





Abstract
Gene retroposition is an important mechanism of genome evolution but the role it plays in dinoflagellates, a critical player in marine ecosystems, is not known. Until recently, when the genomes of two coral-symbiotic dinoflagellate genomes, Symbiodinium kawagutii and S. minutum, were released, it has not been possible to systematically study these retrogenes. Here we examine the abundant retrogenes (∼23% of the total genes) in these species. The hallmark of retrogenes in the genome is the presence of DCCGTAGCCATTTTGGCTCAAG, a spliced leader (DinoSL) constitutively trans-spliced to the 5′-end of all nucleus-encoded mRNAs. Although the retrogenes have often lost part of the 22-nt DinoSL, the putative promoter motif from the DinoSL, TTT(T/G), is consistently retained in the upstream region of these genes, providing an explanation for the high survival rate of retrogenes in dinoflagellates. Our analysis of DinoSL sequence divergence revealed two major bursts of retroposition in the evolutionary history of Symbiodinium, occurring at ∼60 and ∼6 Ma. Reconstruction of the evolutionary trajectory of the Symbiodinium genomes mapped these 2 times to the origin and rapid radiation of this dinoflagellate lineage, respectively. GO analysis revealed differential functional enrichment of the retrogenes between the two episodes, with a broad impact on transport in the first bout and more localized influence on symbiosis-related processes such as cell adhesion in the second bout. This study provides the first evidence of large-scale retroposition as a major mechanism of genome evolution for any organism and sheds light on evolution of coral symbiosis.
Introduction
Gene birth is a major driver of genome evolution and shapes the configuration and function of the genome. A major mechanism of gene birth is gene duplication, and can occur by whole genome duplication (WGD) or segmental duplication (SD) events (Wolfe and Shields 1997; Crow etal. 2006). Retroposition, the integration of an RNA back into the genome (also called mRNA recycling), is also frequent and has been well documented in mammals and fruit flies (Kaessmann etal. 2009). However, because the regulatory elements (i.e., promoters) that drive the expression of the parent genes are not included in the transcripts, most of the retrogenes become “dead on arrival” pseudogenes (Kaessmann etal. 2009). Only those few retrogenes (90 ∼ 100 in mammals or flies) that successfully obtain new promoters can survive (Marques etal. 2005; Bai etal. 2007). Currently known mechanisms of retrogene survival include 1) using the promoter of other genes when the retrocopy is inserted into an intron or an active region of the chromosome; 2) exploiting a distant promoter by transcribing a long 5′-UTR; 3) inheriting a promoter from an alternatively transcribed parental copy; 4) using a “proto-promoter” with promoter potential in CG islands; and 5) accumulating mutations to form a promoter de novo (reviewed in Kaessmann etal. 2009). Despite their infrequent survival, retrogenes play a critical role in shaping genome architecture as they are prone to sub or neofunctionalization and become a valuable source of new genes.
Dinoflagellates, a group of unicellular eukaryotic protists closely related to the Apicomplexa, are important marine primary producers, major contributors to harmful algal blooms (commonly known as “red tides”), and the essential symbionts of reef corals. They display many unusual cytological characteristics (Lin 2011) including generally undetectable levels of histones, permanently condensed chromosomes and widely varied and generally large (∼1–250 Gbp) genomes. Furthermore, the classical TATA box may have been replaced by TTT(T/G) as the core promoter motif. This sequence is one of the few highly conserved motifs in the region upstream of S. kawagutii coding sequences (Shoguchi etal. 2013; Lin etal. 2015). Furthermore, a TBP-like factor with higher affinity to this motif has been found in dinoflagellates (Guillebault 2002) and is the only type of a TATA-box binding protein present. Interestingly, mRNA maturation requires trans-splicing of a 22-nt dinoflagellate spliced leader [DinoSL, DCCGUAGCCAUUUUGGCUCAAG (D = U, A or G)] to their 5′ termini (Zhang etal. 2007).
One of the best studied dinoflagellate lineages is Symbiodinium, which harbors species living endosymbiotically in a wide range of cnidarian hosts including corals and anemone. Due to its importance for coral reef growth and its small genome size (for dinoflagellates, still 50-fold that of Plamodium falciparum), Symbiodinium has become a valuable model for dinoflagellate genome research. No evidence for WGD or SD has been documented in Symbiodinium genomes (Shoguchi etal. 2013; Lin etal. 2015), and thus the major forces driving the rapid expansion of the immense dinoflagellate genomes are still poorly understood. Retrogenes in dinoflagellates, discovered by the presence of relict DinoSL sequences (Slamovits and Keeling 2008), have great potential to contribute to genome evolution as they provide a large source of new genes. However, the genome-wide scale, tempo, and evolutionary as well as ecological implications have not been systematically investigated due to the lack of dinoflagellate genome sequences. Furthermore, as none of the known mechanisms of promoter acquisition can account for a large-scale retention of retrogenes in dinoflagellate genomes, the mechanism facilitating survival of dinoflagellate retrogenes is also an enigma. The recent availability of two dinoflagellate genome assemblies (Symbiodinium kawagutii and S. minutum) (Shoguchi etal. 2013; Lin etal. 2015) has afforded an opportunity to address these questions.
In this study, we used data from the two genomes and identify retrogenes globally by searching for relict DinoSL sequences in the upstream 5′ region of gene coding sequences. We found that >20% of the genes in both genomes have relict DinoSL sequence. We suggest that the unique trans-splicing process which adds TTT(T/G), the potential equivalent of the TATA box in dinoflagellates, may act to introduce a basal promoter into the retrogenes thus facilitating their survival. Extensive analyses of sequence characteristics and functional distribution of the retrogenes reveal that these retrogenes have emerged in two major bouts, which might have promoted the emergence and radiation of Symbiodinium species.
