




Abstract
Retinoid dehydrogenases/reductases catalyze key oxidation–reduction reactions in the visual cycle that converts vitamin A to 11- cis retinal, the chromophore of the rod and cone photoreceptors. It has recently been shown that mutations in RDH12 , encoding a retinol dehydrogenase, result in severe and early-onset autosomal recessive retinal dystrophy (arRD). In a cohort of 1011 individuals diagnosed with arRD, we have now identified 20 different disease-associated RDH12 mutations, of which 16 are novel, in a total of 22 individuals (2.2%). Haplotype analysis suggested a founder mutation for each of the three common mutations: p.L99I, p.T155I and c.806_810delCCCTG. Patients typically presented with early disease that affected the function of both rods and cones and progressed to legal blindness in early adulthood. Eleven of the missense variants identified in our study exhibited profound loss of catalytic activity when expressed in transiently transfected COS-7 cells and assayed for ability to convert all- trans retinal to all- trans retinol. Loss-of-function appeared to result from decreased protein stability, as expression levels were significantly reduced. For the p.T49M variant, differing activity profiles were associated with each of the alleles of the common p.R161Q RDH12 polymorphism, suggesting that genetic background may act as a modifier of mutation effect. A locus ( LCA3 ) for Leber congenital amaurosis, a severe, early-onset form of arRD, maps close to RDH12 on chromosome 14q24. Haplotype analysis in the family in which LCA3 was mapped excluded RDH12 as the LCA3 gene and thus suggests the presence of a novel arRD gene in this region.
INTRODUCTION
The visual cycle consists of a series of enzymatic reactions and transport mechanisms involved in the metabolism of vitamin A needed to sustain the light response of the vertebrate retina (Fig. 1 ). In the dark, the vitamin A analog 11- cis retinal forms a covalent bond with rod and cone opsins to generate the photoreceptor visual pigments (reviewed in 1). Upon illumination, 11- cis retinal is isomerized to all- trans retinal needed to stabilize the active signaling conformation of these proteins. Following decay of the active state, all- trans retinal is released from the apoprotein, reduced to all- trans retinol (vitamin A), transported to the retinal pigment epithelium (RPE) and esterified to phospholipids. The resulting retinyl esters serve as substrates for a concerted isomerization/hydrolysis reaction that regenerates 11- cis retinol, which is subsequently oxidized to reform 11- cis retinal and returned to the photoreceptor cells.
Retinoid dehydrogenases/reductases (or oxidoreductases) located in the photoreceptor cells and the RPE catalyze key oxidation–reduction reactions in the visual cycle. RDH12 (14q23.3–q24.1) (MIM no. 608830) encodes a dual specificity enzyme expressed in the retina that acts on both trans and cis retinoid substrates ( 2 , 3 ). Mutations in human RDH12 were recently shown to be associated with certain forms of severe, childhood-onset autosomal recessive retinal dystrophy (arRD) ( 4 ), establishing a unique requirement for this isoform in visual cycle function. In an initial report of disease-associated RDH12 mutations, we detected a homozygous p.Y226C change in 13 patients of a large consanguineous Austrian family that resulted in loss-of-function in in vitro assays, and four other most likely pathogenic changes (p.T49M, p.R62X, p.Q189X and c.806_810delCCCTG) ( 4 ). Additional disease-associated mutations were reported, including six missense mutations whose functional significance has not yet been established ( 5 ).
Here, we report the identification of 20 novel RDH12 coding sequence variants, and the results of functional analysis of a total of 14 missense mutations with respect to catalytic activity and protein expression. Our studies define the range of RDH12 mutations in patients with childhood-onset severe arRD and suggest that RDH12 loss-of-function disrupts the cycle of synthesis of the visual pigment chromophore, 11- cis retinal, resulting in early-onset and progressive disease. The central role of RDH12 in the visual cycle establishes it as an important retinal dystrophy gene that may be an important future therapeutic target.
RESULTS
RDH12 mutations in individuals with arRD
We analyzed samples from 1011 individuals with retinal dystrophy originating from Germany, Spain, Austria, The Netherlands, Canada and the United States and identified 20 different disease-associated RDH12 mutations, 16 of which represent novel mutations, present in homozygous or compound heterozygous form in a total of 22 probands (Table 1 ). The data from our cohort suggest that RDH12 mutations account for 2.2% of cases of arRD. The majority (12/20, 60%) of sequence changes were missense mutations. Other mutations predicted premature protein termination, including nonsense mutations and rearrangements affecting a few nucleotides. Loss of the methionine initiation codon, c.2T>C (p.M1?) was also observed. Mutations for which a premature termination of translation was predicted or which resulted in loss of enzyme activity in in vitro functional assays (discussed subsequently) were considered most likely pathogenic without further genetic studies. Haplotypes generated by three intragenic SNPs for chromosomes with the three most prevalent RDH12 mutations, p.L99I, p.T155I and c.806_810delCCCTG (discussed subsequently) (Materials and Methods), were consistent with a founder mutation in each case.
In five patients, only one heterozygous RDH12 sequence change was identified, predicting a single amino acid change in four cases (Table 2 ). Variant c.194G>A (p.R65Q) did not segregate with the disease phenotype in Family 16 (data not shown). The mutation c.806_810delCCCTG (p.Ala269GlyfsX1) detected in proband 98 is a most likely functional null allele, but was not likely to be related to the disease in Family 17 as the younger affected sister does not carry this particular change (data not shown). Direct sequencing of all seven coding RDH12 exons did not reveal a second disease-causing change in the three patients carrying c.577C>T (p.R193C), c.617C>T (p.A206V) or c.689C>T (p.P230L) in heterozygous form. Although these variants were not detected in 100 unaffected individuals, and segregated with the disease phenotype in the respective (small) families (data not shown), their role in the pathogenesis of the patients' disease remains to be determined by future studies including functional assays that were not part of this study. Variant c.301G>A (p.D101N; Table 2 ) was detected in heterozygous state in a patient who was not part of the original cohort. The proband was from a three-generation family in which a most likely autosomal dominant form of RD is segregating. However, the mutation was transmitted to the patient by his unaffected mother, making it unlikely to be pathogenic. A number of rare sequence variants were also identified, both in patients and unaffected individuals, which were not likely to be pathogenic, including c.-123C>T (5′-UTR), c.188-31A>T (intron 2), c.188-14insT (intron 2), c.188-14insTTT (intron 2), c.333C>A (p.G111), c.343+101C>T (intron 3), as well as the previously reported variants c.-152A>G (5′-UTR), c.187+54A>T (intron 2), c.187+60G>A (intron 2) and c.195A>C (p.R65) ( 4 ). A previously identified commonly occurring polymorphism, c.482G>A (p.R161Q), was also detected in the present study, for which the minor allele c.482A (p.Q161) is present at a frequency of 0.2 in the general population ( 4 ).
A summary of all RDH12 disease-associated mutations identified to date is shown on a schematic of the gene structure in Figure 2 .
