 2 mm glutamine, 20 mm HEPES, 1% nonessential amino acids and 1% antibiotic/antimycotic (differentiated EB media).
Derivation of iPSCs
DM1 and HD iPSCs derivation followed previous methods with minor deviations (68). Retroviruses were packaged via either Phoenix cells (a gift from the laboratory of W. Balch, The Scripps Research Institute) with Fugene 6 (Roche) or Plat-A cells (a gift from the laboratory of J. Loring, The Scripps Research Institute) with Lipofectamine™ 2000 (Invitrogen). The four reprogramming vectors (Addgene) were packaged individually and pseudotyped with VSV-G (69). DM1 and HD iPSCs were characterized by standard methods (29).
Derivation and differentiation of EB
To form EB, iPSC colonies were cultured in ESC medium without basic FGF overnight when iPSC colonies were ready to passage. Then iPSC colonies were cut out and transferred to ESC medium without basic FGF in 60 mm bacterial grade petri dish (low attachment) to form EB for 8–14 days (42). EB were attached to 0.8% geletin coated 24-well plate and cultured in differentiated EB media.
Derivation of NS and flow cytometry
In vitro differentiation of DM1 iPSCs to NS and flow cytometry was performed as previously described (30,44–47). Induction was performed with LDN-193198 (0.5 μm, Stemgent) and SB431542 (10 μm, Stemgent) in ESC media.
Nucleic acid purification
Total RNA was purified with the RNeasy Plus Mini kit (QIAGEN Inc.). Genomic DNA was purified by isopropanol precipitation (30,70).
Conventional PCR and quantitative RT-PCR
For shorter CTG·CAG triplet-repeat length (less than 130 repeats) conventional PCRs, Taq DNA polymerase (18038-042, Invitrogen) was used according to the manufacturer. Forty nanogram genomic DNA and 0.2 μm primers DM-C and DM-BR were used (26) in 20 μl reactions cycled through the following conditions: 94°C denaturation for 30 s, 60°C annealing for 30 s, 68°C extension for 2 min for 30 cycles with a 5 min initial denaturation and a 10 min final extension. PCRx enhancer solution (from Invitrogen) gave a final concentration of 0.4×.
For longer CTG·CAG triplet-repeat length (more than 130 repeats), Platium Pfx DNA polymerase (11708-013, Invitrogen) was used for conventional PCR, according to the manufacturer. Forty nanogram genomic DNA and 0.3 μm primers DM-C and DM-BR were used (26) in 20 μl reactions cycled through the following conditions: 94°C denaturation for 20 s, 60°C annealing for 30 s, 68°C extension for 3 min for 32 cycles with a 5 min initial denaturation and a 10 min final extension. PCRx enhancer solution gave a final concentration of 1.0×.
For CAG·CTG triplet-repeat length conventional PCRs, Platium Pfx DNA polymerase was used. Forty nanogram genomic DNA and 0.3 μm primers LK-1 and LKH-5 were used (33) in 20 μl reactions cycled through the following conditions: 94°C denaturation for 20 s, 60°C annealing for 30 s, 68°C extension for 1 min for 30 cycles with a 5 min initial denaturation and a 10 min final extension. PCRx enhancer solution gave a final concentration of 1.0×.
Quantification of PCR band size was performed using an inverse power function directly correlating gel migration of a molecular weight ladder to its known sizes (ImageJ software) (71). PCR products with CTG·CAG triplet repeats from the DMPK1 locus contain 147 bp of non-repeat sequences, and PCR products with CAG·CTG triplet repeats from HTT locus contain 421 bp of non-repeat sequences, so triplet-repeat number estimations were adjusted accordingly.
Quantitative RT-PCR analysis was done with the qScript One-Step SYBRGreen qRT-PCR kit (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer's instructions. Primers for pluripotent markers were as described (68). MSH2, MSH3, MSH6 and GAPDH primers have been described (29,30).
Western analysis
Western analysis for MSH2, MSH3, MSH6 and actin in cell lysates was carried out as previously described (30).
Lentiviral shRNA transduction
Lentiviral shRNA for targeting MSH2 has been described (29,30). Lentiviral particles were packaged by co-transfecting HEK293T cells using Lipofectamine™ 2000 transfection reagent (Invitrogen) with shRNA constructs along with psPAX2 (#12260) and pMD2.G (#12259) helper plasmids (Addgene).
Chromatin immunoprecipitation
Cells were cross-linked first with 1.5 mm dithiobis-succinimidyl propionate followed by 1% formaldehyde. Subsequent ChIP procedures were as described (29,72) with MSH2 antibody (SC-494, Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA). The average size of DNA fragments obtained after sonication of chromatin was ∼3 kb. Analysis by qPCR was carried with primers for the DMPK1 intron 7, the region upstream of the CTG·CAG triplet repeats and the region downstream of the repeats as follows: DMPKint7_F, TAGCCAGGCGTGGTAGAGG, DMPKint7_R, CGGAGTCTCGCTCTGTTACC; UPCTG_F2, CAACTCACCGCAGTCTGG, UPCTG_R2, GGCACTCAGTCTTCCAACG; DOWNCTG_F, AAGCTTTCTTGTGCATGACG, DOWNCTG_R, AGAGCAGCGCAAGTGAGG.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by grants from the California Institute for Regenerative Medicine (CIRMRB3-05022 to J.M.G.); NIH. (5U01-NS063953-03 to J.M.G.) and a TSRI Stem Cell Postdoctoral Fellowship (to J.D.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}