Results
Recurring and Widespread Retroposition
As a hallmark of mature mRNA, DinoSL constitutes a unique sequence tag for retroposed genes in the genome (Lin etal. 2015), and its repetition in the upstream region of some genes is an indication that mRNAs of these genes have been retroposed more than once. By searching for the DinoSL sequence or its relicts in the 500-bp upstream of ORFs in S. kawagutii, we confidently identified 9,801 canonical (complete and exact matching) or remnant (partial and slightly varying) DinoSL sequences upstream of 8,564 protein-coding genes (23.24% of the S. kawagutii gene models). The number of DinoSLs in each of these genes varied from one to as many as eight (table 1), implying multiple recycling events for some of these genes (fig. 1a). Roughly 3.02% (1,112) of the S. kawagutii genes have been recycled more than once. Similarly, DinoSL motifs were detected upstream of 22.28% (9,339 of 41,925) of the S. minutum gene models, and 2.41% (1,012) were recycled more than once (table 1). Of all the DinoSLs, 190 in S. kawagutii and 159 in S. minutum are tandemly arranged (i.e., immediately adjacent to one another) in 97 and 74 retrogenes, respectively, and the others were separated from one another in the upstream regions of the retrogenes. The gaps between separated DinoSLs ranges from 6 to ∼450 bp, with a 30-bp spacing most often found. This result is also consistent with earlier observations that 100 out of 500 of cDNAs examined in the dinoflagellates Alexandrium, Heterocapsa, and Prorocentrum contain a second DinoSL relict, suggesting that large scale retroposition is widespread in dinoflagellates (Slamovits and Keeling 2008).
Number of Symbiodinium Genes Harboring Different Number of DinoSL Relicts
| Number of DinoSLs Relicts | S. kawagutii | S. minutum |
|---|---|---|
| 1 | 7,452 | 8,327 |
| 2 | 1,009 | 921 |
| 3 | 91 | 82 |
| 4 | 7 | 6 |
| 5 | 1 | 2 |
| 6 | 3 | 0 |
| 7 | 1 | 0 |
| 8 | 0 | 1 |
| Total | 8,564 | 9,339 |
| Number of DinoSLs Relicts | S. kawagutii | S. minutum |
|---|---|---|
| 1 | 7,452 | 8,327 |
| 2 | 1,009 | 921 |
| 3 | 91 | 82 |
| 4 | 7 | 6 |
| 5 | 1 | 2 |
| 6 | 3 | 0 |
| 7 | 1 | 0 |
| 8 | 0 | 1 |
| Total | 8,564 | 9,339 |
Number of Symbiodinium Genes Harboring Different Number of DinoSL Relicts
| Number of DinoSLs Relicts | S. kawagutii | S. minutum |
|---|---|---|
| 1 | 7,452 | 8,327 |
| 2 | 1,009 | 921 |
| 3 | 91 | 82 |
| 4 | 7 | 6 |
| 5 | 1 | 2 |
| 6 | 3 | 0 |
| 7 | 1 | 0 |
| 8 | 0 | 1 |
| Total | 8,564 | 9,339 |
| Number of DinoSLs Relicts | S. kawagutii | S. minutum |
|---|---|---|
| 1 | 7,452 | 8,327 |
| 2 | 1,009 | 921 |
| 3 | 91 | 82 |
| 4 | 7 | 6 |
| 5 | 1 | 2 |
| 6 | 3 | 0 |
| 7 | 1 | 0 |
| 8 | 0 | 1 |
| Total | 8,564 | 9,339 |
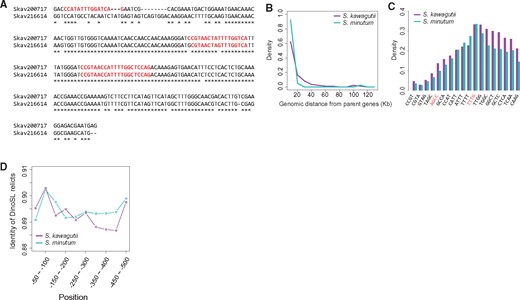
—Retroposition in Symbiodinium genome sequences retaining DinoSL (CCGTAGCCATTTTGGCTCAAG) motifs. (A) Alignment of a retrogene (Skav200717) and its parent (Skav216614), both of which have undergone repeated retroposition events. Red colored sequences show DinoSL relicts. (B) Distance between retrogenes and their parents in S. kawagutii (purple line) and S. minutum (cyan line). (C) DinoSL motif “TTTT,” “TTTG,” and “TTGG” are preferentially retained in all the detected DinoSL relicts in both S. kawagutii (purple bars) and S. minutum (cyan bars). (D) Identities of DinoSL relicts of S. kawagutii (purple line) and S. minutum (cyan line) as a function of upstream distance from the start codon. Higher identities of DinoSLs are seen between −50 and −100 bp where the promoter is expected to be located.
To determine the sequence features of the retrogenes, we compared source genes (without DinoSL) with retrogenes (with DinoSL) with respect to GC ratio, length of coding sequences, intron presence, sequence complexity, and gene expression. As indicated by the Cohen’s d score, a measurement of effect size (Sullivan and Feinn 2012), there is no obvious difference between retro- and source genes in any of the terms tested except that the expression of retrogenes is higher in S. kawagutii (table 2). It has been reported that in human cells, mRNAs encoding soluble proteins are preferentially retroposed because they are translated on free cytoplasmic ribosomes. For example, long interspersed nuclear element 1 (LINE1) mRNAs are translated, and thus are more likely to be reverse transcribed by LINE1 (Roy-Engel 2012). However, most of the retrogenes in Symbiodinium (62.35% in S. kawagutii and 92.31% in S. minutum) were inserted <10 kb from their parents (fig. 1B), suggesting that retroposition process is initiated in situ, immediately after gene transcription. Our results show retrogene-encoded proteins are not more highly soluble (table 2).