In vitro functional analysis of RDH12 missense mutations
To evaluate the effect of RDH12 missense variants on RDH12 function, COS-7 cells were transiently transfected with pcDNA3.1/HIS constructs that express RDH12 as a fusion protein with Xpress epitope. Missense variants identified in individuals with arRD were incorporated on the frequent G allele of the c.482G>A polymorphism (p.R161 variant). When RDH12 activity was assayed in cell lysates using normal phase high performance liquid chromatography (HPLC) under conditions in which retinoid reductase activity was time- and concentration-dependent, 11 missense variants (p.A47T, p.T55M, p.L99I, p.N125K, p.G145E, p.H151D, p.T155I, p.A206D, p.R239W, p.L274P and p.C285Y) showed dramatically reduced ability to convert all- trans retinal to all- trans retinol in the presence of NADPH ( P <0.001), exhibiting 5–18% wild-type levels of activity. (Averaged values from two independent clones assayed in triplicate are shown in Fig. 3 , top panel.) Similar decreases in activity were also seen in reactions with 11- cis retinal as substrate, and also with all- trans retinol and 11- cis retinol in the presence of NADP+, although overall reactivity of the retinoid dehydrogenase reaction was much less than that of the retinoid reductase reaction for both wild-type and missense variants (activity: all- trans retinal>11- cis retinal>11- cis retinol>all- trans retinol) (data not shown).
Analysis of recombinant protein expression on western blots probed with anti-Xpress antibody showed that each of this group of 11 missense variants had significantly decreased expression relative to wild-type. The wild-type protein was seen as immunoreactive bands with apparent molecular weights of 35, 41 and 43 kDa that were not present in cells transfected with empty vector (Fig. 3 , middle panel). For each mutant, decreased expression correlated with loss of the unglycosylated form of the protein (∼35 kDa immunoreactive band) ( 4 ), with equivalent protein loading confirmed by analysis of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) immunoreactivity (Fig. 3 , bottom panel). Immunohistochemical analysis of transfected COS-7 cells was consistent with localization of the 11 missense variants to the same intracellular membrane compartment(s) as the wild-type protein (Fig. 4 ). However, most likely because of higher expression levels, the wild-type protein appeared at higher intensity throughout the cells, often with high intensity focal build-up typical of overexpressed membrane proteins (data not shown).
Different results were obtained for two other missense variants identified in individuals diagnosed with arRD: p.R193C present in an individual in heterozygous form and p.R234H present in another individual in compound heterozygous form with p.N125K. In assays of recombinant protein activity in the presence of all- trans retinal and NADPH, decreased catalytic activity was observed ( P <0.001), however, p.C193 and p.H234 retained 79 and 44% the activity level of wild-type, respectively (Fig. 3 , top panel). This compares with the activity of the p.Q161 polymorphic allele that exhibited 69% of the activity of the p.R161 variant (our ‘wild-type’ cDNA construct). The same trends were seen in assays with 11- cis retinal, all- trans retinol and 11- cis retinol as substrates, although overall reactivity was less as described earlier (data not shown). Expression levels of the p.C193, p.H234 and p.Q161 variants were somewhat variable, appearing similar or less than that of wild-type by western and immunohistochemical analysis (Fig. 3 , middle panel, and Fig. 4 ). Thus, our findings are consistent with the interpretation that the p.R193C and p.R234H sequence changes may not be disease causing. However, in the case of the patient having p.R234H and p.N125K variants in compound heterozygous form, our findings are also consistent with a threshold effect resulting from p.N125K loss-of-function, combined with reduction in p.R234H activity to less than half that of wild-type, with the likely net effect being insufficient RDH12 enzyme levels for normal visual cycle function.
Functional analysis of the p.T49M variant and the effect of genetic background
In previous studies, we identified a patient compound heterozygous for p.T49M and p.R62X. The missense variant p.M49 exhibited elevated catalytic activity in vitro when expressed from a cDNA construct encoding the p.R161 variant ( 4 ). Co-transfection of the p.R62X variant had no effect on the activity of either p.T49 or p.M49 variants (data not shown). By segregation analysis, we found that the p.T49M change in the patient was present on the p.Q161 encoding maternal allele and the p.R62X change was present on the p.R161 encoding paternal allele, thus representing complex alleles. In order to perform a more detailed analysis of the altered enzymatic activity of the p.M49 variant, and possible effect of the genetic background, we generated expression constructs encoding all four possible combinations of p.T49, p.M49, p.R161 and p.Q161 variants. Western and immunohistochemical analyses of transfected COS-7 cells showed that all four proteins were expressed, with the levels of the p.T49/p.R161 recombinant protein (corresponding to the most common allele) consistently somewhat higher than that of the p.M49/p.R161, p.T49/p.Q161 and p.M49/p.Q161 proteins (Fig. 5 C).
In assays of RDH12 retinoid reductase activity with all- trans retinal or 11- cis retinal as substrate, the change from p.T49 to p.M49 on the p.R161 background resulted in a significant stimulation of activity ( P <0.05) that was ∼1.5–2-fold increased (Fig. 5 A and B). The change from p.T49 to p.M49 produced a similar fold stimulation on the p.Q161 background. However, the p.T49/p.Q161 protein corresponding to the less frequent complex allele exhibited reduced retinoid reductase activity relative to the p.T49/p.R161 protein ( P <0.05). Thus, the p.T49 to p.M49 on p.Q161 change did not produce a final level of reductase activity significantly greater than that of the p.T49/p.R161 protein ( P <0.05).
In contrast, the change from p.T49 to p.M49 on the p.R161 background resulted in significant reduction of RDH12 retinoid dehydrogenase activity ( P <0.05) that was ∼0.5-fold decrease in assays with all- trans retinol or 11- cis retinol as substrate (Fig. 5 A and B). The change from p.T49 to p.M49 also resulted in a similar fold reduction in activity on the p.Q161 background. However, as the dehydrogenase activity of the p.T49/p.Q161 protein was less than that of the p.T49/p.R161 protein ( P <0.05), and only ∼10% that of the reductase activity, this situation resulted in very low levels of p.M49/p.Q161 dehydrogenase activity.
Our findings suggest that the p.T49M change shifts the catalytic efficiency of RDH12 to favor the conversion of retinal to retinol, with the final activity level influenced by the p.R161Q polymorphism. The significance of this shift will require establishing the direction and cellular location of the RDH12 reaction in vivo .
RDH12 mutations on a structural model
Although RDH12 is a member of a relatively large protein family, no structural data useful for evaluating RDH12 patient mutations in the context of the protein tertiary structure are currently available. Previously, a model of RDH5 tertiary structure was generated based on its homology (∼24%) with 17β-hydroxysteroid dehydrogenase ( 6 ). In the case of RDH12, however, significantly lower homology (∼12%) limits the usefulness of this strategy. Therefore, to generate a model of RDH12 tertiary structure, a low-resolution solution was predicted from the protein sequence using ab initio methods ( 7 – 9 ). The approach used here is capable of producing roughly correct models with complex topologies ( 10 ), although accuracy diminishes for large structures unassociated with a protein family ( 11 , 12 ).
The resulting structure with predicted coordinates was displayed and annotated to highlight the locations of the amino acid substitutions affected by RDH12 disease-associated missense mutations (Fig. 6 ). The protein has an apparently globular form with a core comprised alpha helical and beta sheet secondary structures. Sites of missense mutations are distributed widely across the predicted protein surface, although in most cases, they do not directly affect secondary structure elements that may participate in, for example, membrane association. The model provides no evidence that p.T49 and p.R161 are in close proximity, suggesting that activity effects resulting from substitutions at these sites are not due to simple steric interactions.