Feature Comparison Between Retroposed and Normal Genes in Symbiodinium Genomes
| S. kawagutii | S. minutum | |||||||
|---|---|---|---|---|---|---|---|---|
| Mean | Statistic Significancea | Mean | Statistic Significancea | |||||
| Retrogene | Normal Gene | P | Cohn’s d | Retrogene | Normal Gene | P | Cohn’s d | |
| No of Introns | 3.74 | 4.17 | 2.20E-16 | 0.11 | 16.74 | 17.1 | 0.06 | 0.02 |
| Length of CDS | 1108.56 | 1021.17 | 4.68E-10 | 0.08 | 1705.54 | 1642.87 | 3.33E-03 | 0.03 |
| GC of CDS | 49.67 | 48.92 | 2.20E-16 | 0.18 | 45.2 | 44.83 | 2.20E-16 | 0.11 |
| Complexity of CDS | 1.45 | 1.44 | 2.20E-16 | 0.18 | 1.45 | 1.44 | 6.91E-15 | 0.11 |
| RPKM | 0.51 | 0.36 | 2.20E-16 | 0.2 | 0.03 | 0.03 | 3.08E-10 | 0.08 |
| Solubility | 0.51 | 0.51 | 0.97 | 0 | 0.51 | 0.51 | 0.02 | 0.03 |
| S. kawagutii | S. minutum | |||||||
|---|---|---|---|---|---|---|---|---|
| Mean | Statistic Significancea | Mean | Statistic Significancea | |||||
| Retrogene | Normal Gene | P | Cohn’s d | Retrogene | Normal Gene | P | Cohn’s d | |
| No of Introns | 3.74 | 4.17 | 2.20E-16 | 0.11 | 16.74 | 17.1 | 0.06 | 0.02 |
| Length of CDS | 1108.56 | 1021.17 | 4.68E-10 | 0.08 | 1705.54 | 1642.87 | 3.33E-03 | 0.03 |
| GC of CDS | 49.67 | 48.92 | 2.20E-16 | 0.18 | 45.2 | 44.83 | 2.20E-16 | 0.11 |
| Complexity of CDS | 1.45 | 1.44 | 2.20E-16 | 0.18 | 1.45 | 1.44 | 6.91E-15 | 0.11 |
| RPKM | 0.51 | 0.36 | 2.20E-16 | 0.2 | 0.03 | 0.03 | 3.08E-10 | 0.08 |
| Solubility | 0.51 | 0.51 | 0.97 | 0 | 0.51 | 0.51 | 0.02 | 0.03 |
P values become misleadingly low when the sample size is sufficiently large giving a false indication of significance. Therefore, we also calculated the effect size measure Cohn’s d to determine the difference between groups. Cohn’s d of 0.2∼0.5 indicates small, 0.5∼0.8 indicates medium and >0.8 indicates large differences (Sullivan and Feinn 2012).
Feature Comparison Between Retroposed and Normal Genes in Symbiodinium Genomes
| S. kawagutii | S. minutum | |||||||
|---|---|---|---|---|---|---|---|---|
| Mean | Statistic Significancea | Mean | Statistic Significancea | |||||
| Retrogene | Normal Gene | P | Cohn’s d | Retrogene | Normal Gene | P | Cohn’s d | |
| No of Introns | 3.74 | 4.17 | 2.20E-16 | 0.11 | 16.74 | 17.1 | 0.06 | 0.02 |
| Length of CDS | 1108.56 | 1021.17 | 4.68E-10 | 0.08 | 1705.54 | 1642.87 | 3.33E-03 | 0.03 |
| GC of CDS | 49.67 | 48.92 | 2.20E-16 | 0.18 | 45.2 | 44.83 | 2.20E-16 | 0.11 |
| Complexity of CDS | 1.45 | 1.44 | 2.20E-16 | 0.18 | 1.45 | 1.44 | 6.91E-15 | 0.11 |
| RPKM | 0.51 | 0.36 | 2.20E-16 | 0.2 | 0.03 | 0.03 | 3.08E-10 | 0.08 |
| Solubility | 0.51 | 0.51 | 0.97 | 0 | 0.51 | 0.51 | 0.02 | 0.03 |
| S. kawagutii | S. minutum | |||||||
|---|---|---|---|---|---|---|---|---|
| Mean | Statistic Significancea | Mean | Statistic Significancea | |||||
| Retrogene | Normal Gene | P | Cohn’s d | Retrogene | Normal Gene | P | Cohn’s d | |
| No of Introns | 3.74 | 4.17 | 2.20E-16 | 0.11 | 16.74 | 17.1 | 0.06 | 0.02 |
| Length of CDS | 1108.56 | 1021.17 | 4.68E-10 | 0.08 | 1705.54 | 1642.87 | 3.33E-03 | 0.03 |
| GC of CDS | 49.67 | 48.92 | 2.20E-16 | 0.18 | 45.2 | 44.83 | 2.20E-16 | 0.11 |
| Complexity of CDS | 1.45 | 1.44 | 2.20E-16 | 0.18 | 1.45 | 1.44 | 6.91E-15 | 0.11 |
| RPKM | 0.51 | 0.36 | 2.20E-16 | 0.2 | 0.03 | 0.03 | 3.08E-10 | 0.08 |
| Solubility | 0.51 | 0.51 | 0.97 | 0 | 0.51 | 0.51 | 0.02 | 0.03 |
P values become misleadingly low when the sample size is sufficiently large giving a false indication of significance. Therefore, we also calculated the effect size measure Cohn’s d to determine the difference between groups. Cohn’s d of 0.2∼0.5 indicates small, 0.5∼0.8 indicates medium and >0.8 indicates large differences (Sullivan and Feinn 2012).
DinoSL Promoter Motif Facilitates Retrogene Survival
Retrogenes will survive as functional genes only if they can be expressed (Kaessmann etal. 2009). None of the currently known mechanisms can adequately explain the extensive retrogene retention in dinoflagellates. However, the DinoSL relicts at the 5′ terminus of a retrogene contain the proposed dinoflagellate core promoter motif TTT(T/G). We thus hypothesize that dinoflagellate retrogenes survive because they are inserted into the genome with a basal promoter element derived from DinoSL already present. In support of this, we found that motif TTT(T/G) is highly conserved in the relicts. A total of 5,834 recycled DinoSLs from 5,334 S. kawagutii retrogenes (62.28% of the retrogenes) and 7,277 DinoSLs from 6,694 S. minutum retrogenes (71.68%) retained either the TTTT or TTTG motif (supplementary table S1, Supplementary Material online). The frequency plot of 4-bp motifs in the retrogene DinoSL shows stepwise increases at motifs “AGCC” and “TTTG” (fig. 1C). AG is a potential acceptor site for another spliced leader trans-splicing of the mRNA (Shoguchi etal. 2013), which could reduce the frequency of the nucleotides before this AG dinucleotide in the retrogene. The higher frequency of TTTG is consistent with the preservation of promoter motifs.
If DinoSL relicts provide promoter motifs, there must be a selective pressure against mutation in these sequences. It follows that if more than one DinoSL relicts are present, the DinoSL relict that acts as the promoter would also be subject to a selective pressure against mutation. We found that the most conserved DinoSL relicts are located between −50- and −100-bp upstream of start codon (fig. 1D), the region in which the promoter core motifs are enriched (Lin etal. 2015). When located farther upstream, the DinoSL relicts become less identical until reaching the distance of −400 bp. This suggests that the DinoSL relicts close to the transcription start sites of retrogenes are more likely to serve as promoters.
In addition to the retrocopy genes, a large number of pseudogenes were also recovered from the genomes of S. kawagutii and S. minutum. However, only 1.82% (4,744 of 260,523) and 2.30% (5,092 of 221,052) of the pseudogenes we identified in S. kawagutii and S. minutum, respectively, contained DinoSL relicts in their upstream regions. If these pseudogenes were retroposed genes, they likely became pseudogenes because they lacked a DinoSL in the upstream region.
These results indicate that retroposition, rather than WGD or SD, plays the predominant role in genome expansion for both the sequenced dinoflagellate genomes (Shoguchi etal. 2013; Lin etal. 2015). Combined, retrogenes and pseudogenes comprise 173.80 Mb (18.6%) of the S. kawagutii genome and 165.51 Mb (26.9%) of the S. minutum genome. These values may even underestimate the contribution of retroposition to genome expansion, as pseudogenized retrogenes that have become undetectable due to extensive mutations would not have been included.
Two Major Bouts of Retroposition in the Evolutionary History of Symbiodinium
We had originally anticipated that retroposition would be both continuous and ongoing. To test this, we identified 843 and 599 parental genes in S. kawagutii and S. minutum, respectively, and calculated the synonymous divergence (Ks) of the retroposed genes as a proxy of gene age (see Methods for details). Surprisingly, the number of gene sequences as a function of Ks shows a bimodal distribution, suggesting that both Symbiodinium genomes have undergone two major bouts of large-scale gene recycling in their evolutionary history (fig. 2A and B). The retrogenes examined were clustered into two groups, one with a mean Ks of 3.02 for S. kawagutii and 3.17 for S. minutum, and another with means of 0.47 and 0.18, respectively. A comparison of orthologs between S. kawagutii and S. minutum, estimated to have diverged ∼32 Ma (Pochon etal. 2006), allowed a calculation of the mutation rate at synonymous sites of 5.16 × 10−8 per site per year. Using this rate, the two bouts of retroposition would have occurred between 61.4 and 58.5 Ma for one and 9.1 and 3.5 Ma for the other (fig. 2C). These times showed a striking correspondence with the emergence of Symbiodinium (>50 Ma) and the expansion of its host range concomitant with radiation of Symbiodinium clades (∼2–15 Ma) (Pochon etal. 2006) (fig. 2C), suggesting a crucial role of retroposition in Symbiodinium evolution.
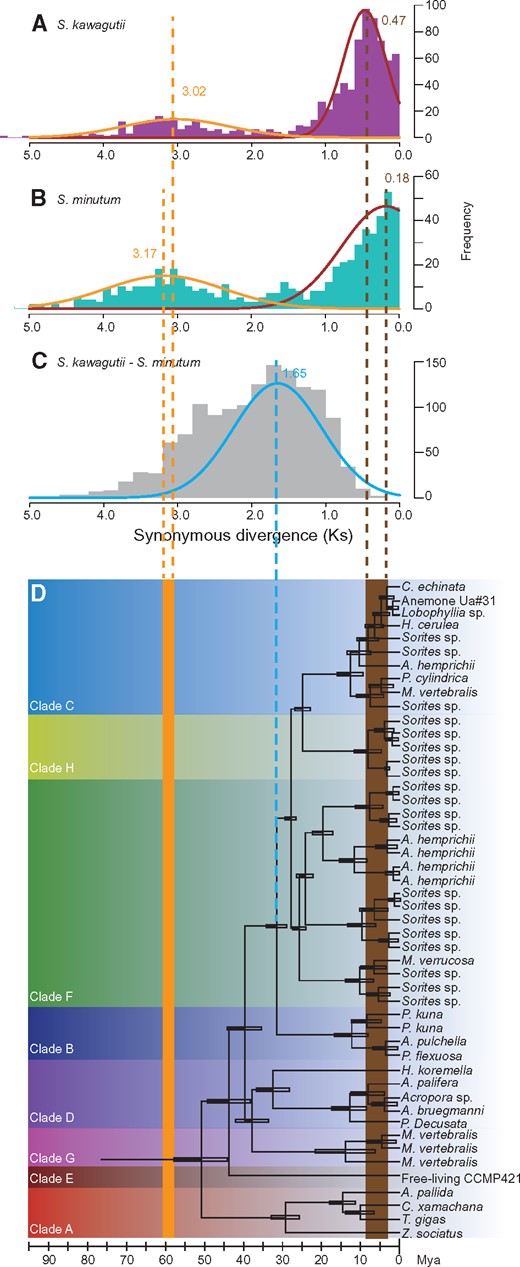
—Two major retroposition events revealed by Ks frequencies of retrogenes in (A)Symbiodinium kawagutii (Ks = 3.02 and 0.47) and (B)S. minutum (Ks = 3.17 and 0.18). (C) Ks frequencies in a comparison of S. kawagutii versus S. minutum (Ks = 1.65). (D) Phylogenetic tree and age of different Symbiodinium strains (adapted from Pochon etal. 2006) named by their coral hosts. The empty box at each node represents ± one standard deviation around divergence age.
Phenotypic Changes Predicted from Retrogene Copies
The probability for a gene to be recycled is largely dependent on levels of its transcript (Pavlicek etal. 2007) (table 2); therefore, the number of retrogenes in a family should reflect the relative activity of the parental gene when the retroposition occurred. We counted the number of retrogenes in each family as a proxy for its ancient expression level and compared it with current expression profiles (Shoguchi etal. 2013; Lin etal. 2015). Our results indicate that for the more recent bout of retroposition, the number of retrogenes in a family is correlated with current levels of gene expression (rho = 0.24, P = 0.0001 for S. kawagutii, and rho= 0.30, P = 0.0005 for S. minutum). However, this correlation does not hold for the more ancient bout of retroposition (table 3), and this may reflect the change from a free living to a symbiotic lifestyle occurring at that time (∼55 Ma). Globally, the most actively expressed genes at the times of peak retroposition are those engaged in processes that are important for the dinoflagellates, including DNA modification (modification methylases and DNA-methyltransferases), DNA mobility, and RNA editing (pentatricopeptide repeat-containing proteins) (supplementary table S2, Supplementary Material online and table 3).