RDH12-associated phenotype
Affected study subjects found to have RDH12 mutations showed a progressive, severe and early-onset retinal dystrophy with visual impairment by the age of 2 years, in many cases with nystagmus, indicative of poor vision before the age of 6 months. In late-stage disease, visual acuity deteriorated to hand movement or light perception. Fundoscopic findings were waxy optic discs, severe attenuation of the arterioles, hyperpigmentation with bone spicule-like pigmentations in the mid-periphery and diffuse RPE/choroid atrophy. Visual fields were concentric restricted, often with eccentric small islands and multiple scotomas. Electroretinogram responses, both photopic and scotopic, were dramatically reduced at the time of first investigation in young patients (5–7 years old), with scotopic responses affected less than photopic ones. Electroretinogram responses were usually absent in older patients.
As mentioned earlier, the p.H234 allele retained 44% activity level of the wild-type enzyme. The proband carrying the p.R234H change (with the mutation p.N125K on the other allele) is 21-year-old and although his ophthalmologic parameters might indicate a somewhat milder overall disease course at this point, a conclusion in this regard would be premature.
LCA3 is not due to an RDH12 mutation
The LCA3 locus corresponds to a form of Leber congenital amaurosis, a severe autosomal recessive retinal degeneration, which maps to chromosome 14q24 within the 10 cM interval bounded by D14S284 and D14S68 ( 13 ). Thus, RDH12 is a candidate gene for LCA3 . To test this hypothesis, several individuals from different branches of the large consanguineous family in which this locus was defined were studied for sequence changes in the RDH12 exons and adjacent intronic sequences. No disease-associated variants were identified. To test for larger deletions or rearrangements, PCR amplification primers were designed within each of the exons to span the intervening introns. All of these yielded the expected size products both in affected, heterozygote, and unaffected family members with the exception of the largest intron (intron 6; 4.4 kb), which could not be amplified from any family member. In addition, no disease-associated mutations were found in RDH11 , which is also present in the critical genetic interval ∼26 kb away from RDH12 . Lastly, the genomic region containing these two genes was tested for co-segregation with the disease phenotype in the family by use of three SNPs within the introns of RDH12 and three microsatellite markers of which two flank the pair of genes and one is between them. This analysis showed lack of sharing of haplotypes both between the two affected siblings in generation VI and between the two parents who are first cousins (Fig. 7 ). On the basis of these results, it seems likely that a mutation in a novel retinal dystrophy disease gene is responsible for the phenotype of the family with the LCA3 locus.
DISCUSSION
Previous studies have shown that a number of different RDHs are present in the retina and RPE, including retSDR1, RDH8 (also named prRDH), RDH10, RDH11, RDH12, RDH13, RDH14, RDH15 and RDH16 ( 2 , 14 – 17 ). RDH5 is an RPE enzyme specific for 11- cis retinoid isomers and is involved in the oxidation of 11- cis retinol to 11- cis retinal ( 18 ). Mutations in human RDH5 cause fundus albipunctatus, a form of autosomal recessive, congenital stationary night blindness that usually does not progress to degenerative disease ( 19 ). These data in aggregate led to the general view that retinoid dehydrogenases/reductases serve in a redundant capacity in the visual cycle. However, the finding that RDH12 mutations are associated with childhood-onset severe arRD established that the encoded protein plays a unique role in retinal physiology. Patient mutations are divided between those predicting premature truncation (and likely absence of protein due to nonsense mediated decay) ( 20 ), and those encoding amino acid substitutions, with both classes of disease-associated variants likely resulting in altered visual cycle throughput.
Our studies of RDH12 function suggest that in the majority of cases, patient missense variants have significantly reduced expression and activity, and appear to represent disease-associated mutations that produce loss-of-function alleles due to disruption of the visual cycle. It is not yet clear whether loss-of-function can be attributed solely to decreased protein levels (most likely due to protein instability) or whether there are also effects on catalytic function, protein–protein interactions, etc. In a few instances, including one in which a second RDH12 change was not found, missense variants exhibited significant levels of activity and expression. However, it is possible that some of these variants have negative effects on in vivo function, alone or in combination, which are not adequately measured by our in vitro assays of catalytic activity. Sites of missense mutations appear to zlocalize on the protein surface in areas of unstructured sequence, suggesting that affected residues may disrupt functional domains involved in protein interactions, protein folding and catalytic activity, with decreased stability seen for the recombinant protein expressed in heterologous cells. Elucidating the relative contribution of these effects to loss-of-function awaits further studies of RDH12 specific activities and interactions, coupled with rigorous structural analysis.
Few studies have systematically addressed the functional consequences of complex alleles wherein two or more mutations occur on one chromosome. In such complex alleles, possibilities for functional interactions exist. For example, one constituent mutation may represent a neutral polymorphic variant with no apparent effect on function, each constituent mutation could contribute to the functional effect ( 21 ), or an apparent polymorphism may modify the functional consequences of a pathogenic mutation by virtue of being in cis ( 22 ). Our functional studies that pair the p.R161Q polymorphism with the p.M49 mutation as a complex allele clearly document the latter possibility. These studies show that the p.T49M change alters the catalytic efficiency of RDH12 to favor the conversion of retinal to retinol, with the level of total activity being influenced by the p.R161Q polymorphism. It remains to be determined how often coding SNPs may have functional consequences in the context of complex alleles.
There is growing evidence to support the notion that RDH12 functions as the key enzyme necessary for the reduction of all- trans retinal released from bleached photopigments in the recovery phase of the visual cycle. RDH12 can use either cis or trans retinoid isomers as substrates for dehydrogenase (retinol to retinal) and reductase (retinal to retinol) activity with varying efficiency ( 2 , 3 ). RDH12 transcripts have been localized to the photoreceptor outer nuclear layer in human and mouse retinas using in situ hybridization ( 2 ). In addition, studies of knockout mice lacking prRDH ( Rdh8 ), a different retinoid dehydrogenase expressed specifically in the photoreceptors, show that its loss has little effect on the murine phenotype, leading the authors to suggest that RDH12 is likely to be the key isoform necessary for the recovery phase ( 23 ). An important remaining question thus pertains to the roles played by other RDH isoforms present in the retina, including retSDR1, RDH13, RDH14 and RDH15. Are their activities in fact redundant or are there differences in, for example, the mechanisms of retinoid processing in rods versus cones? In this vein, RDH15 identified in cone-dominant Nrl -knockout mouse eyecups has been suggested to function in a proposed cone visual cycle ( 17 ). Alternatively, RDH12 may interact with other retinoid dehydrogenase/reductase isoforms to modulate function. Recent studies of Rdh5; Rdh11 double knockout mice suggest that such interactions are involved in regulating the activity of these two isoforms expressed in the RPE ( 24 ). Further insight into issues surrounding the function of RDH12 will require establishing whether RDH12 is expressed in rods or cones, or both, and defining the substrates and direction of the in vivo reaction(s). RDH12 mutations associated with arRD may provide important clues in this regard, in the form of differential effects on the oxidizing or reducing activity of the enzyme.