Number of Retrogenes in the Two Major Retroposition Bouts and the Correlation Between the Number of Family Members and Their Current Expression Levels
| Bout | S. kawagutii | S. minutum | ||
|---|---|---|---|---|
| No. of Genes (Family)a | Rho (P value)b | No. of Genes (Family)a | Rho (P value)b | |
| 1 | 195 (88) | −0.06 (0.63) | 214 (145) | 0.11 (0.14) |
| 2 | 648 (361) | 0.24 (0.0001) | 385 (202) | 0.30 (0.0005) |
| Bout | S. kawagutii | S. minutum | ||
|---|---|---|---|---|
| No. of Genes (Family)a | Rho (P value)b | No. of Genes (Family)a | Rho (P value)b | |
| 1 | 195 (88) | −0.06 (0.63) | 214 (145) | 0.11 (0.14) |
| 2 | 648 (361) | 0.24 (0.0001) | 385 (202) | 0.30 (0.0005) |
Number of genes that can be accurately assigned into these two events based on Ks values; numbers in parentheses depict the number of families these genes were clustered into.
Rho, the Spearman’s rank correlation coefficient between the copy number of retrogenes and their expression levels (RPKM) as recently measured for genes in each family (Lin etal. 2015).
Number of Retrogenes in the Two Major Retroposition Bouts and the Correlation Between the Number of Family Members and Their Current Expression Levels
| Bout | S. kawagutii | S. minutum | ||
|---|---|---|---|---|
| No. of Genes (Family)a | Rho (P value)b | No. of Genes (Family)a | Rho (P value)b | |
| 1 | 195 (88) | −0.06 (0.63) | 214 (145) | 0.11 (0.14) |
| 2 | 648 (361) | 0.24 (0.0001) | 385 (202) | 0.30 (0.0005) |
| Bout | S. kawagutii | S. minutum | ||
|---|---|---|---|---|
| No. of Genes (Family)a | Rho (P value)b | No. of Genes (Family)a | Rho (P value)b | |
| 1 | 195 (88) | −0.06 (0.63) | 214 (145) | 0.11 (0.14) |
| 2 | 648 (361) | 0.24 (0.0001) | 385 (202) | 0.30 (0.0005) |
Number of genes that can be accurately assigned into these two events based on Ks values; numbers in parentheses depict the number of families these genes were clustered into.
Rho, the Spearman’s rank correlation coefficient between the copy number of retrogenes and their expression levels (RPKM) as recently measured for genes in each family (Lin etal. 2015).
To gain insight into the cellular phenotypes at the time of the first or the second retroposition bout, we assigned the retrogenes to different biological processes by GO enrichment analyses. We found that general housekeeping processes (such as translation and DNA replication) and reactions essential for retroposition (RNA-dependent DNA replication) are shared between these two bouts. However, some enriched GO terms are distinct in each of these two major retroposition episodes suggesting the two events punctuate substantial changes in phenotype (fig. 3). For example, the first bout shows enrichment for many enzymes of sugar and fatty acid metabolism, response to oxidative stress, and regulation of protein synthesis and activity (fig. 3A). Consistent with development of a symbiotic lifestyle, a number of transport processes (including “carbohydrate transport,” “intracellular protein transport,” and “sodium/potassium ion transport;” fig. 3A) were enriched at that time. This suggests that stimulation of the expression of these genes might have promoted the emergence of symbiotic capacity. In contrast, processes related to photosynthesis and carbohydrate metabolism were particularly enriched during the second bout (fig. 3B), implying high photosynthetic activity at that time. These results are consistent with the conclusion that the second retroposition took place at a symbiotic stage where enhanced carbon fixation activity would be beneficial to feed both the Symbiodinium and host cells. Processes related to the establishment of symbiosis, such as “cell adhesion” and “protein N-linked glycosylation” were also highly expressed at this stage.
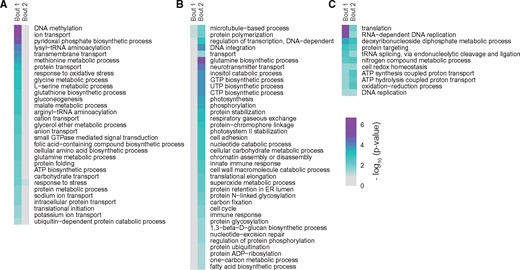
—Differential enrichment of sequences in biological processes during the first and second bout of retroposition. The enrichment P value is shown for GO processes at (A) the first or (B) the second episode only, or (C) for GO processes shared by both. Color scale represents the −log10 value of P values, with purple cells indicating higher enrichment. More details are shown in supplementary table S4, Supplementary Material online.
Discussion
The birth of genes is important in shaping the genome and driving the evolution of species. In most of the known eukaryotic genomes, WGD and SD efficiently generate new gene copies. As chromosome fragments are duplicated, the genes on it are also doubled. The regulatory elements are always duplicated together with the coding regions, and as a result, a new copy initially has an expression pattern similar to the parental copy. The gene copy can gradually change its function or become silenced by pseudogenization or posttranscriptional regulation, such as by small RNAs. In contrast, generation of new gene copies by retroposition is a very different process. Retroposition is an mRNA-mediated gene birth process similar to retrotransposition but differing in that the gene that is being copied does not encode the endonuclease and reverse transcriptase activities required for retrotransposition. The endonuclease and reverse transcriptase activities required for retroposition are presumably provided by LINEs (Pavlicek etal. 2007). In most organisms, retrogenes are very often “dead on arrival” due to the lack of regulatory elements (i.e., promoter), and are termed pseudogenes. Only the few retrogenes that integrate close enough to an existing promoter to be activated by it will survive. The retrogenes that are expressed typically play critical roles in shaping the genome and phenotypes because their expression pattern will be different from the parental gene copies, leading to dramatic changes in phenotypes.
Retroposition Is Widespread in Dinoflagellate Genomes
Dinoflagellate nucleus-encoded mRNAs are capped by a 22-nt leader sequence before maturation (Zhang etal. 2007). This character allows easy identification of retrogenes in dinoflagellate genomes by searching for the DinoSL or its relicts in the upstream region of genes. The search results show the retrogenes account for >20% of the total genes in both the Symbiodinium genomes, with some of them (2 ∼ 3%) having been recycled multiple times (fig. 1A). Besides Symbiodinium, retrogenes have also been found in ESTs from many other dinoflagellates including Alexandrium, Oxyrrhis, Heterocapsa, and Prorocentrum (Slamovits and Keeling 2008; Jaeckisch etal. 2011; Lee etal. 2014). These results suggest large scale retroposition is widespread in dinoflagellates. These larger dinoflagellate genomes may have experienced more extensive retroposition than Symbiodinium because ∼12% of their cDNAs, ∼4–6 times greater than what is found in Symbiodinium genomes, have more than three DinoSLs (Slamovits and Keeling 2008) which is indicative of multiple recycling events (table 1).
The large number of retrogenes in both the Symbiodinium genomes allows their character and evolution to be analyzed in more detail. Surprisingly, this analysis indicates that retroposition occurred in two major bouts during evolution, whose timing corresponds first to a time before the emergence of Symbiodinium and second to the radiation of Symbiodinium species. It is interesting that the times of retroposition estimated from the two different Symbiodinium genomes are virtually identical (fig. 2). The first bout of retroposition occurred ∼60 Ma. This time is very close to the time (∼55–75 Ma, the Cretaceous–Paleogene boundary) when WGD events frequently occurred in plant genomes (Van de Peer etal. 2017). The Cretaceous–Paleogene boundary is marked by a number of catastrophic events, which had led to major climate changes (Petersen etal. 2016). A mass extinction of species (60–70% of all plant and animal) including nonavian dinosaur occurred at this time. These environmental changes whose effects are so noticeable in the terrestrial organisms may thus be mirrored in the burst of retroposition in marine dinoflagellates. The second retroposition occurred at ∼6 Ma, after the split of S. kawagutii and S. minutum, suggesting that the large-scale retroposition process is not species-specific but rather may also have been due to another major episode of environmental changes. If true, it suggests that retroposition may also have occurred at these times in other dinoflagellate genomes. As the interval between these two bouts of retroposition is rather small, and retroposition seems to be easily triggered by environmental factors, we speculate that there might be more than two bouts of retroposition during dinoflagellate evolution. Although this is certainly possible it seems likely they will be undetectable using our current data due to several limitations, including that 1) the SL is rather short, hence very old retrogenes would mutate beyond recognition; 2) the loss of parental copies would result in fewer or false pairs for calculation of Ks; and 3) the saturation of synonymous mutations may limit detection of old bouts. Despite these caveats, the frequency of retroposition identified in Symbiodinium genomes clearly indicates that large-scale retroposition events have occurred several times during dinoflagellate evolution.
The Survival of Dinoflagellate Retrogenes Is Facilitated by DinoSL
As mentioned earlier, most retrogenes in other genomes have become pseudogenes and their sequences have degenerated. We thus asked what mechanisms might have facilitated survival of dinoflagellate retrogenes. We noticed that DinoSL, which is constitutively trans-spliced to the 5′ end of mRNA, has a TTT(T/G) motif, proposed to be the dinoflagellate equivalent of a TATA box. This leader sequence is retroposed together with mRNA and might be able to promote the transcription of retrogene. If this hypothesis is true, we predicted that 1) the DinoSLs located close to the promoter region (probably within the first 100-bp upstream from the start codon) would be more conserved than the others regardless of their ages; 2) the TTT(T/G) motif in particular would be more conserved; 3) there would be a limited number of multiply recycled retrogenes. In a multi-recycled retrogene, it seems reasonable to assume that the DinoSL closest to the coding region represents an early retroposition event while the one further away represents a more recent retroposition event—upstream DinoSL sequences would thus be younger than the downstream ones. We find DinoSLs located at −50 to −100 bp are the most highly conserved (fig. 1C), as predicted, and note that this pattern of sequence conservation is thus not related to their ages. Furthermore, we also find an increased conservation of TTT(T/G) motifs. If the TTT(G) in the recycled DinoSL served as a promoter, the primary transcripts from the retrogenes would lack a DinoSL, and secondary recycled retrogenes would thus also contain only one DinoSL upstream of its coding sequence. Only when a retrogene was transcribed from a site upstream of the recycled DinoSL would a secondary retrogene with multiple DinoSLs be observed. This might be expected to be a relatively infrequent event, and indeed, the number of retrogenes with multiple DinoSLs is very small in both Symbiodinium genomes (table 1).
Our result regarding the conservation of DinoSL motifs appears different from what has been described in earlier reports conducted on ESTs (Slamovits and Keeling 2008; Jaeckisch etal. 2011), in which the authors found that the degree of conservation increases as a function of the distance between the DinoSL and the start codon. This difference is presumably attributable to the different data sets used in the two types of studies. In EST studies, the analyses were performed on 5′-UTRs, which are necessarily downstream of the promoter, and any DinoSLs upstream of the transcriptional start site are simply not present. Genomic sequence, on the other hand, allows these upstream DinoSLs to be observed. Since the sequence actually used as a promoter is likely to be more conserved independent of where it is located relative to the start codon, we suggest that the poorly conserved DinoSL sequences further upstream in the genome are not used as a promoter element and are thus less conserved (supplementary fig. S1, Supplementary Material online). Of the two factors affecting the degree of DinoSL sequence conservation, one the age of the sequence and the other the selective pressure exerted by its ability to function as a promoter, only the age of the sequence can be observed in the EST data set. We found the highest degree of conservation in DinoSLs located between −50 and −100 (fig. 1D), a location where dinoflagellate promoters are likely to be found based on the usual length of 5′-UTR (Zhang etal. 2007; Kim etal. 2011).
Our analysis of dinoflagellate retrogenes supports the initially described model of dinoflagellate retroposition (Slamovits and Keeling 2008), a model which differs from that in humans and other model organisms. In dinoflagellates, genes are transcribed from active regions and the transcripts are rapidly capped with DinoSL. Although most of these DinoSL-capped transcripts subsequently have the introns removed and are exported to the cytoplasm for translation, some appear to have been rapidly reverse transcribed and integrated into the genome. The TTT(T/G) motif in the DinoSL cap may then serve as a basal promoter to facilitate the survival of retrogenes in the genome (fig. 4A).