RDH12 is one of several retinal dystrophy genes that disrupt the cycling of vitamin A analogs between the photoreceptor cells and the adjacent RPE (reviewed in 25). These include RPE65 that has been recently shown to encode the retinoid isomerohydrolase ( 26 – 29 ), RLBP1 that encodes the RPE cellular retinal binding protein CRALBP ( 30 , 31 ), RGR that encodes the retinal G protein-coupled receptor that enhances isomerohydrolase activity ( 32 – 34 ), and LRAT that encodes the retinyl ester synthase, lecithin retinol acyl transferase ( 35 , 36 ). In each case, the autosomal recessive mode of inheritance suggests that pathogenicity of disease-associated mutations in these genes results from loss-of-function. Considerable effort is now focused on developing strategies for therapeutic intervention in patients with RPE65 defects, involving both gene and retinoid replacement approaches ( 37 – 39 ). Because the diseases associated with RPE65 and RDH12 mutations share similarities in phenotype, and potentially in molecular pathology (chromophore loss), replacement strategies similar to those under development for RPE65 may be appropriate for RDH12 , especially gene replacement therapy. However, strategies for retinoid replacement therapy in the case of RDH12 may be less straightforward in that it is not yet known whether the associated pathology results from deficits in the production of chromophore, or in the build-up of toxic intermediates (e.g. free aldehydes), or both. In the latter case, addition of exogenous chromophore (11- cis retinal) that would continue to push the visual cycle forward could, in the long term, exacerbate rather than ameliorate associated pathology and functional loss. Therefore, definition of the specific role of RDH12 in the visual cycle will be an essential prerequisite for moving toward therapeutic intervention. The hope is that targeted strategies can be developed, that will benefit the majority of patients with mutations in visual cycle genes.
MATERIALS AND METHODS
Patients and families
One hundred and eleven individuals with a clinical diagnosis of (juvenile) retinitis pigmentosa, Leber congenital amaurosis or unclassified retinal dystrophy were enrolled in this study, including simplex cases and cases with evidence for autosomal recessive inheritance. Informed consent was obtained from all participants, and protocols were approved by the institutional review boards of University of Michigan, Universität Innsbruck, Universitätsklinikum Hamburg-Eppendorf, Universität Tübingen, University of Pennsylvania, Columbia University, Radboud University Nijmegen Medical Center, Baylor College of Medicine, Hospitales Universitarios Virgen del Rocío, La Fundación Jiménez Díaz, Hospital San Pau, and McGill University Health Center. DNA was purified from venous blood or buccal smears. Patients underwent ophthalmoscopy and electroretinography.
Mutation screening and genotyping
The seven exons of RDH12 were amplified using standard PCR conditions and oligonucleotide primers positioned in the intronic regions. Mutation screening was performed using denaturing HPLC on a WAVE DNA Fragment Analysis System (Transgenomic, Crewe, UK) or by single-strand conformation analysis with 8% glycerol. Fragments exhibiting an abnormal retention/electrophoresis pattern were sequenced on an ABI 310 DNA sequencer using BigDye terminator mix (Applied Biosystems). Patient haplotypes were determined by analysis of intragenic SNPs for chromosomes with the three most prevalent RDH12 mutations, p.L99I (c.187/IVS2+54A–c.187/IVS2+60G–c.482A/p.161Q), p.T155I (c.187/IVS2+54A–c.187/IVS2+60A–c.482G/p.161R) and c.806_810delCCCTG (c.187/IVS2+54A–c.187/IVS2+60A–c.482G/p.161R). Primer sequences and screening conditions are available upon request.
In vitro mutagenesis and expression analysis
RDH12 cDNA [c.482G allele (p.R161) including 10 bp 5′- and 32 bp 5′-untranslated sequence] was cloned into pcDNA3.1/HIS which expresses the recombinant protein as a fusion with Xpress epitope (Invitrogen). RDH12 sequence variants identified in individuals with arRD were introduced using the QuikChange Kit (Stratagene) and constructs verified by DNA sequencing. COS-7 cells in six-well plates or on chamber slides were transiently transfected with mutant or wild-type RDH12 constructs, or empty vector, using FuGene6 (1 µg DNA/3 µl reagent) according to the manufacturer's instructions (Roche), and were harvested at 44 h post-transfection. A β-galactosidase encoding expression construct (pCMV-βgal) was included as 5% of total DNA and assayed using o -nitrophenyl- d -galacto-pyranoside as substrate ( A420 nm ) and showed that all constructs were transfected with approximately equal efficiency.
For western analysis of RDH12 fusion-protein expression, transfected cells were harvested in 10 m m Tris–HCl, pH 7.4, 250 m m sucrose, and protein concentrations determined by Lowry assay. Samples in SDS sample buffer were electrophoresed on NuPage ® Novex Bis–Tris acrylamide gels (4–12%) (Invitrogen) alongside SeeBlue ® Plus2 standards (Invitrogen), transferred to nitrocellulose, incubated with Xpress antibody (Invitrogen), and then with alkaline phosphatase-conjugated anti-mouse IgG (Molecular Probes). Equivalent protein loading was verified by Coomassie blue staining and analysis of GAPDH immunoreactivity (Ambion).
For immunocytochemical analysis of protein localization, transfected cells were fixed using 4% paraformaldehyde, 0.1 m sodium phosphate, pH 7.2 for 10 min at room temperature, incubated with the Anti-Xpress antibody (1 : 250) (Invitrogen) for 2 h at room temperature, washed, then incubated with Alexa-Fluor 555 anti-mouse IgG secondary antibody (1 : 500) (Molecular Probes) for 1 h at room temperature. A negative control was done for each transfection, excluding the primary antibody incubation step (data not shown). Slides were viewed under oil immersion (100×) and photographed on a Nikon Eclipse E800 microscope with fluorescence filter cube using a Nikon DMX1200 digital camera with the manufacturer's data acquisition software.
Enzyme assay
In vitro analysis of recombinant RDH12 activity in COS-7 cell extracts was performed as described previously ( 4 ), using at least two independent clones of each mutation, assayed in triplicate. Briefly, cells were harvested, disrupted by sonication, and enzyme activity assayed in reactions containing 6 µg protein, 200 µ m NADPH, 100 µ m all- trans retinal or 11- cis retinal (for reductase activity) or 6 µg protein, 400 µ m NADP + , 200 µ m all- trans retinol or 11- cis retinol (for dehydrogenase activity) according to published methods ( 2 ), with retinoid concentrations calculated using extinction coefficients in Garwin and Saari ( 40 ). Retinoids were extracted into organic solvents and identified and quantitated by normal phase HPLC analysis using a Waters 2695 Alliance Separation Module and 2996 Photodiode Array Detector with a Supelcosil LC-31 column (25×4.6 mm 2 , 3 µm) developed with 5% 1,4-dioxane in hexane. Peak identification was by comparison to retention times of retinoid standards and evaluation of wavelength maxima. Quantitative analysis was by comparison of peak areas at 325 nm for all- trans retinol, 318 nm for 11- cis retinol, 368 nm for all- trans retinal and 362.5 nm for 11- cis retinal. Data were analyzed using ANOVA (α=0.05; Excel software), with significant statistical difference determined as P <0.05. Assay conditions were optimized such that retinoid concentrations were not rate limiting, and product formation by RDH12 wild-type protein was linear during the reaction period (10–20-fold above background in 30 min), as well as with respect to amount of added protein.
RDH12 tertiary structure prediction
A low-resolution model of the tertiary structure of the RDH12 protein was derived, ab initio , by submitting the human RDH12 amino acid sequence (GenBank no. AAA99012) to the automated I-sites/HMMSTR/Rosetta server ( http://www.bioinfo.rpi.edu/~bystrc/hmmstr/server.php ) ( 7 ). The server automates a process of modeling tertiary structure from amino acid sequence using HMMSTR, a hidden Markov model based on protein structures in the invariant or initiation folding sites (I-sites) library of non-redundant short sequence motifs (supersecondary structures) that correlate with local structures ( 8 ), coupled with the Rosetta program to build structures from protein fragments using a Monte Carlo simulated annealing algorithm ( 9 ). The resulting tertiary structure with predicted coordinates was visualized and displayed with Discover Studio ViewerPro 5.0 (Accelrys, San Diego, CA, USA).
ACKNOWLEDGEMENTS
We thank Jana Schroth for expert technical assistance and David M. Reed for structural modeling. Grant support: National Institutes of Health Grants R01-EY12298 (D.A.T.), P30-EY07003 (UM-Vision Research Core Grant), M01-RR00042 (UM-General Clinical Research Center), RO1-EY13385 (S.G.J.); EVI-GENORET LSHG-CT-2005-512036 (A.G., B.W., EZ., C.A.); FWF grant 17174-B05 (A.R.J.); Foundation Fighting Blindness (P.A.S., D.A.T., S.G.J.); Research to Prevent Blindness (D.A.T.); Tiroler Medizinischer Forschungsfond (A.R.J., G.U.); Deutsche Forschungsgemeinschaft/KFO134 Erbliche Netzhauterkrankungen (B.W., E.Z.); Flieringa/Stichting Wetenschappelijk Onderzoek Oogziekenhuis Rotterdam, the Algemene Nederlandse Vereniging ter Voorkoming van Blindheid, the Gelderse Blinden Vereniging, Stichting Blindenhulp, Stichting Dondersfonds and Stichting Simonsfonds (S.Y.).
Conflict of Interest statement . None declared.
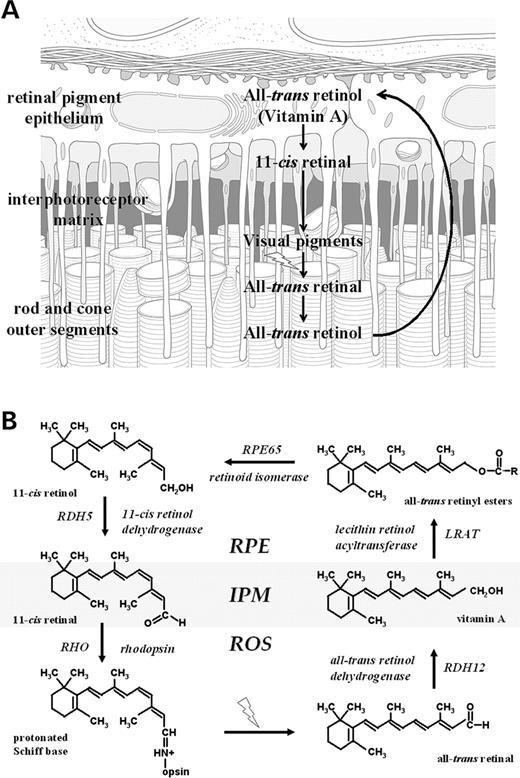
Figure 1. Schematic of the reactions of the visual cycle (A) involved in the interconversion of vitamin A and 11- cis retinal and (B) showing the proteins and enzymes (together with the corresponding genes) and retinoids present in photoreceptors and RPE. IPM, interphotoreceptor matrix; ROS, rod outer segments. The likely site of RDH12 action is indicated.
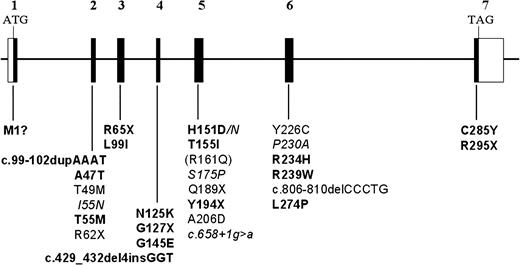
Figure 2.RDH12 gene structure and patient mutations associated with childhood-onset severe retinal dystrophy. The coding regions of the seven exons are shown as filled boxes and untranslated regions are shown as open boxes. Nucleotide numbering starts with the A of ATG as number 1. Mutations newly identified in the present study are in bold, mutations identified in our previous study ( 4 ) are in regular font and mutations unique to the study by Perrault et al . ( 5 ) are in italics. The commonly occurring p.R161Q polymorphism is shown in parentheses.
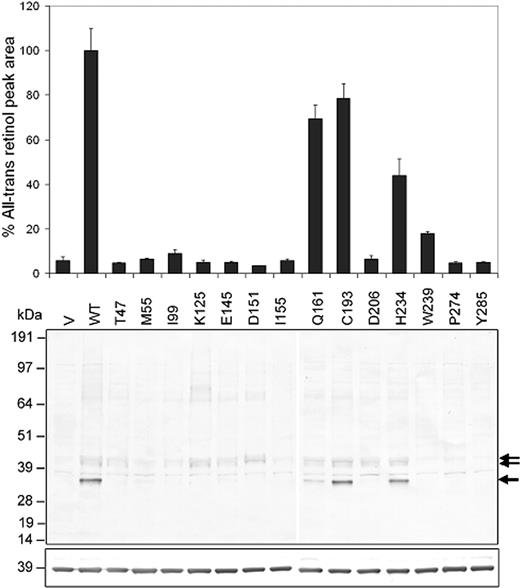
Figure 3. (Top) HPLC analysis of recombinant RDH12 catalytic activity in lysates from COS-7 cells transfected with RDH12 wild-type and missense cDNA variants in pcDNA3.1/HIS constructs, supplemented with all- trans retinal and NADPH, and assayed as described in Materials and Methods. All- trans retinol formation was quantitated by integrating the area under the A325 nm peak (µmV s) at the standard retention time, and assay data were normalized with respect to that for the wild-type construct (p.T49/p.R161) shown as 100%. Averaged data from two independent clones assayed in triplicate with standard deviations are shown and are representative of the results obtained in at least two independent transfection experiments. (Middle) Western analysis of the expression of wild-type and mutant RDH12 in transfected COS-7 cells. Cell lysates (10 µg total protein) were electrophoresed on SDS–polyacrylamide gels and transferred to nitrocellulose and probed with Xpress primary antibody and alkaline phosphatase-conjugated anti-mouse IgG secondary antibody. Arrows mark the positions of RDH12 protein bands. (Bottom) Western analysis using GAPDH primary antibody and alkaline phosphatase-conjugated anti-mouse IgG secondary antibody. Positions of molecular weight markers are indicated. V, empty vector; WT, wild-type. Variants are named by the predicted amino acid substitution.
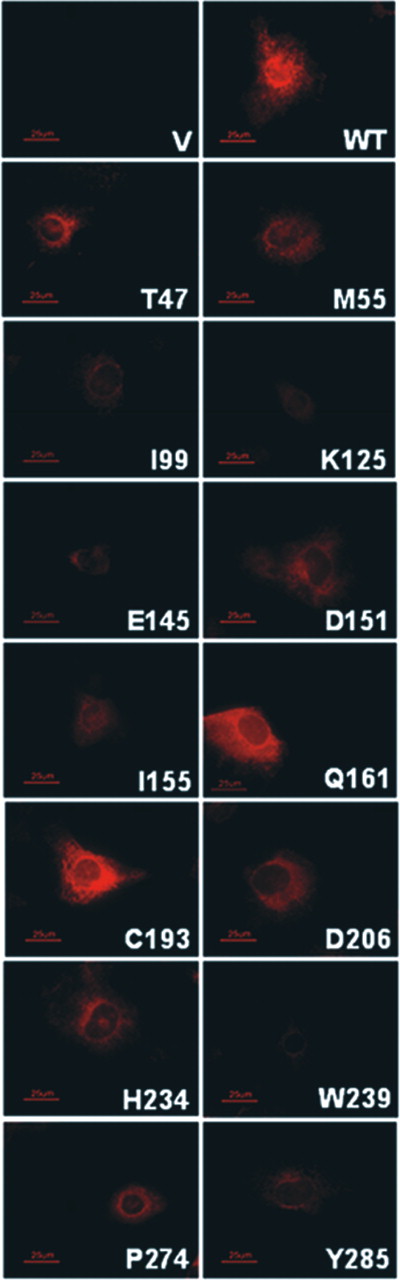
Figure 4. Immunohistochemistry of transfected COS-7 cells expressing recombinant RDH12 wild-type and mutant proteins. Cells grown in chamber slides were paraformaldehyde fixed, incubated with Anti-Xpress antibody, washed and incubated with Alexa-Fluor 555 anti-mouse IgG secondary antibody. Slides were viewed under oil immersion (100×) and photographed as described in Materials and Methods. Abbreviations are as in Figure 3 .