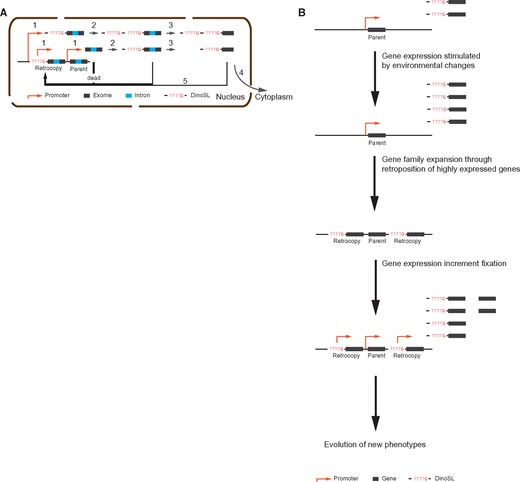
—The retroposition process in dinoflagellate. (A) Gene retroposition process inferred from the characteristics of retrogenes. A gene transcript (step 1) is trans-spliced through which the DinoSL sequence is added to the 5′ terminus of a nascent pre-mRNA (step 2), followed by cis-splicing (step 3) before they were exported to cytoplasm for translation (step 4); Reverse transcription and genome integration (step 5) can occur at either of these steps inside nucleus (step 1, 2, 3 and 4); in dinoflagellates, the DinoSL sequence harboring core promoter motif “TTTT” and “TTTG” potentially serves as a promoter enabling the survival of retrogenes; therefore, most retrocopies stemmed from nascent transcripts (step 1) would be dead upon their birth due to the lack of promoter; the whole procedure is limited in the nucleus. (B) A scheme showing the self-enforcing model enabling the fixation of the increased gene activities.
It is unknown what protein or mechanism mediated the reverse transcription. The GO category “RNA-dependent DNA replication” was enriched during both the retroposition bouts, suggesting retrotransposons might be the major driver of retroposition. Lee etal. (2014) proposed the LTR-retrotransposon, Ty1/copia, as a candidate but direct evidence is still lacking (Lee etal. 2014). Retroposition in the human genome is mediated by LINE1 (Pavlicek etal. 2007). Pavlicek et al. proposed that mRNA with higher stability, smaller length, and which is translated on free as opposed to bound ribosomes will have a higher chance of retroposition. However, we did not find that any of these terms affect dinoflagellate retrogenes suggesting that machinery mediating retroposition differs from that in humans. Furthermore, we found that some retrogenes appeared to have retained the introns from their parents. Although these retrogenes were filtered out in our downstream analysis in order to exclude possible paralog duplicates in the genome, the presence of these genes suggests that retroposition could happen before or during the splicing process of introns. This could also account for the fact that retrogenes frequently insert into a locus near their parents. If true, the retroposition process in dinoflagellate is likely to occur before the transcripts are exported to the cytoplasm.
Ancient Gene Activities Predicted by the Number of Retrogenes
Both a previous report (Pavlicek etal. 2007) and our own data (table 2) indicate that more highly expressed genes have a greater chance to be retroposed. As a result, more retrocopies in a gene family born, for example, during the second retroposition bout, suggest that this family was actively transcribed at that time. This, in principle, provides an estimate of the transcript profiles at the time of the first and second retroposition bout. To attempt to predict cell behavior from these transcript profiles, we categorized the retroposed genes into GO biological processes and performed an enrichment analysis. These results showed that biological processes related to retroposition, such as “RNA-dependent DNA replication” and “DNA replication,” were activated at both the bouts, as expected. There is also enrichment in the “response to stress” at both the bouts of retroposition, supporting the idea that retroposition may have been stimulated by environmental changes. In line with our conclusion that the second retroposition occurred after the emergence of Symbiodinium, processes related to photosynthesis, carbon fixation (i.e., photosystem II stabilization, ATP synthesis coupled proton transport), and symbiosis establishment were enriched at the second bout. The expansion and diversification of these families may have facilitated the radiation of Symbiodinium strains into new hosts (fig. 2B). We have previously reported (Lin etal. 2015) that the biosynthesis pathway of the N-Glycans essential for host recognition had diverged between S. kawagutii and S. minutum. Interestingly, several important transport processes including “transmembrane transport” and “ion transport” were enriched during the first bout. This suggests the cell may have been ready for the evolution of symbiotic capacities.
A Self-Enforcing Mechanism Is Adopted by the Dinoflagellate Genome
The retroposition of highly expressed genes may have had the effect of “fixing” an increased level of gene expression by increasing its gene copy in the genome. This is likely to be particularly acute for any genes stimulated by the same factors that activate the retroposition process. Our data implies that some dramatic environmental changes had led to the increased levels of retroposition, and other genes whose expression levels increased in response to these changes would thus also have a higher chance of retroposition. The increased abundance of transcripts of these genes, when fixed in the genome, could then promote the evolution of new adaptive phenotypes (fig. 4B). This mechanism presumably enabled dinoflagellate genomes to accumulate genes engaged in the resistance against environmental stresses, as observed in the two sequenced dinoflagellate genomes.
In summary, gene retroposition is widespread in the two Symbiodinium genomes examined here. We suggest that preservation of the retroposed genes may be facilitated by trans-splicing of a leader sequence containing a basal transcriptional promoter element to the 5′-UTR of the mRNAs. Our analysis of the time when retroposition was most active during the evolutionary history of Symbiodinium genomes reveals two major bouts of gene retroposition, one during the emergence of Symbiodinium and the second during its radiation among different hosts. The retroposition events and the retrogenes enriched during these processes likely played critical roles in driving genome evolution, especially genome expansion and shaping of the phenotype. This unusual evolutionary mechanism for dinoflagellate genome evolution may have been driven by environmental factors stimulating gene expression, followed by fixation of the increased transcript levels due to an increase in the number of gene copies per gene family (fig. 4B). It appears that large-scale retroposition, instead of WGD or SD, has been the major mechanism driving the evolution of dinoflagellate genomes.
Data and Methods
Genome and Transcriptome Data
Two dinoflagellate genomes (Shoguchi etal. 2013; Lin etal. 2015) were released recently, enabling us to perform a deep analysis of this group using comparative genomic approaches. We collected their genomic and transcriptomic data from the NCBI database (https://www.ncbi.nlm.nih.gov/; last accessed August 3, 2017). The S. kawagutii genomic and transcriptomic data (Lin etal. 2015) are available from the NCBI database under the accession of SRA148697 and SRR1300303. The genome assembly and transcriptome reads of S. minutum (Shoguchi etal. 2013) are available from NCBI using accession PRJDB2108.