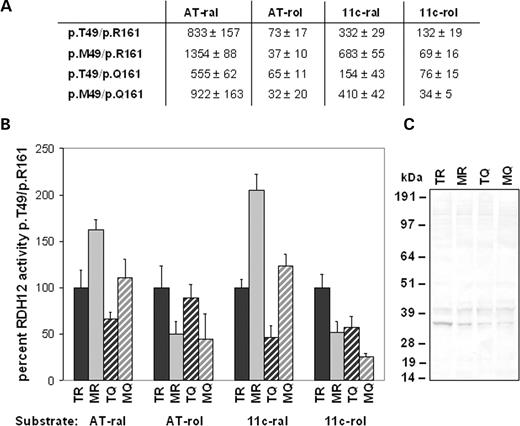
Figure 5. Retinoid dehydrogenase (retinol to retinal) and reductase (retinal to retinol) activity of p.T49 and p.M49 RDH12 variants on p.R161 and p.Q161 alleles. COS-7 cells were transfected with RDH12 missense variants in pcDNA3.1/HIS constructs and cell lysates assayed using HPLC analysis. Product formation was quantitated by integration of absorbance peak areas (µmV s/1000) at 325 nm for all- trans retinol, 318 nm for 11- cis retinol, 368 nm for all- trans retinal, 362.5 nm for 11- cis retinal and 363 nm for 13- cis retinal, minus values from transfections with empty vector. (A) Averaged integrations of product peaks±standard deviation for two independent clones of each variant assayed in triplicate in two independent transfections. In assays with 11- cis retinol as substrate, 13- cis and all- trans retinal were seen as products likely resulting from thermal interconversion of 11- cis retinal during the reaction incubation, therefore, the sum of all three isomers is reported. The same trends were observed when only the data for 11- cis retinal were included in the analysis. (B) Data from (A) normalized to p.T49/p.R161 assay data for each substrate as percent activity. (C) Western analysis of recombinant protein expression. Abbreviations are: AT-ral, all- trans retinal; AT-rol, all- trans retinol; 11c-ral, 11- cis retinal; 11c-rol, 11- cis retinol; TR, p.T49/p.R161; MR, p.M49/p.R161; TQ, p.T49/p.Q161; MQ, p.M49/p.Q161.
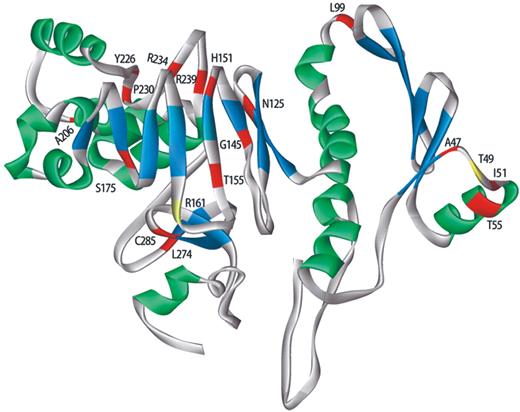
Figure 6. Model of RDH12 tertiary structure predicted using the ab initio method of Bystroff and Shao ( 7 ). The ribbon representing the peptide backbone is color coded according to structural components: alpha-helices, green; beta-pleated sheets, blue; random coil, gray. Sites of amino acid residues corresponding to the p.R161Q polymorphism and the activating p.T49M missense mutation are shown in yellow, with the remaining sites of arRD-associated missense mutations shown in red and labelled according to the wild type sequence.
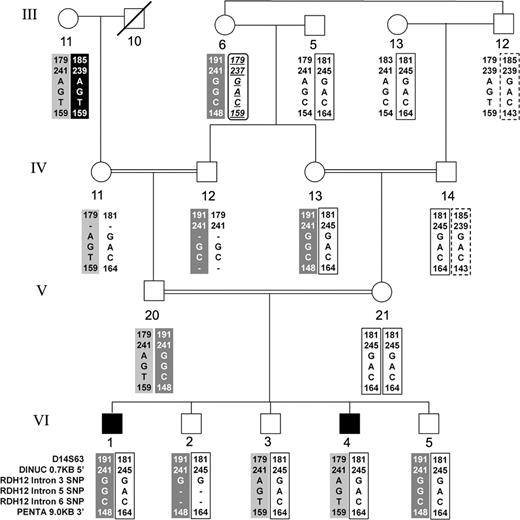
Figure 7. Haplotype analysis in Family KKESH-019 in which the autosomal recessive LCA3 locus was mapped to chromosome 14q24 ( 13 ). The most informative portion of the family is shown with the original numbering of generations and individuals retained. Physical map and order of loci in the region the haplotypes span is D14S63 – 3.5 Mb – 5′- RDH11 -3′ – 26 kb – 5′- RDH12 – dinucleotide repeat (DINUC) – 0.7 kb – RDH12 -Intron 3 SNP – RDH12 -Intron 5 SNP – RDH12 -Intron 6 SNP – 3′- RDH12 – 9.0 kb – pentanucleotide repeat (PENTA). Note that in generation VI, the two affected offspring (1 and 4) from a consanguineous marriage carry different paternal haplotypes consistent with exclusion of both RDH11 and RDH12 as disease genes in this family.