Detection and Analysis of SL Relicts in Symbiodinium Genomes
The canonical DinoSL sequence has 22 base pairs (DCCGUAGCCAUUUUGGCUCAAG, [D = U, A or G]) (Zhang etal. 2007). The first nucleotide is degenerate and hence was trimmed off to search for DinoSL relicts. This search was performed by BLASTn against the 500-bp sequences upstream of the ORFs with the parameters empirically set as “-e 20,000 -W 4 -G 0 -E 2 -q -1 -n T” to detect as the shortest hit a 9-mer DinoSL sequence with no mismatches or gaps. A 9-bp-long DinoSL relict appears in a 500-bp region at a probability of 0.023, which is sufficiently small to reject the null hypothesis that the relict SL is a random array of nucleotides.
Pseudogene fragments were identified by BLASTx searches. Briefly, intact gene models were masked before the genome sequence was queried by protein sequences of both S. kawagutii and S. minutum using BLASTx with an e-value cutoff of 1e-5. Sequences with >75% identity were retained. Fragments were linked together as a pseudogene if they were unidirectionally oriented and were separated by <1 kb. Since we could not accurately determine the transcription start points of these pseudogenes, a longer sequence (1,000 bp) was retrieved from the upstream of each pseudogene as its promoter region. Potential presence of DinoSL relicts in these pseudo-promoters was examined as described earlier.
Identification of Parental Genes and the Estimation of Synonymous Divergence (Ks)
An all-against-all BLASTn search was performed with each of the analyzed genomes to find the best hit for each gene. Reciprocal best hit genes were paired and parental genes were identified according to the following criteria: 1) the identity between retrogene and the parental copy is >60% and the aligned length longer than 50% of the shorter sequence; 2) the parental gene has fewer DinoSL relicts than the retrogene; 3) the retrogene should not retain >40% of the parental introns. Introns from retrogenes and parent genes were blasted against each other. Retrogene introns with >60% identity to any of the parental introns was counted as a retained intron. The number of retained introns was <40% of the number of parental and retrogene introns.
Once the retrogene and its parental copy was successfully paired, the synonymous divergence (the number of substitutions per synonymous site, Ks) for each pair was estimated using kaks_calculator (Wang etal. 2010). Any Ks with a P > 0.01 was excluded from further analyses. A total of 843 S. kawagutii and 599 S. minutum gene pairs with synonymous divergence information were retained. We plotted the distribution of Ks for both S. kawagutii and S. minutum gene pairs, and the two peaks apparent in this plot suggested two major episodes of retroposition during the genome evolution (fig. 2A and B). The retrogenes were then categorized into two groups according to their Ks values using the mclust package of R.
To estimate the time of these two episodes, we calculated the mutation speed at synonymous sites according to the timeline of the emergence of Symbiodinium species. Orthologous genes between S. kawagutii and S. minutum were clustered into syntenic blocks by MCScanX (Wang etal. 2012) after an all-vs-all BLAST, followed by the calculation of synonymous divergence between gene pairs using the kaks_calculator with the same settings as above. The substitution speed was then calculated based on the time of divergence between S. kawagutii (clade F) and S. minutum (clade B), estimated at ∼32 Ma (Pochon etal. 2006). This yielded a substitution rate of 5.16 × 10−8 Ks per site per year, which was subsequently used to calculate the time of the two retroposition episodes.
Functional Analysis
Because only a small fraction of the retrogenes found can be accurately categorized into either of the episodes (due to the lack of identified parent copies for most retrogenes), to perform the functional analysis of the gene retroposition events, genes from both the Symbiodinium genomes were pooled and clustered from reciprocal BLAST (blastp, e-value cutoff, 10−5) results using OrthoMCL with default parameters (Li etal. 2003). For each family, the copy number of retrogenes in group 1 or 2 were counted and normalized by the total number of retrogenes in each episode to represent its ancient transcriptional activity when retroposition occurred. The present activity for each family was estimated by reads per kilobase per million mapped reads (RPKM) calculated from RNA-seq reads (Shoguchi etal. 2013; Lin etal. 2015). The Spearman’s rank correlation coefficient between ancient and present gene activities was computed using a package in R.
To identify the biological processes that were active when gene retroposition occurred, we retrieved Gene Ontology (GO) terms for each gene from its corresponding InterPro entry. The ancient (bout 1 or 2) activity for a given GO biological process was measured by the P value of enrichment analyses, with GO processes with P values <0.05 assigned as active processes. The GO enrichment analysis was conducted using a hypergeometric test followed by a Bonferroni correction to filter out potential false enrichments.
Comparison of Structure Features of Retrogenes
Where P(N) represents the frequency of base N in a given sequence. Protein solubility was predicted using SOLpro, a sequence-based predictor (Magnan etal. 2009).
The differences of the tested terms were analyzed by Student’s t test after a Shapiro–Wilk test using the t.test and shapiro.test R packages, respectively. Effect sizes (Sullivan and Feinn 2012) were also calculated because a large sampling size tends to demonstrate a significant difference. The effect size Cohen’s d was calculated following the formula: d = (M1−M2)/s, where M1 and M2 are means of the two compared groups and s is the standard deviation of either group.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Author Contributions
B.S. and S.L. conceived the work. B.S., S.L., and D.M. wrote and revised the manuscript. B.S. conducted the search and analyses of retrogenes. Y.S. and Y.F. collected the data and participated in the evolutionary analyses of retrogenes. X.L., W.W., S.C., W.C., and X.L. participated in retrogene functional analyses.
Acknowledgments
We thank anonymous referees for their critical suggestions. This study was supported by National Natural Science Foundation of China under the grant #31601042 (to B.S.), #31661143029 and #41330959 (to S.L.), National Key R&D Program of China 2016YFA0601202 and 2017YFC1404302 (to S.L.), Shenzhen Municipal Government of China (JCYJ20151015162041454 to W.C. and JCYJ20150529150505656 to X.L.), and China Postdoctoral Science Foundation under the grant #2017M610542 (to B.S.).
Literature Cited
Author notes
Associate editor: John Archibald

{kind=link}
{kind=link}
{kind=link}
{kind=link}