Disease-associated RDH12 mutations identified in the present study in patients carrying the changes in homozygous or compound heterozygous form
| Exon | Mutation | Predicted effect | Patient | Alleles a |
|---|---|---|---|---|
| Exon 1 | c.2T>C | p.M1? | 261 (m) | 1 |
| Exon 2 | c.99_102dupAAAT | p.Val35LysfsX27 | 237 (m) | 1 |
| c.139G>A | p.A47T | 3069 (p) | 1 | |
| c.164C>T | p.T55M | 1047 (p) | 1 | |
| Exon 3 | c.193C>T | p.R65X | 915 (p) | 1 |
| c.295C>A | p.L99I | 237 (p) | 11 | |
| 203 (m, p) | ||||
| 131 (m) | ||||
| 16 (m, p) | ||||
| 447 (m, p) | ||||
| 3069 (m) | ||||
| 02 (m, p) | ||||
| Exon 4 | c.375T>A | p.N125K | 0379 (p) | 1 |
| c.379G>T | p.G127X | 84 (m, p) | 2 | |
| c.429_432del4insGGT | p.His143GlnfsX19 | 679 (m, p) | 2 | |
| c.434G>A | p.G145E | 261 (p) | 1 | |
| Exon 5 | c.451C>G | p.H151D | 434 (p) | 2 |
| 603 (p) | ||||
| c.464C>T | p.T155I | 19 (m, p) | 5 | |
| 434 (m) | ||||
| 217 (m, p) | ||||
| c.582C>A | p.Y194X | 2975 (p) | 1 | |
| c.617C>A | p.A206D | 2975 (m) | 1 | |
| Exon 6 | c.701G>A | p.R234H | 0379 (m) | 1 |
| c.715C>G | p.R239W | 1921 (nd) | 1 | |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 131 (p) | 6 | |
| 1921 (nd) | ||||
| 603 (m) | ||||
| 915 (m) | ||||
| 93 (m, p) | ||||
| c.821T>C | p.L274P | 74 (m, p) | 2 | |
| Exon 7 | c.854G>A | p.C285Y | 496 (m, p) | 2 |
| c.883C>T | p.R295X | 1047 (m) | 1 | |
| Total | 44 |
| Exon | Mutation | Predicted effect | Patient | Alleles a |
|---|---|---|---|---|
| Exon 1 | c.2T>C | p.M1? | 261 (m) | 1 |
| Exon 2 | c.99_102dupAAAT | p.Val35LysfsX27 | 237 (m) | 1 |
| c.139G>A | p.A47T | 3069 (p) | 1 | |
| c.164C>T | p.T55M | 1047 (p) | 1 | |
| Exon 3 | c.193C>T | p.R65X | 915 (p) | 1 |
| c.295C>A | p.L99I | 237 (p) | 11 | |
| 203 (m, p) | ||||
| 131 (m) | ||||
| 16 (m, p) | ||||
| 447 (m, p) | ||||
| 3069 (m) | ||||
| 02 (m, p) | ||||
| Exon 4 | c.375T>A | p.N125K | 0379 (p) | 1 |
| c.379G>T | p.G127X | 84 (m, p) | 2 | |
| c.429_432del4insGGT | p.His143GlnfsX19 | 679 (m, p) | 2 | |
| c.434G>A | p.G145E | 261 (p) | 1 | |
| Exon 5 | c.451C>G | p.H151D | 434 (p) | 2 |
| 603 (p) | ||||
| c.464C>T | p.T155I | 19 (m, p) | 5 | |
| 434 (m) | ||||
| 217 (m, p) | ||||
| c.582C>A | p.Y194X | 2975 (p) | 1 | |
| c.617C>A | p.A206D | 2975 (m) | 1 | |
| Exon 6 | c.701G>A | p.R234H | 0379 (m) | 1 |
| c.715C>G | p.R239W | 1921 (nd) | 1 | |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 131 (p) | 6 | |
| 1921 (nd) | ||||
| 603 (m) | ||||
| 915 (m) | ||||
| 93 (m, p) | ||||
| c.821T>C | p.L274P | 74 (m, p) | 2 | |
| Exon 7 | c.854G>A | p.C285Y | 496 (m, p) | 2 |
| c.883C>T | p.R295X | 1047 (m) | 1 | |
| Total | 44 |
m, maternal allele; p, paternal allele; nd, parental origin unknown. Italics indicate change identified in this and our previous study ( 4 ).
a How many alleles among the 22 individuals had each change.
Disease-associated RDH12 mutations identified in the present study in patients carrying the changes in homozygous or compound heterozygous form
| Exon | Mutation | Predicted effect | Patient | Alleles a |
|---|---|---|---|---|
| Exon 1 | c.2T>C | p.M1? | 261 (m) | 1 |
| Exon 2 | c.99_102dupAAAT | p.Val35LysfsX27 | 237 (m) | 1 |
| c.139G>A | p.A47T | 3069 (p) | 1 | |
| c.164C>T | p.T55M | 1047 (p) | 1 | |
| Exon 3 | c.193C>T | p.R65X | 915 (p) | 1 |
| c.295C>A | p.L99I | 237 (p) | 11 | |
| 203 (m, p) | ||||
| 131 (m) | ||||
| 16 (m, p) | ||||
| 447 (m, p) | ||||
| 3069 (m) | ||||
| 02 (m, p) | ||||
| Exon 4 | c.375T>A | p.N125K | 0379 (p) | 1 |
| c.379G>T | p.G127X | 84 (m, p) | 2 | |
| c.429_432del4insGGT | p.His143GlnfsX19 | 679 (m, p) | 2 | |
| c.434G>A | p.G145E | 261 (p) | 1 | |
| Exon 5 | c.451C>G | p.H151D | 434 (p) | 2 |
| 603 (p) | ||||
| c.464C>T | p.T155I | 19 (m, p) | 5 | |
| 434 (m) | ||||
| 217 (m, p) | ||||
| c.582C>A | p.Y194X | 2975 (p) | 1 | |
| c.617C>A | p.A206D | 2975 (m) | 1 | |
| Exon 6 | c.701G>A | p.R234H | 0379 (m) | 1 |
| c.715C>G | p.R239W | 1921 (nd) | 1 | |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 131 (p) | 6 | |
| 1921 (nd) | ||||
| 603 (m) | ||||
| 915 (m) | ||||
| 93 (m, p) | ||||
| c.821T>C | p.L274P | 74 (m, p) | 2 | |
| Exon 7 | c.854G>A | p.C285Y | 496 (m, p) | 2 |
| c.883C>T | p.R295X | 1047 (m) | 1 | |
| Total | 44 |
| Exon | Mutation | Predicted effect | Patient | Alleles a |
|---|---|---|---|---|
| Exon 1 | c.2T>C | p.M1? | 261 (m) | 1 |
| Exon 2 | c.99_102dupAAAT | p.Val35LysfsX27 | 237 (m) | 1 |
| c.139G>A | p.A47T | 3069 (p) | 1 | |
| c.164C>T | p.T55M | 1047 (p) | 1 | |
| Exon 3 | c.193C>T | p.R65X | 915 (p) | 1 |
| c.295C>A | p.L99I | 237 (p) | 11 | |
| 203 (m, p) | ||||
| 131 (m) | ||||
| 16 (m, p) | ||||
| 447 (m, p) | ||||
| 3069 (m) | ||||
| 02 (m, p) | ||||
| Exon 4 | c.375T>A | p.N125K | 0379 (p) | 1 |
| c.379G>T | p.G127X | 84 (m, p) | 2 | |
| c.429_432del4insGGT | p.His143GlnfsX19 | 679 (m, p) | 2 | |
| c.434G>A | p.G145E | 261 (p) | 1 | |
| Exon 5 | c.451C>G | p.H151D | 434 (p) | 2 |
| 603 (p) | ||||
| c.464C>T | p.T155I | 19 (m, p) | 5 | |
| 434 (m) | ||||
| 217 (m, p) | ||||
| c.582C>A | p.Y194X | 2975 (p) | 1 | |
| c.617C>A | p.A206D | 2975 (m) | 1 | |
| Exon 6 | c.701G>A | p.R234H | 0379 (m) | 1 |
| c.715C>G | p.R239W | 1921 (nd) | 1 | |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 131 (p) | 6 | |
| 1921 (nd) | ||||
| 603 (m) | ||||
| 915 (m) | ||||
| 93 (m, p) | ||||
| c.821T>C | p.L274P | 74 (m, p) | 2 | |
| Exon 7 | c.854G>A | p.C285Y | 496 (m, p) | 2 |
| c.883C>T | p.R295X | 1047 (m) | 1 | |
| Total | 44 |
m, maternal allele; p, paternal allele; nd, parental origin unknown. Italics indicate change identified in this and our previous study ( 4 ).
a How many alleles among the 22 individuals had each change.
RDH12 variants identified in the present study in patients carrying the changes in heterozygous form
| Mutation (heterozygous) | Predicted effect | Patient | Alleles a | |
|---|---|---|---|---|
| Exon 3 | c.194G>A | p.R65Q | 0874 (p) | 1 |
| c.301G>A | p.D101N | 377 (m) | 1 | |
| Exon 5 | c.577C>T | p.R193C | 99 (p) | 1 |
| c.617C>T | p.A206V | 0613 (p) | 1 | |
| Exon 6 | c.689C>T | p.P230L | 242 (p) | 1 |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 98 (p) | 1 | |
| Total | 6 |
| Mutation (heterozygous) | Predicted effect | Patient | Alleles a | |
|---|---|---|---|---|
| Exon 3 | c.194G>A | p.R65Q | 0874 (p) | 1 |
| c.301G>A | p.D101N | 377 (m) | 1 | |
| Exon 5 | c.577C>T | p.R193C | 99 (p) | 1 |
| c.617C>T | p.A206V | 0613 (p) | 1 | |
| Exon 6 | c.689C>T | p.P230L | 242 (p) | 1 |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 98 (p) | 1 | |
| Total | 6 |
a How many alleles among the 22 individuals had each change.
RDH12 variants identified in the present study in patients carrying the changes in heterozygous form
| Mutation (heterozygous) | Predicted effect | Patient | Alleles a | |
|---|---|---|---|---|
| Exon 3 | c.194G>A | p.R65Q | 0874 (p) | 1 |
| c.301G>A | p.D101N | 377 (m) | 1 | |
| Exon 5 | c.577C>T | p.R193C | 99 (p) | 1 |
| c.617C>T | p.A206V | 0613 (p) | 1 | |
| Exon 6 | c.689C>T | p.P230L | 242 (p) | 1 |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 98 (p) | 1 | |
| Total | 6 |
| Mutation (heterozygous) | Predicted effect | Patient | Alleles a | |
|---|---|---|---|---|
| Exon 3 | c.194G>A | p.R65Q | 0874 (p) | 1 |
| c.301G>A | p.D101N | 377 (m) | 1 | |
| Exon 5 | c.577C>T | p.R193C | 99 (p) | 1 |
| c.617C>T | p.A206V | 0613 (p) | 1 | |
| Exon 6 | c.689C>T | p.P230L | 242 (p) | 1 |
| c.806_810delCCCTG | p.Ala269GlyfsX1 | 98 (p) | 1 | |
| Total | 6 |
a How many alleles among the 22 individuals had each change.
References
Saari, J.C. (
Haeseleer, F., Jang, G.F., Imanishi, Y., Driessen, C.A., Matsumura, M., Nelson, P.S. and Palczewski, K. (
Belyaeva, O.V., Korkina, O.V., Stetsenko, A.V., Kim, T., Nelson, P.S. and Kedishvili, N.Y. (
Janecke, A.R., Thompson, D.A., Utermann, G., Becker, C., Hubner, C.A., Schmid, E., McHenry, C.L., Nair, A.R., Ruschendorf, F., Heckenlively, J. et al. (
Perrault, I., Hanein, S., Gerber, S., Barbet, F., Ducroq, D., Dollfus, H., Hamel, C., Dufier, J.L., Munnich, A., Kaplan, J. et al. (
Liden, M., Romert, A., Tryggvason, K., Persson, B. and Eriksson, U. (
Bystroff, C. and Shao, Y. (
Bystroff, C., Thorsson, V. and Baker, D. (
Simons, K.T., Bonneau, R., Ruczinski, I. and Baker, D. (
Rohl, C.A., Strauss, C.E., Misura, K.M. and Baker, D. (
Bonneau, R., Strauss, C.E., Rohl, C.A., Chivian, D., Bradley, P., Malmstrom, L., Robertson, T. and Baker, D. (
Bradley, P., Chivian, D., Meiler, J., Misura, K.M., Rohl, C.A., Schief, W.R., Wedemeyer, W.J., Schueler-Furman, O., Murphy, P., Schonbrun, J. et al. (
Stockton, D.W., Lewis, R.A., Abboud, E.B., Al-Rajhi, A., Jabak, M., Anderson, K.L. and Lupski, J.R. (
Haeseleer, F., Huang, J., Lebioda, L., Saari, J.C. and Palczewski, K. (
Rattner, A., Smallwood, P.M. and Nathans, J. (
Wu, B.X., Chen, Y., Fan, J., Rohrer, B., Crouch, R.K. and Ma, J.X. (
Jin, M., Mata, N.L., Koag, M.C., Reid, S.N.M., Lee, J.S., Garcia, J., Farber, D.B. and Travis, G.H. (
Simon, A., Hellman, U., Wernstedt, C. and Eriksson, U. (
Yamamoto, H., Simon, A., Eriksson, U., Harris, E., Berson, E.L. and Dryja, T.P. (
Frischmeyer, P.A. and Dietz, H.C. (
Shroyer, N.F., Lewis, R.A., Yatsenko, A.N. and Lupski, J.R. (
Yasuda, M., Shabbeer, J., Benson, S.D., Maire, I., Burnett, R.M. and Desnick, R.J. (
Maeda, A., Maeda, T., Imanishi, Y., Kuksa, V., Alekseev, A., Bronson, D.J., Zhang, H., Zhu, L., Sun, W., Saperstein, D.A. et al. (
Kim, T.S., Maeda, A., Maeda, T., Heinlein, C., Kedishvili, N., Palczewski, K. and Nelson, P.S. (
Thompson, D.A. and Gal, A. (
Gu, S.M., Thompson, D.A., Srikumari, C.R., Lorenz, B., Finckh, U., Nicoletti, A., Murthy, K.R., Rathmann, M., Kumaramanickavel, G., Denton, M.J. et al. (
Redmond, T.M., Yu, S., Lee, E., Bok, D., Hamasaki, D., Chen, N., Goletz, P., Ma, J.X., Crouch, R.K. and Pfeifer, K. (
Ma, J.-X., Chen, Y., Takahashi, Y. and Moiseyev, G. (
Jin, M., Li, S., Moghrabi, W.N., Sun, H. and Travis, G.H. (
Intres, R., Goldflam, S., Cook, J.R. and Crabb, J.W. (
Morimura, H., Berson, E.L. and Dryja, T.P. (
Chen, P., Hao, W., Rife, L., Wang, X.P., Shen, D., Chen, J., Ogden, T., Van Boemel, G.B., Wu, L., Yang, M. et al. (
Morimura, H., Saindelle-Ribeaudeau, F., Berson, E.L. and Dryja, T.P. (
Wenzel, A., Oberhauser, V., Pugh, E.N., Jr, Lamb, T.D., Grimm, C., Samardzija, M., Fahl, E., Seeliger, M.W., Reme, C.E. and von Lintig, J. (
Ruiz, A., Winston, A., Lim, Y.H., Gilbert, B.A., Rando, R.R. and Bok, D. (
Thompson, D.A., Li, Y., McHenry, C.L., Carlson, T.J., Ding, X., Sieving, P.A., Apfelstedt-Sylla, E. and Gal, A. (
Acland, G.M., Aguirre, G.D., Ray, J., Zhang, Q., Aleman, T.S., Cideciyan, A.V., Pearce-Kelling, S.E., Anand, V., Zeng, Y., Maguire, A.M. et al. (
Van Hooser, J.P., Aleman, T.S., He, Y.G., Cideciyan, A.V., Kuksa, V., Pittler, S.J., Stone, E.M., Jacobson, S.G. and Palczewski, K. (
Jacobson, S.G., Aleman, T.S., Cideciyan, A.V., Sumaroka, A., Schwartz, S.B., Windsor, E.A., Traboulsi, E.I., Heon, E., Pittler, S.J., Milam, A.H. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}